
DNA Damage by Radiopharmaceuticals and Mechanisms of Cellular Repair

 ,
,  , ,
, ,
Abstract
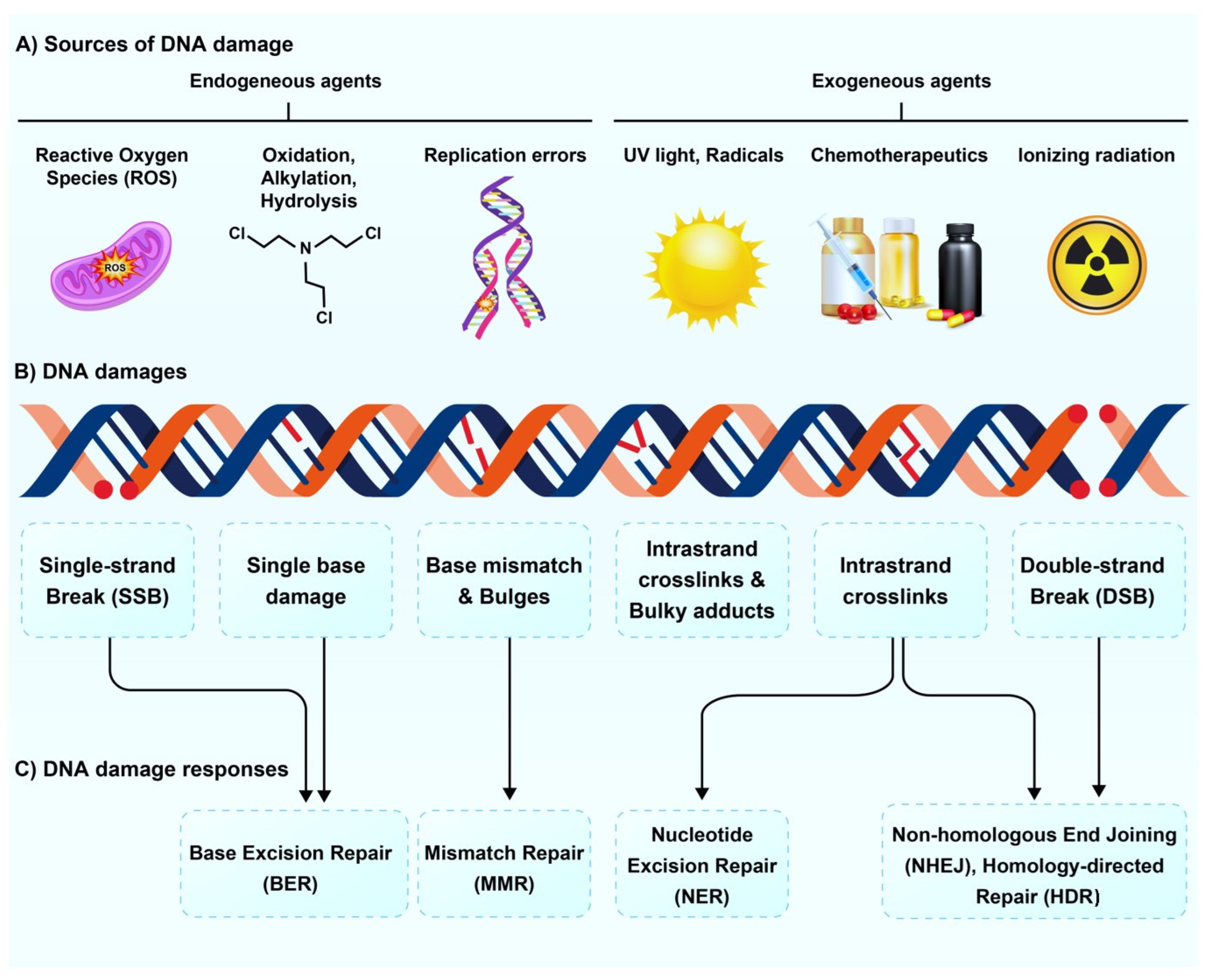
:1. Introduction
2. Effect of Radioisotopes on DNA Damage and Repair Pathways
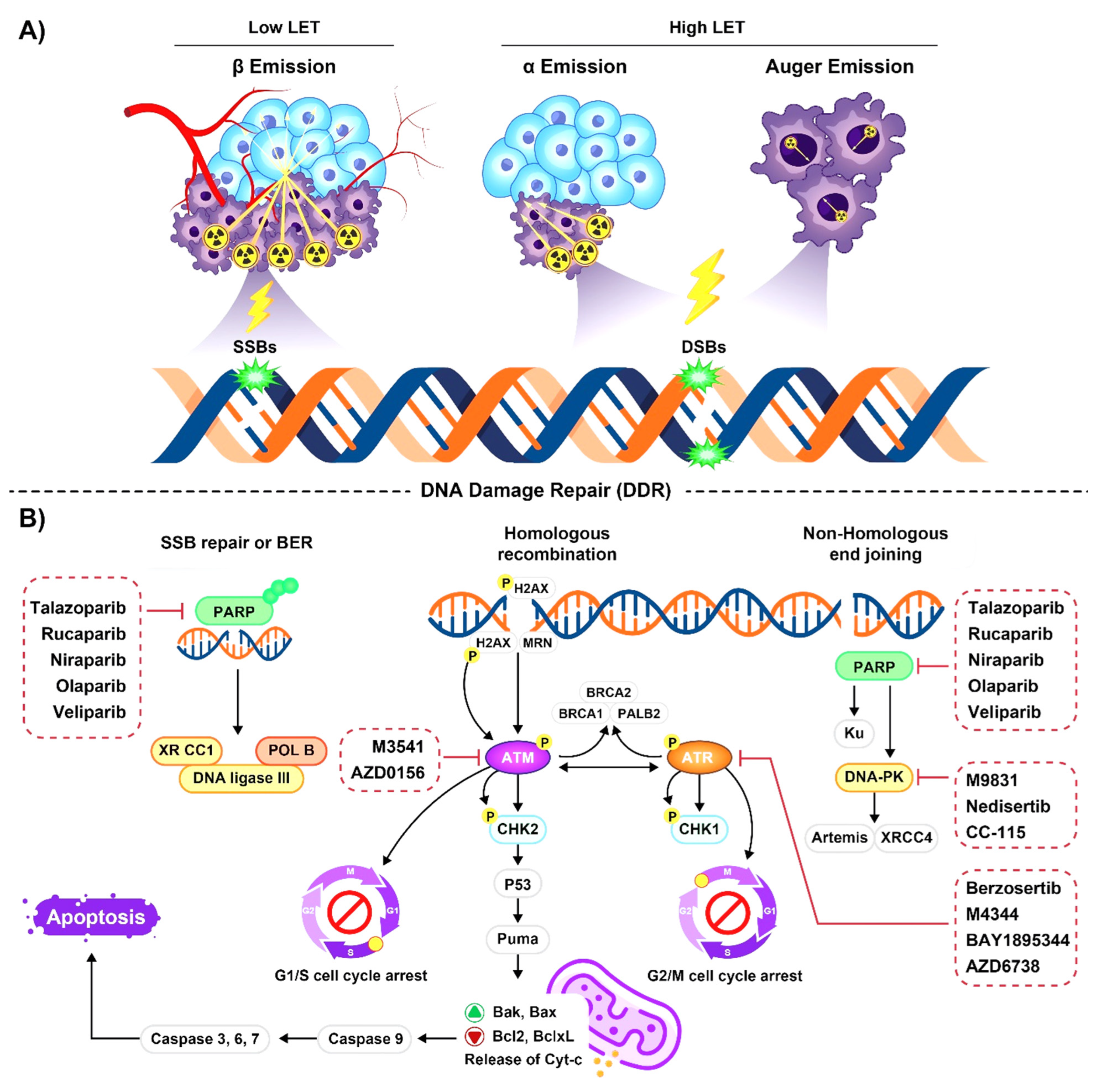
2.1. Emission Properties of Radioisotopes Used for Radiopharmaceutical Therapy
2.2. Radioisotopes Emitting High-LET Particles
2.2.1. Alpha-Particle Emitters
2.2.2. Auger Emitters
2.3. Radioisotopes Emitting Low-LET Particles
Beta Particle Emitters

{kind=link}
{kind=link}
| Radioisotopes | Emitting | Labeled | Mechanism of DNA Damage | DNA Repair Pathways | Biomarkers | Ref. |
|---|---|---|---|---|---|---|
| 223Ra | α-emitter | - | DNA DSB and clustered DNA damage | NHEJ |
| [52,53,54,58] |
| 212Pb | α-emitter | - | DNA DSB | HR |
| [59,60,61] |
| HER2 | ||||||
| TCMC | ||||||
| 213Bi | α-emitter | E-cadherin | - | - |
| [62,63,64] |
| CD20 |
| |||||
| CD45 | Irreversible DNA DSB | NHEJ |
| |||
| 125I | AE | - | DNA DSB | - | - | [100,108,109] |
| CEA | DNA DSB and ROS-mediated pathway | - | Enhanced formation of 53BP1 and γ-H2AX foci | |||
| 111In | AE | γH2AX | Lethal DNA DSB damage | - | Enhanced formation of ɤ-H2AX foci | [103,104,106,107] |
| Anti-CD33 | ||||||
| anti-HER2 | Induced significant DNA DSBs | |||||
| 99mTc | AE | HYNIC-DAPI | Induced SSBs and DSBs via a direct interaction with DNA | - | - | [99] |
| 177Lu | β-emitter | DOTATATE | Induction of indirect DNA damage through ROS generation and formation of SSBs | - | Slightly increased γH2AX and pATM | [137] |
| DOTATATE and DOTATOC | time- and dose-dependent induction of DNA-DSBs | Enhanced formation of γ-H2AX and 53BP1 nuclear foci | [126,135,136] | |||
| PSMA | ||||||
| HER2 | DNA DSBs | NEHJ |
| [138] | ||
| DOTA-JR11, SSTR antagonist DOTA-octreotide, SSTR agonist | Reversable DNA DSBs | - | Enhanced the formation of 53BP1 and γ-H2AX foci | [132] | ||
| EDTMP and DOTMP | - |
| [143] | |||
| Minigastrin analog | - | Activation of DNA damage response by p53 | - | [145] | ||
| 90Y | β-emitter | - | Induction of indirect DNA damage through ROS generation and formation of SSBs | NEHJ | - | [68,123] |
| 131I | β-emitter | - | Induction of indirect DNA damage through ROS | -- |
| [153,154] |
| 89Sr | β-emitter | - | - | - |
| [141,151] |
3. The Role of DNA Damage Repair Pathways in Response to Radiopharmaceuticals
4. Combination of Radiopharmaceuticals and DNA Damage Repair Inhibitors
5. Impact of Dose Rate on DNA Damage and Repair
6. Knowledge Gap
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef]
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage—How and why we age? eLife 2021, 10, e62852. [Google Scholar] [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- De Bont, R.; Van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef]
- Theodorakis, C. Mutagenesis. In Encyclopedia of Ecology; Elsevier: Oxford, UK, 2008; Volume 5, pp. 2475–2484. [Google Scholar] [CrossRef]
- Friedberg, E.C. DNA damage and repair. Nature 2003, 421, 436–440. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.-E.; Malki, M.I. DNA damage/repair management in cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef]
- Santivasi, W.L.; Xia, F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid. Redox Signal. 2014, 21, 251–259. [Google Scholar] [CrossRef]
- Abdulrahman, G.O.; Curtin, N. Targeting DNA damage response pathways in cancer. In Comprehensive Medicinal Chemistry III; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Asadian, S.; Mirzaei, H.; Kalantari, B.A.; Davarpanah, M.R.; Mohamadi, M.; Shpichka, A.; Nasehi, L.; Es, H.A.; Timashev, P.; Najimi, M. β-radiating radionuclides in cancer treatment, novel insight into promising approach. Pharmacol. Res. 2020, 160, 105070. [Google Scholar] [CrossRef]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- Salih, S.; Alkatheeri, A.; Alomaim, W.; Elliyanti, A. Radiopharmaceutical treatments for cancer therapy, radionuclides characteristics, applications, and challenges. Molecules 2022, 27, 5231. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves first PSMA-targeted radiopharmaceutical. Nat. Rev. Drug Discov. 2022, 21, 327. [Google Scholar] [CrossRef]
- Menegakis, A.; De Colle, C.; Yaromina, A.; Hennenlotter, J.; Stenzl, A.; Scharpf, M.; Fend, F.; Noell, S.; Tatagiba, M.; Brucker, S. Residual γH2AX foci after ex vivo irradiation of patient samples with known tumour-type specific differences in radio-responsiveness. Radiother. Oncol. 2015, 116, 480–485. [Google Scholar] [CrossRef]
- Löbrich, M.; Shibata, A.; Beucher, A.; Fisher, A.; Ensminger, M.; Goodarzi, A.A.; Barton, O.; Jeggo, P.A. γH2AX foci analysis for monitoring DNA double-strand break repair: Strengths, limitations and optimization. Cell Cycle (Georget. Tex.) 2010, 9, 662–669. [Google Scholar] [CrossRef]
- Redon, C.E.; Nakamura, A.J.; Sordet, O.; Dickey, J.S.; Gouliaeva, K.; Tabb, B.; Lawrence, S.; Kinders, R.J.; Bonner, W.M.; Sedelnikova, O.A. γ-H2AX detection in peripheral blood lymphocytes, splenocytes, bone marrow, xenografts, and skin. In DNA Damage Detection In Situ, Ex Vivo, and In Vivo; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2011; pp. 249–270. [Google Scholar]
- Stenvall, A.; Larsson, E.; Holmqvist, B.; Strand, S.-E.; Jönsson, B.-A. Quantitative γ-H2AX immunofluorescence method for DNA double-strand break analysis in testis and liver after intravenous administration of 111InCl3. EJNMMI Res. 2020, 10, 22. [Google Scholar] [CrossRef]
- Barbieri, S.; Baiocco, G.; Babini, G.; Morini, J.; Friedland, W.; Buonanno, M.; Grilj, V.; Brenner, D.J.; Ottolenghi, A. Modelling γ-H2AX foci induction to mimic limitations in the scoring technique. Radiat. Prot. Dosim. 2019, 183, 121–125. [Google Scholar] [CrossRef]
- Wilde, S.; Dambowsky, M.; Hempt, C.; Sutter, A.; Queisser, N. Classification of in vitro genotoxicants using a novel multiplexed biomarker assay compared to the flow cytometric micronucleus test. Environ. Mol. Mutagen. 2017, 58, 662–677. [Google Scholar] [CrossRef]
- Ji, J.; Zhang, Y.; Redon, C.E.; Reinhold, W.C.; Chen, A.P.; Fogli, L.K.; Holbeck, S.L.; Parchment, R.E.; Hollingshead, M.; Tomaszewski, J.E. Phosphorylated fraction of H2AX as a measurement for DNA damage in cancer cells and potential applications of a novel assay. PLoS ONE 2017, 12, e0171582. [Google Scholar] [CrossRef]
- Kumar, C.; Shetake, N.; Desai, S.; Kumar, A.; Samuel, G.; Pandey, B.N. Relevance of radiobiological concepts in radionuclide therapy of cancer. Int. J. Radiat. Biol. 2016, 92, 173–186. [Google Scholar] [CrossRef]
- Ferrier, M.G.; Radchenko, V. An appendix of radionuclides used in targeted alpha therapy. J. Med. Imaging Radiat. Sci. 2019, 50, S58–S65. [Google Scholar] [CrossRef]
- Yeong, C.-H.; Cheng, M.-h.; Ng, K.-H. Therapeutic radionuclides in nuclear medicine: Current and future prospects. J. Zhejiang Univ. Sci. B 2014, 15, 845. [Google Scholar] [CrossRef]
