
Uses and Challenges of Antiviral Polyclonal and Monoclonal Antibody Therapies
 ,
,
Abstract
:1. Introduction
2. Antibodies as Therapeutics
2.1. Production and Characterization of Antibody Therapies
2.1.1. Specific Polyclonal Antibody Therapies
Treatment Timing and Dosing for SpIG
2.1.2. Monoclonal Antibodies
Engineering of mAbs
Development of mAb Combinations
2.2. Advantages and Disadvantages of Polyclonal and Monoclonal Antibodies
3. Evaluation of Antiviral Activity
3.1. Types of Potency Assays
3.2. Cells for Potency Assays
3.3. Resistance
3.4. Animal Studies
4. Combining Antiviral Antibodies and Other Therapies
4.1. Combinations of Specific Polyclonal Antibodies with Vaccines or Drugs
4.2. Combinations of Monoclonal Antibodies
4.2.1. Potential Benefits of Monoclonal Antibody Combinations
4.2.2. Potential Challenges of Monoclonal Antibody Combinations
4.3. Combinations of Monoclonal Antibodies with Other Types of Antivirals
5. Future Directions and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Disclaimer
References
- Armitage, C. The high burden of infectious disease. Nature 2021, 598, S9. [Google Scholar] [CrossRef]
- Parra, D.; Takizawa, F.; Sunyer, J.O. Evolution of B Cell Immunity. Annu. Rev. Anim. Biosci. 2013, 1, 65–97. [Google Scholar] [CrossRef] [PubMed]
- CBER. Science and the Regulation of Biological Products. Available online: https://www.fda.gov/about-fda/histories-product-regulation/science-and-regulation-biological-products (accessed on 3 May 2023).
- Chen, W.C.; Murawsky, C.M. Strategies for Generating Diverse Antibody Repertoires Using Transgenic Animals Expressing Human Antibodies. Front. Immunol. 2018, 9, 460. [Google Scholar] [CrossRef]
- Sheehan, J.; Marasco, W.A. Phage and Yeast Display. Microbiol. Spectr. 2015, 3, 103–127. [Google Scholar] [CrossRef] [PubMed]
- Wrammert, J.; Smith, K.; Miller, J.; Langley, W.A.; Kokko, K.; Larsen, C.; Zheng, N.-Y.; Mays, I.; Garman, L.; Helms, C.; et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008, 453, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Karuna, S.T.; Corey, L. Broadly Neutralizing Antibodies for HIV Prevention. Annu. Rev. Med. 2020, 71, 329–346. [Google Scholar] [CrossRef]
- Corti, D.; Lanzavecchia, A. Efficient Methods to Isolate Human Monoclonal Antibodies from Memory B Cells and Plasma Cells. Microbiol. Spectr. 2014, 2, 129–139. [Google Scholar] [CrossRef]
- Corti, D.; Misasi, J.; Mulangu, S.; Stanley, D.A.; Kanekiyo, M.; Wollen, S.; Ploquin, A.; Doria-Rose, N.A.; Staupe, R.P.; Bailey, M.; et al. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science 2016, 351, 1339–1342. [Google Scholar] [CrossRef]
- Marasco, W.A.; Sui, J. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat. Biotechnol. 2007, 25, 1421–1434. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Trkola, A. Humoral immunity to HIV-1: Neutralization and beyond. J. Intern. Med. 2007, 262, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Klasse, P.J. Neutralization of Virus Infectivity by Antibodies: Old Problems in New Perspectives. Adv. Biol. 2014, 2014, 157895. [Google Scholar] [CrossRef] [PubMed]
- Reading, S.A.; Dimmock, N.J. Neutralization of animal virus infectivity by antibody. Arch. Virol. 2007, 152, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Gunn, B.M.; Yu, W.-H.; Karim, M.M.; Brannan, J.M.; Herbert, A.S.; Wec, A.Z.; Halfmann, P.J.; Fusco, M.L.; Schendel, S.L.; Gangavarapu, K.; et al. A Role for Fc Function in Therapeutic Monoclonal Antibody-Mediated Protection against Ebola Virus. Cell Host Microbe 2018, 24, 221–233.e5. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Klein, F.; Pietzsch, J.; Seaman, M.S.; Nussenzweig, M.C.; Ravetch, J.V. Broadly Neutralizing Anti-HIV-1 Antibodies Require Fc Effector Functions for In Vivo Activity. Cell 2014, 158, 1243–1253. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Palese, P.; Wilson, P.C.; Ravetch, J.V. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Investig. 2016, 126, 605–610. [Google Scholar] [CrossRef]
- Lu, L.L.; Suscovich, T.J.; Fortune, S.M.; Alter, G. Beyond binding: Antibody effector functions in infectious diseases. Nat. Rev. Immunol. 2017, 18, 46–61. [Google Scholar] [CrossRef]
- Phelps, M.; Balazs, A.B. Contribution to HIV Prevention and Treatment by Antibody-Mediated Effector Function and Advances in Broadly Neutralizing Antibody Delivery by Vectored Immunoprophylaxis. Front. Immunol. 2021, 12, 734304. [Google Scholar] [CrossRef]
- Taylor, A.; Foo, S.-S.; Bruzzone, R.; Dinh, L.V.; King, N.J.C.; Mahalingam, S. Fc receptors in antibody-dependent enhancement of viral infections. Immunol. Rev. 2015, 268, 340–364. [Google Scholar] [CrossRef]
- Halstead, S.B. Dengue Antibody-Dependent Enhancement: Knowns and Unknowns. Microbiol. Spectr. 2014, 2, 249–271. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Screaton, G.; Mongkolsapaya, J.; Yacoub, S.; Roberts, C. New insights into the immunopathology and control of dengue virus infection. Nat. Rev. Immunol. 2015, 15, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Singh, G.; Acklin, J.; Lee, S.; Duehr, J.; Chokola, A.; Frere, J.; Hoffman, K.W.; Foster, G.A.; Krysztof, D.; et al. Dengue Virus Immunity Increases Zika Virus-Induced Damage during Pregnancy. Immunity 2019, 50, 751–762.e5. [Google Scholar] [CrossRef] [PubMed]
- Martín-Acebes, M.A.; Saiz, J.-C.; de Oya, N.J. Antibody-Dependent Enhancement and Zika: Real Threat or Phantom Menace? Front. Cell. Infect. Microbiol. 2018, 8, 44. [Google Scholar] [CrossRef]
- Ramadhany, R.; Hirai, I.; Sasaki, T.; Ono, K.-I.; Ramasoota, P.; Ikuta, K.; Kurosu, T. Antibody with an engineered Fc region as a therapeutic agent against dengue virus infection. Antivir. Res. 2015, 124, 61–68. [Google Scholar] [CrossRef]
- Kotaki, T.; Kurosu, T.; Grinyo-Escuer, A.; Davidson, E.; Churrotin, S.; Okabayashi, T.; Puiprom, O.; Mulyatno, K.C.; Sucipto, T.H.; Doranz, B.J.; et al. An affinity-matured human monoclonal antibody targeting fusion loop epitope of dengue virus with in vivo therapeutic potency. Sci. Rep. 2021, 11, 12987. [Google Scholar] [CrossRef]
- Lu, J.; Chen, L.; Du, P.; Guo, J.; Wang, X.; Jiang, Y.; Yu, Y.; Wang