- Goldsmith, S.J. Targeted radionuclide therapy: A historical and personal review. Semin. Nucl. Med. 2020, 50, 87–97. [Google Scholar] [CrossRef]
- Widel, M.; Przybyszewski, W.M.; Cieslar-Pobuda, A.; Saenko, Y.V.; Rzeszowska-Wolny, J. Bystander normal human fibroblasts reduce damage response in radiation targeted cancer cells through intercellular ROS level modulation. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2012, 731, 117–124. [Google Scholar] [CrossRef]
- Thamboo, T.P.; Tan, K.B.; Wang, S.C.; Salto-Tellez, M. Extra-hepatic embolisation of Y-90 microspheres from selective internal radiation therapy (SIRT) of the liver. Pathology 2003, 35, 351–353. [Google Scholar]
- Arslan, N.; Emi, M.; Alagöz, E.; Üstünsöz, B.; Oysul, K.; Arpacı, F.; Uğurel, Ş.; Beyzadeoğlu, M.; Özgüven, A.M. Selective intraarterial radionuclide therapy with Yttrium-90 (Y-90) microspheres for hepatic neuroendocrine metastases: Initial experience at a single center. Vojnosanit. Pregl. 2011, 68, 341–348. [Google Scholar] [CrossRef]
- Brady, D.; O’Sullivan, J.M.; Prise, K.M. What is the role of the bystander response in radionuclide therapies? Front. Oncol. 2013, 3, 215. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; de Toledo, S.M.; Gooding, T.; Little, J.B. Intercellular communication is involved in the bystander regulation of gene expression in human cells exposed to very low fluences of alpha particles. Radiat. Res. 1998, 150, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Persaud, R.; Zhou, H.; Hei, T.K.; Hall, E.J. Demonstration of a radiation-induced bystander effect for low dose low LET β-particles. Radiat. Environ. Biophys. 2007, 46, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Mamlouk, O.; Balagurumoorthy, P.; Wang, K.; Adelstein, S.J.; Kassis, A.I. Bystander effect in tumor cells produced by Iodine-125 labeled human lymphocytes. Int. J. Radiat. Biol. 2012, 88, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.; Sorensen, A.; McCluskey, A.G.; Mairs, R.J. Radiation quality-dependent bystander effects elicited by targeted radionuclides. J. Pharm. Pharmacol. 2008, 60, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Mairs, R.J.; Fullerton, N.E.; Zalutsky, M.R.; Boyd, M. Targeted radiotherapy: Microgray doses and the bystander effect. Dose-Response 2007, 5. [Google Scholar] [CrossRef]
- Ault, M.R. Gamma emitting isotopes of medical origin detected in sanitary waste samples. Radiat. Prot. Manag. 1989, 6, 48–52. [Google Scholar]
- International Commission on Radiation Units and Measurements. Stopping Powers and Ranges for Protons and Alpha Particles; International Commission on Radiation Units and Measurements: Bethesda, MD, USA, 1993. [Google Scholar]
- Kassis, A.; Harris, C.; Adelstein, S. The in vitro radiobiology of astatine-211 decay. Radiat. Res. 1986, 105, 27–36. [Google Scholar] [CrossRef]
- Parker, C.; Heidenreich, A.; Nilsson, S.; Shore, N. Current approaches to incorporation of radium-223 in clinical practice. Prostate Cancer Prostatic Dis. 2018, 21, 37–47. [Google Scholar] [CrossRef]
- Dandapani, S.V.; Wong, J.; Twardowski, P. Review of Radium-223 and Metastatic Castration-Sensitive Prostate Cancer. Cancer Biother. Radiopharm. 2020, 35, 490–496. [Google Scholar] [CrossRef]
- Etchebehere, E.; Brito, A.E.; Rezaee, A.; Langsteger, W.; Beheshti, M. Therapy assessment of bone metastatic disease in the era of 223radium. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 84–96. [Google Scholar] [CrossRef]
- Ng, T.S.C.; Gao, X.; Salari, K.; Zlatev, D.V.; Heidari, P.; Kamran, S.C. Incorporating PSMA-Targeting Theranostics Into Personalized Prostate Cancer Treatment: A Multidisciplinary Perspective. Front. Oncol. 2021, 11, 722277. [Google Scholar] [CrossRef] [PubMed]
- Roots, R.; Holley, W.; Chatterjee, A.; Irizarry, M.; Kraft, G. The formation of strand breaks in DNA after high-LET irradiation: A comparison of data from in vitro and cellular systems. Int. J. Radiat. Biol. 1990, 58, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Bennett, P.; Oza, U.D. Metastatic Bone Tumor Therapy. In Diagnostic Imaging: Nuclear Medicine, 2nd ed.; Bennett, P., Oza, U.D., Trout, A.T., Mintz, A., Eds.; Elsevier: Philadelphia, PA, USA, 2016; pp. 482–485. [Google Scholar]
- Mulford, D.A.; Scheinberg, D.A.; Jurcic, J.G. The promise of targeted α-particle therapy. J. Nucl. Med. 2005, 46, 199S–204S. [Google Scholar] [PubMed]
- Sgouros, G. Dosimetry, radiobiology and synthetic lethality: Radiopharmaceutical therapy (RPT) with alpha-particle-emitters. Semin. Nucl. Med. 2020, 50, 124–132. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Bigner, D.D. Radioimmunotherapy with α-particle emitting radioimmunoconjugates. Acta Oncol. 1996, 35, 373–379. [Google Scholar] [CrossRef]
- Narayanan, P.; Goodwin, E.H.; Lehnert, B. α particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res. 1997, 57, 3963–3971. [Google Scholar]
- Suominen, M.I.; Fagerlund, K.M.; Rissanen, J.P.; Konkol, Y.M.; Morko, J.P.; Peng, Z.; Alhoniemi, E.J.; Laine, S.K.; Corey, E.; Mumberg, D. Radium-223 inhibits osseous prostate cancer growth by dual targeting of cancer cells and bone microenvironment in mouse models. Clin. Cancer Res. 2017, 23, 4335–4346. [Google Scholar] [CrossRef]
- Schumann, S.; Eberlein, U.; Lapa, C.; Müller, J.; Serfling, S.; Lassmann, M.; Scherthan, H. α-Particle-induced DNA damage tracks in peripheral blood mononuclear cells of [223Ra]RaCl2-treated prostate cancer patients. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2761–2770. [Google Scholar] [CrossRef]
- Abramenkovs, A.; Hariri, M.; Spiegelberg, D.; Nilsson, S.; Stenerlöw, B. Ra-223 induces clustered DNA damage and inhibits cell survival in several prostate cancer cell lines. Transl. Oncol. 2022, 26, 101543. [Google Scholar] [CrossRef]
- Franken, N.A.; Hovingh, S.; Ten Cate, R.; Krawczyk, P.; Stap, J.; Hoebe, R.; Aten, J.; Barendsen, G.W. Relative biological effectiveness of high linear energy transfer α-particles for the induction of DNA-double-strand breaks, chromosome aberrations and reproductive cell death in SW-1573 lung tumour cells. Oncol. Rep. 2012, 27, 769–774. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Kashino, G.; Ono, K.; Watanabe, M. Phosphorylated H2AX foci in tumor cells have no correlation with their radiation sensitivities. J. Radiat. Res. 2009, 50, 151–160. [Google Scholar] [CrossRef]
- Schumann, S.; Eberlein, U.; Müller, J.; Scherthan, H.; Lassmann, M. Correlation of the absorbed dose to the blood and DNA damage in leukocytes after internal ex-vivo irradiation of blood samples with Ra-224. Ejnmmi Res. 2018, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Schumann, S.; Eberlein, U.; Muhtadi, R.; Lassmann, M.; Scherthan, H. DNA damage in leukocytes after internal ex-vivo irradiation of blood with the α-emitter Ra-223. Sci. Rep. 2018, 8, 2286. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.J.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W. 212Pb-Radioimmunotherapy Induces G2 Cell-Cycle Arrest and Delays DNA Damage Repair in Tumor Xenografts in a Model for Disseminated Intraperitoneal Disease. 212Pb-RIT-Induced Apoptosis in Tumor Xenografts. Mol. Cancer Ther. 2012, 11, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.J.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W. Sensitization of tumor to 212Pb radioimmunotherapy by gemcitabine involves initial abrogation of G2 arrest and blocked DNA damage repair by interference with Rad51. Int. J. Radiat. Oncol.*Biol.*Phys. 2013, 85, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.J.; Milenic, D.E.; Baidoo, K.E.; Kim, Y.S.; Brechbiel, M.W. Gene expression profiling upon 212Pb-TCMC-trastuzumab treatment in the LS-174T i.p. xenograft model. Cancer Med. 2013, 2, 646–653. [Google Scholar] [CrossRef]
- Seidl, C.; Port, M.; Gilbertz, K.-P.; Morgenstern, A.; Bruchertseifer, F.; Schwaiger, M.; Roper, B.; Senekowitsch-Schmidtke, R.; Abend, M. 213Bi-induced death of HSC45-M2 gastric cancer cells is characterized by G2 arrest and up-regulation of genes known to prevent apoptosis but induce necrosis and mitotic catastrophe. Mol. Cancer Ther. 2007, 6, 2346–2359. [Google Scholar] [CrossRef] [PubMed]
- Friesen, C.; Glatting, G.; Koop, B.; Schwarz, K.; Morgenstern, A.; Apostolidis, C.; Debatin, K.-M.; Reske, S.N. Breaking chemoresistance and radioresistance with [213Bi]anti-CD45 antibodies in leukemia cells. Cancer Res. 2007, 67, 1950–1958. [Google Scholar] [CrossRef]
- Roscher, M.; Hormann, I.; Leib, O.; Marx, S.; Moreno, J.; Miltner, E.; Friesen, C. Targeted alpha-therapy using [Bi-213] anti-CD20 as novel treatment option for radio-and chemoresistant non-Hodgkin lymphoma cells. Oncotarget 2013, 4, 218. [Google Scholar] [CrossRef]
- Rizvi, S.M.A.; Henniker, A.; Goozee, G.; Allen, B. In vitro testing of the leukaemia monoclonal antibody WM-53 labeled with alpha and beta emitting radioisotopes. Leuk. Res. 2002, 26, 37–43. [Google Scholar] [CrossRef]
- Zwicker, F.; Hauswald, H.; Debus, J.; Huber, P.E.; Weber, K.J. Impact of dimethyl sulfoxide on irradiation-related DNA double-strand-break induction, -repair and cell survival. Radiat. Environ. Biophys. 2019, 58, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Bajinskis, A.; Natarajan, A.T.; Erixon, K.; Harms-Ringdahl, M. DNA double strand breaks induced by the indirect effect of radiation are more efficiently repaired by non-homologous end joining compared to homologous recombination repair. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2013, 756, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Bajinskis, A.; Olsson, G.; Harms-Ringdahl, M. The indirect effect of radiation reduces the repair fidelity of NHEJ as verified in repair deficient CHO cell lines exposed to different radiation qualities and potassium bromate. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2012, 731, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; ten Cate, R.; Krawczyk, P.M.; Stap, J.; Haveman, J.; Aten, J.; Barendsen, G.W. Comparison of RBE values of high-LET α-particles for the induction of DNA-DSBs, chromosome aberrations and cell reproductive death. Radiat. Oncol. 2011, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988, 35, 95–125. [Google Scholar]
- Gollapalle, E.; Wang, R.; Adetolu, R.; Tsao, D.; Francisco, D.; Sigounas, G.; Georgakilas, A.G. Detection of oxidative clustered DNA lesions in X-irradiated mouse skin tissues and human MCF-7 breast cancer cells. Radiat. Res. 2007, 167, 207–216. [Google Scholar] [CrossRef]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2011, 711, 123–133. [Google Scholar] [CrossRef]
- Magnander, K.; Hultborn, R.; Claesson, K.; Elmroth, K. Clustered DNA damage in irradiated human diploid fibroblasts: Influence of chromatin organization. Radiat. Res. 2010, 173, 272–282. [Google Scholar] [CrossRef]
- Nikjoo, H.; O′Neill, P.; Goodhead, D.T.; Terrissol, M. Computational modelling of low-energy electron-induced DNA damage by early physical and chemical events. Int. J. Radiat. Biol. 1997, 71, 467–483. [Google Scholar] [CrossRef]
- Nikjoo, H.; O′Neill, P.; Wilson, W.E.; Goodhead, D.T. Computational approach for determining the spectrum of DNA damage induced by ionizing radiation. Radiat. Res. 2001, 156, 577–583. [Google Scholar] [CrossRef]
- Gillman, R.; Lopes Floro, K.; Wankell, M.; Hebbard, L. The role of DNA damage and repair in liver cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188493. [Google Scholar] [CrossRef]
- Watanabe, R.; Rahmanian, S.; Nikjoo, H. Spectrum of Radiation-Induced Clustered Non-DSB Damage—A Monte Carlo Track Structure Modeling and Calculations. Radiat. Res. 2015, 183, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.J.; Nickson, C.M.; Thompson, J.M.; Kacperek, A.; Hill, M.A.; Parsons, J.L. Complex DNA Damage Induced by High Linear Energy Transfer Alpha-Particles and Protons Triggers a Specific Cellular DNA Damage Response. Int. J. Radiat. Oncol.*Biol.*Phys. 2018, 100, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Aghamohammadi, S.; Goodhead, D.; Savage, J. Induction of sister chromatid exchanges (SCE) in G0 lymphocytes by plutonium-238 α-particles. Int. J. Radiat. Biol. 1988, 53, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Charlton, D.; Goodhead, D.; Wilson, W.; Paretzke, H. The deposition of energy in small cylindrical targets by high LET radiations. Radiat. Prot. Dosim. 1985, 13, 123–125. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Geuting, V.; Löbrich, M. The role of homologous recombination in radiation-induced double-strand break repair. Radiother. Oncol. 2011, 101, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat. Res./Rev. Mutat. Res. 2012, 751, 158–246. [Google Scholar] [CrossRef]
- Zhao, L.; Bao, C.; Shang, Y.; He, X.; Ma, C.; Lei, X.; Mi, D.; Sun, Y. The determinant of DNA repair pathway choices in ionising radiation-induced DNA double-strand breaks. BioMed Res. Int. 2020, 2020, 4834965. [Google Scholar] [CrossRef]
- Cucinotta, F.A.; Pluth, J.M.; Anderson, J.A.; Harper, J.V.; O’Neill, P. Biochemical kinetics model of DSB repair and induction of γ-H2AX foci by non-homologous end joining. Radiat. Res. 2008, 169, 214–222. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Aten, J.A.; Stap, J.; Krawczyk, P.M.; van Oven, C.H.; Hoebe, R.A.; Essers, J.; Kanaar, R. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science 2004, 303, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhang, Y.; Wang, J.; Wang, X.; Fan, D.; He, L.; Zhang, X.; Gao, Y.; Li, Q.; Chen, H. γ-H2AX/53BP1/pKAP-1 foci and their linear tracks induced by in vitro exposure to radon and its progeny in human peripheral blood lymphocytes. Sci. Rep. 2016, 6, 38295. [Google Scholar] [CrossRef]
- Goodhead, D.; Charlton, D. Analysis of high-LET radiation effects in terms of local energy deposition. Radiat. Prot. Dosim. 1985, 13, 253–258. [Google Scholar] [CrossRef]
- Auger, P. Sur les rayons β secondaires produits dans un gaz par des rayons X. CR Acad. Sci.(F) 1923, 177, 169. [Google Scholar]
- Ku, A.; Facca, V.J.; Cai, Z.; Reilly, R.M. Auger electrons for cancer therapy–a review. EJNMMI Radiopharm. Chem. 2019, 4, 27. [Google Scholar] [CrossRef]
- Kassis, A.I. Cancer therapy with Auger electrons: Are we almost there? J. Nucl. Med. 2003, 44, 1479–1481. [Google Scholar] [PubMed]
- Auger, P. The auger effect. Surf. Sci. 1975, 48, 1–8. [Google Scholar] [CrossRef]
- Valerie, K.; Povirk, L.F. Regulation and mechanisms of mammalian double-strand break repair. Oncogene 2003, 22, 5792–5812. [Google Scholar] [CrossRef]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef]
- Panyutin, I.G.; Neumann, R.D. Radioprobing of DNA: Distribution of DNA breaks produced by decay of 125I incorporated into a triplex-forming oligonucleotide correlates with geometry of the triplex. Nucleic Acids Res. 1997, 25, 883–887. [Google Scholar] [CrossRef]
- Reske, S.N.; Deisenhofer, S.; Glatting, G.; Zlatopolskiy, B.D.; Morgenroth, A.; Vogg, A.T.; Buck, A.K.; Friesen, C. 123I-ITdU–mediated nanoirradiation of DNA efficiently induces cell kill in HL60 leukemia cells and in doxorubicin-, β-, or γ-radiation–resistant cell lines. J. Nucl. Med. 2007, 48, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Lobachevsky, P.N.; Karagiannis, T.C.; Martin, R.F. Plasmid DNA breakage by decay of DNA-associated Auger electron emitters: Approaches to analysis of experimental data. Radiat. Res. 2004, 162, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Lobachevsky, P.; Martin, R. Iodine-125 decay in a synthetic oligodeoxynucleotide. II. The role of Auger electron irradiation compared to charge neutralization in DNA breakage. Radiat. Res. 2000, 153, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Kotzerke, J.; Punzet, R.; Runge, R.; Ferl, S.; Oehme, L.; Wunderlich, G.; Freudenberg, R. 99mTc-labeled HYNIC-DAPI causes plasmid DNA damage with high efficiency. PLoS ONE 2014, 9, e104653. [Google Scholar] [CrossRef] [PubMed]
- Kassis, A.; Sastry, K.; Adelstein, S. Kinetics of uptake, retention, and radiotoxicity of 125IUdR in mammalian cells: Implications of localized energy deposition by Auger processes. Radiat. Res. 1987, 109, 78–89. [Google Scholar] [CrossRef]
- Makrigiorgos, G.; Kassis, A.; Baranowska-Kortylewicz, J.; McElvany, K.; Welch, M.; Sastry, K.; Adelstein, S. Radiotoxicity of in V79 Cells: A Comparison with. Radiat. Res. 1989, 118, 532–544. [Google Scholar] [CrossRef]
- Chan, P.; Lisco, E.; Lisco, H.; Adelstein, S. The radiotoxicity of iodine-125 in mammalian cells II. A comparative study on cell survival and cytogenetic responses to 125IUdR, 131TUdR, and 3HTdR. Radiat. Res. 1976, 67, 332–343. [Google Scholar] [CrossRef]
- Costantini, D.L.; Hu, M.; Reilly, R.M. Peptide motifs for insertion of radiolabeled biomolecules into cells and routing to the nucleus for cancer imaging or radiotherapeutic applications. Cancer Biother. Radiopharm. 2008, 23, 3–24. [Google Scholar] [CrossRef]
- Cornelissen, B.; Darbar, S.; Kersemans, V.; Allen, D.; Falzone, N.; Barbeau, J.; Smart, S.; Vallis, K.A. Amplification of DNA damage by a γH2AX-targeted radiopharmaceutical. Nucl. Med. Biol. 2012, 39, 1142–1151. [Google Scholar] [CrossRef]
- Chen, P.; Wang, J.; Hope, K.; Jin, L.; Dick, J.; Cameron, R.; Brandwein, J.; Minden, M.; Reilly, R.M. Nuclear localizing sequences promote nuclear translocation and enhance the radiotoxicity of the anti-CD33 monoclonal antibody HuM195 labeled with 111In in human myeloid leukemia cells. J. Nucl. Med. 2006, 47, 827–836. [Google Scholar]
- Costantini, D.L.; Chan, C.; Cai, Z.; Vallis, K.A.; Reilly, R.M. 111In-labeled trastuzumab (Herceptin) modified with nuclear localization sequences (NLS): An Auger electron-emitting radiotherapeutic agent for HER2/neu-amplified breast cancer. J. Nucl. Med. 2007, 48, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Chen, Z.; Bailey, K.E.; Scollard, D.A.; Reilly, R.M.; Vallis, K.A. Relationship between induction of phosphorylated H2AX and survival in breast cancer cells exposed to 111In-DTPA-hEGF. J. Nucl. Med. 2008, 49, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Paillas, S.; Ladjohounlou, R.; Lozza, C.; Pichard, A.; Boudousq, V.; Jarlier, M.; Sevestre, S.; Le Blay, M.; Deshayes, E.; Sosabowski, J. Localized irradiation of cell membrane by auger electrons is cytotoxic through oxidative stress-mediated nontargeted effects. Antioxid. Redox Signal. 2016, 25, 467–484. [Google Scholar] [CrossRef]
- Paillas, S.; Boudousq, V.; Piron, B.; Kersual, N.; Bardiès, M.; Chouin, N.; Bascoul-Mollevi, C.; Arnaud, F.-X.; Pèlegrin, A.; Navarro-Teulon, I. Apoptosis and p53 are not involved in the anti-tumor efficacy of 125I-labeled monoclonal antibodies targeting the cell membrane. Nucl. Med. Biol. 2013, 40, 471–480. [Google Scholar] [CrossRef]
- Piroozfar, B.; Alirezapoor, B.; Sedeh, F.M.; Jalilian, A.R.; Mirzaei, M.; Raisali, G. Evaluation of DNA damage in a Her2+ cell line induced by an Auger-emitting immunoconjugate. Iran. J. Nucl. Med. 2016, 24, 107. [Google Scholar]
- Yasui, L.S.; Chen, K.; Wang, K.; Jones, T.P.; Caldwell, J.; Guse, D.; Kassis, A.I. Using Hoechst 33342 to target radioactivity to the cell nucleus. Radiat. Res. 2007, 167, 167–175. [Google Scholar] [CrossRef]
- Balagurumoorthy, P.; Wang, K.; Adelstein, S.J.; Kassis, A.I. DNA double-strand breaks induced by decay of 123I-labeled Hoechst 33342: Role of DNA topology. Int. J. Radiat. Biol. 2008, 84, 976–983. [Google Scholar] [CrossRef]
- Tanious, F.A.; Veal, J.M.; Buczak, H.; Ratmeyer, L.S.; Wilson, W.D. DAPI (4′,6-diamidino-2-phenylindole) binds differently to DNA and RNA: Minor-groove binding at AT sites and intercalation at AU sites. Biochemistry 1992, 31, 3103–3112. [Google Scholar] [CrossRef]
- Onoue, R.; Watanabe, H.; Ono, M. Hoechst-tagged radioiodinated BODIPY derivative for Auger-electron cancer therapy. Chem. Commun. 2023, 59, 928–931. [Google Scholar] [CrossRef]
- Åberg, T. Unified theory of Auger electron emission. Phys. Scr. 1992, 1992, 71. [Google Scholar] [CrossRef]
- Goorley, T.; Zamenhof, R.; Nikjoo, H. Calculated DNA damage from gadolinium Auger electrons and relation to dose distributions in a head phantom. Int. J. Radiat. Biol. 2004, 80, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Adelstein, S.J.; Kassis, A.I. Strand breaks in plasmid DNA following positional changes of Auger-electron-emitting radionuclides. Acta Oncol. 1996, 35, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; Kalen, J.D.; Hall, J. Report of Meeting Held to Discuss Existing and Future Radionuclide Requirements for the National Cancer Institute. Science Applications International Corporation. 2008. Available online: https://www.osti.gov/biblio/1299088 (accessed on 5 October 2023).
- Nordberg, E.; Orlova, A.; Friedman, M.; Tolmachev, V.; Ståhl, S.; Nilsson, F.Y.; Glimelius, B.; Carlsson, J. In vivo and in vitro uptake of 111In, delivered with the affibody molecule (ZEGFR:955)2, in EGFR expressing tumour cells. Oncol. Rep. 2008, 19, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Sabongi, J.G.; Gonçalves, M.C.P.; Alves, C.D.C.; Alves, J.; Scapulatempo-Neto, C.; Moriguchi, S.M. Lutetium 177-DOTA-TATE therapy for esthesioneuroblastoma: A case report. Exp. Ther. Med. 2016, 12, 3078–3082. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, S.V.; Shilyagina, N.Y.; Vodeneev, V.A.; Zvyagin, A.V. Targeted radionuclide therapy of human tumors. Int. J. Mol. Sci. 2015, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Rao, D.V.; Bouchet, L.G.; Bolch, W.E.; Howell, R.W. Protection by DMSO against cell death caused by intracellularly localized iodine-125, iodine-131 and polonium-210. Radiat. Res. 2000, 153, 416–427. [Google Scholar] [CrossRef]
- Filippi, L.; Schillaci, O.; Cianni, R.; Bagni, O. Yttrium-90 resin microspheres and their use in the treatment of intrahepatic cholangiocarcinoma. Future Oncol. 2018, 14, 809–818. [Google Scholar] [CrossRef]
- Lassmann, M.; Hänscheid, H.; Gassen, D.; Biko, J.; Meineke, V.; Reiners, C.; Scherthan, H. In vivo formation of γ-H2AX and 53BP1 DNA repair foci in blood cells after radioiodine therapy of differentiated thyroid cancer. J. Nucl. Med. 2010, 51, 1318–1325. [Google Scholar] [CrossRef]
- Eberlein, U.; Lassmann, M.; Bluemel, C.; Buck, A.; Nowak, C.; Meineke, V.; Scherthan, H. Investigation of early DNA damage after radioiodine therapy in patients with thyroid cancer using the gamma-H2AX focus assay. In Proceedings of the EANM’13: Annual Congress of the European Association of Nuclear Medicine, Lyon, France, 19–23 October 2013. [Google Scholar]
- Schumann, S.; Scherthan, H.; Lapa, C.; Serfling, S.; Muhtadi, R.; Lassmann, M.; Eberlein, U. DNA damage in blood leucocytes of prostate cancer patients during therapy with 177Lu-PSMA. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1723–1732. [Google Scholar] [CrossRef]
- Eberlein, U.; Nowak, C.; Bluemel, C.; Buck, A.K.; Werner, R.A.; Scherthan, H.; Lassmann, M. DNA damage in blood lymphocytes in patients after 177Lu peptide receptor radionuclide therapy. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1739–1749. [Google Scholar] [CrossRef]
- Octavia, M.G.; Oliveira, N.; Rodrigues, A.; Rueff, J.; Margarida, G.; Antonio, L.; Ferreira, T. Persistence of micronuclei in peripheral blood lymphocytes of thyroid cancer patients 24 months after treatment with 131I. In Proceedings of the LOWRAD International Conference on Low Dose Radiation Effects on Human Health and Environment, Budapest, Hungary, 17–20 October 2007. [Google Scholar]
- Stott Reynolds, T.; P Bandari, R.; Jiang, Z.; J Smith, C. Lutetium-177 labeled bombesin peptides for radionuclide therapy. Curr. Radiopharm. 2016, 9, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Panigone, S.; Nunn, A. Lutetium-177-labeled gastrin releasing peptide receptor binding analogs: A novel approach to radionuclide therapy. Q. J. Nucl. Med. Mol. Imaging 2006, 50, 310. [Google Scholar] [PubMed]
- Purohit, N.K.; Shah, R.G.; Adant, S.; Hoepfner, M.; Shah, G.M.; Beauregard, J.-M. Potentiation of 177Lu-octreotate peptide receptor radionuclide therapy of human neuroendocrine tumor cells by PARP inhibitor. Oncotarget 2018, 9, 24693. [Google Scholar] [CrossRef] [PubMed]
- Dalm, S.U.; Nonnekens, J.; Doeswijk, G.N.; de Blois, E.; van Gent, D.C.; Konijnenberg, M.W.; de Jong, M. Comparison of the therapeutic response to treatment with a 177Lu-labeled somatostatin receptor agonist and antagonist in preclinical models. J. Nucl. Med. 2016, 57, 260–265. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, A.; Iravani, A.; Saboury, B.; Jadvar, H.; Catalano, O.; Mahmood, U.; Heidari, P. Integrating Theranostics Into Patient Care Pathways: AJR Expert Panel Narrative Review. Am. J. Roentgenol. 2023, 220, 619–629. [Google Scholar] [CrossRef]
- Graf, F.; Fahrer, J.; Maus, S.; Morgenstern, A.; Bruchertseifer, F.; Venkatachalam, S.; Fottner, C.; Weber, M.M.; Huelsenbeck, J.; Schreckenberger, M. DNA double strand breaks as predictor of efficacy of the alpha-particle emitter Ac-225 and the electron emitter Lu-177 for somatostatin receptor targeted radiotherapy. PLoS ONE 2014, 9, e88239. [Google Scholar] [CrossRef]
- O’Neill, E.; Kersemans, V.; Allen, P.D.; Terry, S.Y.; Torres, J.B.; Mosley, M.; Smart, S.; Lee, B.Q.; Falzone, N.; Vallis, K.A. Imaging DNA damage repair in vivo after 177Lu-DOTATATE therapy. J. Nucl. Med. 2020, 61, 743–750. [Google Scholar] [CrossRef]
- Denoyer, D.; Lobachevsky, P.; Jackson, P.; Thompson, M.; Martin, O.A.; Hicks, R.J. Analysis of 177Lu-DOTA-octreotate therapy–induced DNA damage in peripheral blood lymphocytes of patients with neuroendocrine tumors. J. Nucl. Med. 2015, 56, 505–511. [Google Scholar] [CrossRef]
- Delbart, W.; Karabet, J.; Marin, G.; Penninckx, S.; Derrien, J.; Ghanem, G.E.; Flamen, P.; Wimana, Z. Understanding the Radiobiological Mechanisms Induced by 177Lu-DOTATATE in Comparison to External Beam Radiation Therapy. Int. J. Mol. Sci. 2022, 23, 12369. [Google Scholar] [CrossRef]
- Yong, K.J.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W. Mechanisms of cell killing response from low linear energy transfer (LET) radiation originating from 177Lu radioimmunotherapy targeting disseminated intraperitoneal tumor xenografts. Int. J. Mol. Sci. 2016, 17, 736. [Google Scholar] [CrossRef]
- Kumar, C. Beta radiation induces apoptosis in human histiocytic lymphoma cells. J. Radiat. Cancer Res. 2017, 8, 168. [Google Scholar] [CrossRef]
- Chinol, M.; Vallabhajosula, S.; Goldsmith, S.J.; Klein, M.J.; Deutsch, K.F.; Chinen, L.K.; Brodack, J.W.; Deutsch, E.A.; Watson, B.A.; Tofe, A.J. Chemistry and biological behavior of samarium-153 and rhenium-186-labeled hydroxyapatite particles: Potential radiopharmaceuticals for radiation synovectomy. J. Nucl. Med. 1993, 34, 1536–1542. [Google Scholar]
- Wang, C.; Wang, J.; Jiang, H.; Zhu, M.; Chen, B.; Bao, W. In vitro study on apoptosis induced by strontium-89 in human breast carcinoma cell line. J. Biomed. Biotechnol. 2011, 2011, 541487. [Google Scholar] [CrossRef]
- Semont, A.; Nowak, E.B.; Silva Lages, C.; Mathieu, C.; Mouthon, M.-A.; May, E.; Allemand, I.; Millet, P.; Boussin, F.D. Involvement of p53 and Fas/CD95 in murine neural progenitor cell response to ionizing irradiation. Oncogene 2004, 23, 8497–8508. [Google Scholar] [CrossRef]
- Kumar, C.; Sharma, R.; Das, T.; Korde, A.; Sarma, H.; Banerjee, S.; Dash, A. 177Lu-DOTMP induces G2/M cell cycle arrest and apoptosis in MG63 cell line. J. Label. Compd. Radiopharm. 2018, 61, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.; Korde, A.; V Kumari, K.; Das, T.; Samuel, G. Cellular toxicity and apoptosis studies in osteocarcinoma cells, a comparison of 177Lu-EDTMP and Lu-EDTMP. Curr. Radiopharm. 2013, 6, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Grzmil, M.; Boersema, P.; Sharma, A.; Blanc, A.; Imobersteg, S.; Pruschy, M.; Picotti, P.; Schibli, R.; Behe, M. Comparative analysis of cancer cell responses to targeted radionuclide therapy (TRT) and external beam radiotherapy (EBRT). J. Hematol. Oncol. 2022, 15, 123. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Blomberg, J.; Lindgren, T.; Löfroth, P.-O.; Johansson, L.; Riklund, K.; Stigbrand, T. Iodine-131 induces mitotic catastrophes and activates apoptotic pathways in HeLa Hep2 cells. Cancer Biother. Radiopharm. 2008, 23, 541–550. [Google Scholar] [PubMed]
- Khanna, A. DNA damage in cancer therapeutics: A boon or a curse? Cancer Res. 2015, 75, 2133–2138. [Google Scholar] [CrossRef]
- Friesen, C.; Lubatschofski, A.; Kotzerke, J.; Buchmann, I.; Reske, S.N.; Debatin, K.-M. Beta-irradiation used for systemic radioimmunotherapy induces apoptosis and activates apoptosis pathways in leukaemia cells. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1251–1261. [Google Scholar] [CrossRef]
- Szperka, M.; Connor, E.; Paape, M.; Williams, J.; Bannerman, D. Characterization of bovine FAS-associated death domain gene 1. Anim. Genet. 2005, 36, 63–66. [Google Scholar] [CrossRef]
- Narita, M.; Shimizu, S.; Ito, T.; Chittenden, T.; Lutz, R.J.; Matsuda, H.; Tsujimoto, Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 14681–14686. [Google Scholar] [CrossRef]
- Junning, Z.; Chengjiao, H.; Shoupeng, Z. The characteristics and mechanism of apoptosis in K562 cells induced by radionuclides strontium-89. J. Radiat. Res. Radiat. Process. 2003, 21, 77–80. [Google Scholar]
- Lee, J.; Yang, W.; Lee, M.-G.; Ryu, Y.; Park, J.; Shin, K.-H.; Kim, G.; Suh, C.; Seong, J.; Han, B. Effective local control of malignant melanoma by intratumoural injection of a beta-emitting radionuclide. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.; Jayakumar, S.; Pandey, B.N.; Samuel, G.; Venkatesh, M. Cellular and molecular effects of beta radiation from I-131 on human tumor cells: A comparison with gamma radiation. Curr. Radiopharm. 2014, 7, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, R.; Yu, Y.; Guo, K.; Hou, P.; Yu, M.; Liu, Y.; Yang, A. Iodine-131 induces apoptosis in HTori-3 human thyrocyte cell line and G2/M phase arrest in a p53-independent pathway. Mol. Med. Rep. 2015, 11, 3148–3154. [Google Scholar] [CrossRef]
- van der Doelen, M.J.; Velho, P.I.; Slootbeek, P.H.; Naga, S.P.; Bormann, M.; van Helvert, S.; Kroeze, L.I.; van Oort, I.M.; Gerritsen, W.R.; Antonarakis, E.S. Impact of DNA damage repair defects on response to radium-223 and overall survival in metastatic castration-resistant prostate cancer. Eur. J. Cancer 2020, 136, 16–24. [Google Scholar] [CrossRef]
- Velho, P.I.; Qazi, F.; Hassan, S.; Carducci, M.A.; Denmeade, S.R.; Markowski, M.C.; Thorek, D.L.; DeWeese, T.L.; Song, D.Y.; Tran, P.T. Efficacy of radium-223 in bone-metastatic castration-resistant prostate cancer with and without homologous repair gene defects. Eur. Urol. 2019, 76, 170–176. [Google Scholar] [CrossRef]
- Liu, A.J.; Kosiorek, H.E.; Ueberroth, B.E.; Jaeger, E.; Ledet, E.; Kendi, A.T.; Tzou, K.; Quevedo, F.; Choo, R.; Moore, C.N. The impact of genetic aberrations on response to radium-223 treatment for castration-resistant prostate cancer with bone metastases. Prostate 2022, 82, 1202–1209. [Google Scholar] [CrossRef]
- Paschalis, A.; Sheehan, B.; Riisnaes, R.; Rodrigues, D.N.; Gurel, B.; Bertan, C.; Ferreira, A.; Lambros, M.B.K.; Seed, G.; Yuan, W.; et al. Prostate-specific Membrane Antigen Heterogeneity and DNA Repair Defects in Prostate Cancer. Eur. Urol. 2019, 76, 469–478. [Google Scholar] [CrossRef]
- van der Doelen, M.J.; Mehra, N.; van Oort, I.M.; Looijen-Salamon, M.G.; Janssen, M.J.R.; Custers, J.A.E.; Slootbeek, P.H.J.; Kroeze, L.I.; Bruchertseifer, F.; Morgenstern, A.; et al. Clinical outcomes and molecular profiling of advanced metastatic castration-resistant prostate cancer patients treated with 225Ac-PSMA-617 targeted alpha-radiation therapy. Urol. Oncol. Semin. Orig. Investig. 2021, 39, 729.e7–729.e16. [Google Scholar] [CrossRef] [PubMed]
- Crumbaker, M.; Emmett, L.; Horvath, L.G.; Joshua, A.M. Exceptional response to 177Lutetium prostate-specific membrane antigen in prostate cancer harboring DNA repair defects. JCO Precis. Oncol. 2019, 3, 1–5. [Google Scholar] [CrossRef]
- Chen, S.; Xiong, G.; Wu, S.; Mo, J. Downregulation of apurinic/apyrimidinic endonuclease 1/redox factor-1 enhances the sensitivity of human pancreatic cancer cells to radiotherapy in vitro. Cancer Biother. Radiopharm. 2013, 28, 169–176. [Google Scholar] [PubMed]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Haro, K.J.; Scott, A.C.; Scheinberg, D.A. Mechanisms of resistance to high and low linear energy transfer radiation in myeloid leukemia cells. Blood J. Am. Soc. Hematol. 2012, 120, 2087–2097. [Google Scholar] [CrossRef]
- Yard, B.D.; Gopal, P.; Bannik, K.; Siemeister, G.; Hagemann, U.B.; Abazeed, M.E. Cellular and genetic determinants of the sensitivity of cancer to α-particle irradiation. Cancer Res. 2019, 79, 5640–5651. [Google Scholar] [CrossRef]
- King, A.P.; Lin, F.I.; Escorcia, F.E. Why bother with alpha particles? Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Berlin, A.; Lalonde, E.; Sykes, J.; Zafarana, G.; Chu, K.C.; Ramnarine, V.R.; Ishkanian, A.; Sendorek, D.H.; Pasic, I.; Lam, W.L. NBN gain is predictive for adverse outcome following image-guided radiotherapy for localized prostate cancer. Oncotarget 2014, 5, 11081. [Google Scholar] [CrossRef]
- Wilkins, A.; Dearnaley, D.; Somaiah, N. Genomic and histopathological tissue biomarkers that predict radiotherapy response in localised prostate cancer. BioMed Res. Int. 2015, 2015, 238757. [Google Scholar] [CrossRef]
- Bourton, E.C.; Ahorner, P.-A.; Plowman, P.N.; Zahir, S.A.; Al-Ali, H.; Parris, C.N. The PARP-1 inhibitor Olaparib suppresses BRCA1 protein levels, increases apoptosis and causes radiation hypersensitivity in BRCA1+/− lymphoblastoid cells. J. Cancer 2017, 8, 4048. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Heussel, C.-P.; Kazdal, D.; Endris, V.; Nientiedt, C.; Bruchertseifer, F.; Kippenberger, M.; Rathke, H.; Leichsenring, J. Patients resistant against PSMA-targeting α-radiation therapy often harbor mutations in DNA damage-repair–associated genes. J. Nucl. Med. 2020, 61, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, A.J. Science in focus: Combining radiotherapy with inhibitors of the DNA damage response. Clin. Oncol. 2016, 28, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Barnieh, F.M.; Loadman, P.M.; Falconer, R.A. Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100017. [Google Scholar] [CrossRef] [PubMed]
- Wickstroem, K.; Hagemann, U.B.; Cruciani, V.; Wengner, A.M.; Kristian, A.; Ellingsen, C.; Siemeister, G.; Bjerke, R.M.; Karlsson, J.; Ryan, O.B. Synergistic effect of a mesothelin-targeted 227Th conjugate in combination with DNA damage response inhibitors in ovarian cancer xenograft models. J. Nucl. Med. 2019, 60, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Wickstroem, K.; Hagemann, U.B.; Kristian, A.; Ellingsen, C.; Sommer, A.; Ellinger-Ziegelbauer, H.; Wirnitzer, U.; Hagelin, E.-M.; Larsen, A.; Smeets, R.; et al. Preclinical Combination Studies of an FGFR2 Targeted Thorium-227 Conjugate and the ATR Inhibitor BAY 1895344. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Wickstroem, K.; Karlsson, J.; Ellingsen, C.; Cruciani, V.; Kristian, A.; Hagemann, U.B.; Bjerke, R.M.; Ryan, O.B.; Linden, L.; Mumberg, D.; et al. Synergistic Effect of a HER2 Targeted Thorium-227 Conjugate in Combination with Olaparib in a BRCA2 Deficient Xenograft Model. Pharmaceuticals 2019, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Makvandi, M.; Lee, H.; Puentes, L.N.; Reilly, S.W.; Rathi, K.S.; Weng, C.-C.; Chan, H.S.; Hou, C.; Raman, P.; Martinez, D. Targeting PARP-1 with Alpha-Particles Is Potently Cytotoxic to Human Neuroblastoma in Preclinical ModelsAlpha-Particle Therapy for the Treatment of Neuroblastoma. Mol. Cancer Ther. 2019, 18, 1195–1204. [Google Scholar] [CrossRef]
- Bannik, K.; Madas, B.; Jarke, S.; Sutter, A.; Siemeister, G.; Schatz, C.; Mumberg, D.; Zitzmann-Kolbe, S. DNA repair inhibitors sensitize cells differently to high and low LET radiation. Sci. Rep. 2021, 11, 23257. [Google Scholar] [CrossRef]
- Quinn, Z.; Leiby, B.; Sonpavde, G.; Choudhury, A.D.; Sweeney, C.; Einstein, D.; Szmulewitz, R.; Sartor, O.; Knudsen, K.; Yang, E.S.; et al. Phase I Study of Niraparib in Combination with Radium-223 for the Treatment of Metastatic Castrate-Resistant Prostate Cancer. Clin. Cancer Res. 2023, 29, 50–59. [Google Scholar] [CrossRef]
- Mello, B.P.; Revskaya, E.; Scheinberg, D.A.; McDevitt, M.R. Abstract 3056: Alpha particle radiobiology and the treatment of prostate adenocarcinoma. Cancer Res. 2021, 81, 3056. [Google Scholar] [CrossRef]
- Qin, Y.; Imobersteg, S.; Frank, S.; Blanc, A.; Chiorazzo, T.; Berger, P.; Schibli, R.; Béhé, M.P.; Grzmil, M. Signaling Network Response to α-Particle–Targeted Therapy with the 225Ac-Labeled Minigastrin Analog 225Ac-PP-F11N Reveals the Radiosensitizing Potential of Histone Deacetylase Inhibitors. J. Nucl. Med. 2023, 64, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Riad, A.; Martorano, P.; Mansfield, A.; Samanta, M.; Batra, V.; Mach, R.H.; Maris, J.M.; Pryma, D.A.; Makvandi, M. PARP-1–Targeted Auger Emitters Display High-LET Cytotoxic Properties In Vitro but Show Limited Therapeutic Utility in Solid Tumor Models of Human Neuroblastoma. J. Nucl. Med. 2020, 61, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Riad, A.; Gitto, S.B.; Lee, H.; Winters, H.D.; Martorano, P.M.; Hsieh, C.-J.; Xu, K.; Omran, D.K.; Powell Jr, D.J.; Mach, R.H. Parp theranostic auger emitters are cytotoxic in brca mutant ovarian cancer and viable tumors from ovarian cancer patients enable ex-vivo screening of tumor response. Molecules 2020, 25, 6029. [Google Scholar] [CrossRef] [PubMed]
- Pirovano, G.; Jannetti, S.A.; Carter, L.M.; Sadique, A.; Kossatz, S.; Guru, N.; Demétrio De Souza França, P.; Maeda, M.; Zeglis, B.M.; Lewis, J.S. Targeted brain tumor radiotherapy using an Auger emitter. Clin. Cancer Res. 2020, 26, 2871–2881. [Google Scholar] [CrossRef] [PubMed]
- Koosha, F.; Eynali, S.; Eyvazzadeh, N.; Kamalabadi, M.A. The effect of iodine-131 beta-particles in combination with A-966492 and Topotecan on radio-sensitization of glioblastoma: An in-vitro study. Appl. Radiat. Isot. 2021, 177, 109904. [Google Scholar] [CrossRef] [PubMed]
- Jannetti, S.A.; Carlucci, G.; Carney, B.; Kossatz, S.; Shenker, L.; Carter, L.M.; Salinas, B.; Brand, C.; Sadique, A.; Donabedian, P.L. PARP-1–targeted radiotherapy in mouse models of glioblastoma. J. Nucl. Med. 2018, 59, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Cullinane, C.; Waldeck, K.; Kirby, L.; Rogers, B.E.; Eu, P.; Tothill, R.W.; Hicks, R.J. Enhancing the anti-tumour activity of 177Lu-DOTA-octreotate radionuclide therapy in somatostatin receptor-2 expressing tumour models by targeting PARP. Sci. Rep. 2020, 10, 10196. [Google Scholar] [CrossRef]
- Nonnekens, J.; van Kranenburg, M.; Beerens, C.E.M.T.; Suker, M.; Doukas, M.; van Eijck, C.H.J.; de Jong, M.; van Gent, D.C. Potentiation of Peptide Receptor Radionuclide Therapy by the PARP Inhibitor Olaparib. Theranostics 2016, 6, 1821–1832. [Google Scholar] [CrossRef]
- Fu, J.; Qiu, F.; Stolniceanu, C.R.; Yu, F.; Zang, S.; Xiang, Y.; Huang, Y.; Matovic, M.; Stefanescu, C.; Tang, Q.; et al. Combined use of 177Lu-DOTATATE peptide receptor radionuclide therapy and fluzoparib for treatment of well-differentiated neuroendocrine tumors: A preclinical study. J. Neuroendocrinol. 2022, 34, e13109. [Google Scholar] [CrossRef]
- Ruigrok, E.A.M.; Verkaik, N.S.; de Blois, E.; de Ridder, C.M.A.; Stuurman, D.C.; Roobol, S.J.; van Gent, D.C.; de Jong, M.; van Weerden, W.M.; Nonnekens, J. Preclinical Assessment of the Combination of PSMA-Targeting Radionuclide Therapy with PARP Inhibitors for Prostate Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 8037. [Google Scholar] [CrossRef]
- Esfahani, S.A.; Ferreira, C.D.A.; Summer, P.; Mahmood, U.; Heidari, P. Addition of Peptide Receptor Radiotherapy to Immune Checkpoint Inhibition Therapy Improves Outcomes in Neuroendocrine Tumors. J. Nucl. Med. 2023. [Google Scholar] [CrossRef]
- de Aguiar Ferreira, C.; Heidari, P.; Ataeinia, B.; Sinevici, N.; Granito, A.; Kumar, H.M.; Wehrenberg-Klee, E.; Mahmood, U. Immune Checkpoint Inhibitor-Mediated Cancer Theranostics with Radiolabeled Anti-Granzyme B Peptide. Pharmaceutics 2022, 14, 1460. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Lozza, C.; Deshayes, E.; Boudousq, V.; Navarro-Teulon, I. Introduction to radiobiology of targeted radionuclide therapy. Front. Med. 2015, 2, 12. [Google Scholar] [CrossRef]
- Joiner, M.C.; van der Kogel, A.J. Basic Clinical Radiobiology; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Solanki, J.H.; Tritt, T.; Pasternack, J.B.; Kim, J.J.; Leung, C.N.; Domogauer, J.D.; Colangelo, N.W.; Narra, V.R.; Howell, R.W. Cellular response to exponentially increasing and decreasing dose rates: Implications for treatment planning in targeted radionuclide therapy. Radiat. Res. 2017, 188, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Olipitz, W.; Wiktor-Brown, D.; Shuga, J.; Pang, B.; McFaline, J.; Lonkar, P.; Thomas, A.; Mutamba, J.T.; Greenberger, J.S.; Samson, L.D. Integrated molecular analysis indicates undetectable change in DNA damage in mice after continuous irradiation at ~400-fold natural background radiation. Environ. Health Perspect. 2012, 120, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; De Toledo, S.M.; Spitz, D.R.; Little, J.B. Oxidative metabolism modulates signal transduction and micronucleus formation in bystander cells from α-particle-irradiated normal human fibroblast cultures. Cancer Res. 2002, 62, 5436–5442. [Google Scholar]
- Brooks, A.L.; Hoel, D.G.; Preston, R.J. The role of dose rate in radiation cancer risk: Evaluating the effect of dose rate at the molecular, cellular and tissue levels using key events in critical pathways following exposure to low LET radiation. Int. J. Radiat. Biol. 2016, 92, 405–426. [Google Scholar] [CrossRef]
- Brooks, A.L. Chromosome damage in liver cells from low dose rate alpha, beta, and gamma irradiation: Derivation of RBE. Science 1975, 190, 1090–1092. [Google Scholar] [CrossRef] [PubMed]
- Penninckx, S.; Cekanaviciute, E.; Degorre, C.; Guiet, E.; Viger, L.; Lucas, S.; Costes, S.V. Dose, LET and strain dependence of radiation-induced 53BP1 foci in 15 mouse strains ex vivo introducing novel DNA damage metrics. Radiat. Res. 2019, 192, 1–12. [Google Scholar] [CrossRef]
- Neumaier, T.; Swenson, J.; Pham, C.; Polyzos, A.; Lo, A.T.; Yang, P.; Dyball, J.; Asaithamby, A.; Chen, D.J.; Bissell, M.J. Evidence for formation of DNA repair centers and dose-response nonlinearity in human cells. Proc. Natl. Acad. Sci. USA 2012, 109, 443–448. [Google Scholar] [CrossRef]
- Rothkamm, K.; Löbrich, M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low X-ray doses. Proc. Natl. Acad. Sci. USA 2003, 100, 5057–5062. [Google Scholar] [CrossRef]
- Grudzenski, S.; Raths, A.; Conrad, S.; Rübe, C.E.; Löbrich, M. Inducible response required for repair of low-dose radiation damage in human fibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 14205–14210. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; He, M.; Dong, C.; Xie, Y.; Ye, S.; Yuan, D.; Shao, C. Dose response of micronuclei induced by combination radiation of α-particles and γ-rays in human lymphoblast cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2013, 741, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Kabacik, S.; Finnon, P.; Bouffler, S.; Badie, C. High and low dose responses of transcriptional biomarkers in ex vivo X-irradiated human blood. Int. J. Radiat. Biol. 2013, 89, 512–522. [Google Scholar] [CrossRef]
- Ghandhi, S.A.; Smilenov, L.B.; Elliston, C.D.; Chowdhury, M.; Amundson, S.A. Radiation dose-rate effects on gene expression for human biodosimetry. BMC Med. Genom. 2015, 8, 22. [Google Scholar] [CrossRef]
- Brooks, A.; Mead, D.K.; Peters, R. Effect of chronic exposure to 60Co on the frequency of metaphase chromosome aberrations in the liver cells of the Chinese hamster (in vivo). Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1971, 20, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Barnard, S.; Moquet, J.; Ellender, M.; Rana, Z.; Burdak-Rothkamm, S. DNA damage foci: Meaning and significance. Environ. Mol. Mutagen. 2015, 56, 491–504. [Google Scholar] [CrossRef]
- Tubiana, M.; Aurengo, A.; Averbeck, D.; Masse, R. The debate on the use of linear no threshold for assessing the effects of low doses. J. Radiol. Prot. 2006, 26, 317. [Google Scholar] [CrossRef]
- Löbrich, M.; Rief, N.; Kühne, M.; Heckmann, M.; Fleckenstein, J.; Rübe, C.; Uder, M. In vivo formation and repair of DNA double-strand breaks after computed tomography examinations. Proc. Natl. Acad. Sci. USA 2005, 102, 8984–8989. [Google Scholar] [CrossRef]
- Penninckx, S.; Pariset, E.; Cekanaviciute, E.; Costes, S.V. Quantification of radiation-induced DNA double strand break repair foci to evaluate and predict biological responses to ionizing radiation. NAR Cancer 2021, 3, zcab046. [Google Scholar] [CrossRef]
- Blyth, B.J.; Sykes, P.J. Radiation-induced bystander effects: What are they, and how relevant are they to human radiation exposures? Radiat. Res. 2011, 176, 139–157. [Google Scholar] [CrossRef]
- Nagasawa, H.; Little, J.B. Induction of sister chromatid exchanges by extremely low doses of α-particles. Cancer Res. 1992, 52, 6394–6396. [Google Scholar] [PubMed]
- Widel, M. Radionuclides in radiation-induced bystander effect; may it share in radionuclide therapy. Neoplasma 2017, 64, 641–654. [Google Scholar] [CrossRef]
- Gerashchenko, B.I.; Howell, R.W. Proliferative response of bystander cells adjacent to cells with incorporated radioactivity. Cytom. Part A J. Int. Soc. Anal. Cytol. 2004, 60, 155–164. [Google Scholar] [CrossRef]
- Painter, R.B.; Drew, R.M.; Hughes, W.L. Inhibition of HeLa growth by intranuclear tritium. Science 1958, 127, 1244–1245. [Google Scholar] [CrossRef] [PubMed]
- Drew, R.M.; Painter, R.B. Action of tritiated thymidine on the clonal growth of mammalian cells. Radiat. Res. 1959, 11, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Azzam, E.; Howell, R. Bystander responses in three-dimensional cultures containing radiolabelled and unlabelled human cells. Radiat. Prot. Dosim. 2006, 122, 252–255. [Google Scholar] [CrossRef]
- Persaud, R.; Zhou, H.; Baker, S.E.; Hei, T.K.; Hall, E.J. Assessment of low linear energy transfer radiation–induced bystander mutagenesis in a three-dimensional culture model. Cancer Res. 2005, 65, 9876–9882. [Google Scholar] [CrossRef] [PubMed]
- Howell, R.W.; Bishayee, A. Bystander effects caused by nonuniform distributions of DNA-incorporated 125I. Micron 2002, 33, 127–132. [Google Scholar] [CrossRef]
- Akudugu, J.M.; Azzam, E.I.; Howell, R.W. Induction of lethal bystander effects in human breast cancer cell cultures by DNA-incorporated Iodine-125 depends on phenotype. Int. J. Radiat. Biol. 2012, 88, 1028–1038. [Google Scholar] [CrossRef]
- Sedelnikova, O.A.; Nakamura, A.; Kovalchuk, O.; Koturbash, I.; Mitchell, S.A.; Marino, S.A.; Brenner, D.J.; Bonner, W.M. DNA double-strand breaks form in bystander cells after microbeam irradiation of three-dimensional human tissue models. Cancer Res. 2007, 67, 4295–4302. [Google Scholar] [CrossRef]
- Fu, J.; Yuan, D.; Xiao, L.; Tu, W.; Dong, C.; Liu, W.; Shao, C. The crosstalk between α-irradiated Beas-2B cells and its bystander U937 cells through MAPK and NF-κB signaling pathways. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2016, 783, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lam, R.; Han, W.; Yu, K. Unirradiated cells rescue cells exposed to ionizing radiation: Activation of NF-κB pathway in irradiated cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2015, 782, 23–33. [Google Scholar] [CrossRef]
- Boyd, M.; Ross, S.C.; Dorrens, J.; Fullerton, N.E.; Tan, K.W.; Zalutsky, M.R.; Mairs, R.J. Radiation-induced biologic bystander effect elicited in vitro by targeted radiopharmaceuticals labeled with α-, β-, and Auger electron–emitting radionuclides. J. Nucl. Med. 2006, 47, 1007–1015. [Google Scholar]
- Kishikawa, H.; Wang, K.; Adelstein, S.J.; Kassis, A.I. Inhibitory and stimulatory bystander effects are differentially induced by iodine-125 and iodine-123. Radiat. Res. 2006, 165, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.Y.; Butler, N.J.; Makrigiorgos, G.M.; Adelstein, S.J.; Kassis, A.I. Bystander effect produced by radiolabeled tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 13765–13770. [Google Scholar] [CrossRef] [PubMed]
- Rudqvist, N.; Parris, T.Z.; Schüler, E.; Helou, K.; Forssell-Aronsson, E. Transcriptional response of BALB/c mouse thyroids following in vivo astatine-211 exposure reveals distinct gene expression profiles. EJNMMI Res. 2012, 2, 32. [Google Scholar] [CrossRef]
- Mitrofanova, E.; Hagan, C.; Qi, J.; Seregina, T.; Link, C., Jr. Sodium iodide symporter radioactive iodine system has more efficient antitumor effect in three-dimensional spheroids. Anticancer Res. 2003, 23, 2397–2404. [Google Scholar]


| Alpha Particle | Beta Particle | Auger Electron | |
|---|---|---|---|
| Type of particles | 4He nuclei | Energetic electrons | Low-energy electrons; electron capture and/or internal conversion |
| Energy range | 4–9 MeV | 50–2300 KeV | 25–80 KeV |
| Emission range in tissues | 28–100 µm | 0.5–10 mm | <0.5 µm |
| LET (KeV/µm) | ~50–230 | ~0.1–1.0 | ~4–26 |
| Main mechanism of damage | At high doses: widespread DNA damage, leads to significant cellular damage and reduced repair capability to induce cell death or mutations with potential long-term effects. At low to moderate doses: DSBs, less reparable by cellular mechanisms. | At high doses: exponential relationship with tumor survival. The rate of DNA damage may exceed the cell’s repair capacity, leading to the accumulation of unrepaired or misrepaired DNA lesions. At low to moderate doses: linear relationship with tumor survival. Primarily involves SSBs and minor chemical modifications to DNA bases. Damage is more likely to be repaired by the cell’s repair mechanisms. | At high doses: multiple DSBs, lead to increased genetic instability and potential cell death. At low to moderate doses: clustered DNA damage, leads to complex lesions that overwhelm repair systems. |
| Tissue damage size | Small (a number of cells) | Higher volume solid tumor | Micro (a few cells) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khazaei Monfared, Y.; Heidari, P.; Klempner, S.J.; Mahmood, U.; Parikh, A.R.; Hong, T.S.; Strickland, M.R.; Esfahani, S.A. DNA Damage by Radiopharmaceuticals and Mechanisms of Cellular Repair. Pharmaceutics 2023, 15, 2761. https://doi.org/10.3390/pharmaceutics15122761
Khazaei Monfared Y, Heidari P, Klempner SJ, Mahmood U, Parikh AR, Hong TS, Strickland MR, Esfahani SA. DNA Damage by Radiopharmaceuticals and Mechanisms of Cellular Repair. Pharmaceutics. 2023; 15(12):2761. https://doi.org/10.3390/pharmaceutics15122761
Chicago/Turabian StyleKhazaei Monfared, Yousef, Pedram Heidari, Samuel J. Klempner, Umar Mahmood, Aparna R. Parikh, Theodore S. Hong, Matthew R. Strickland, and Shadi A. Esfahani. 2023. "DNA Damage by Radiopharmaceuticals and Mechanisms of Cellular Repair" Pharmaceutics 15, no. 12: 2761. https://doi.org/10.3390/pharmaceutics15122761
APA StyleKhazaei Monfared, Y., Heidari, P., Klempner, S. J., Mahmood, U., Parikh, A. R., Hong, T. S., Strickland, M. R., & Esfahani, S. A. (2023). DNA Damage by Radiopharmaceuticals and Mechanisms of Cellular Repair. Pharmaceutics, 15(12), 2761. https://doi.org/10.3390/pharmaceutics15122761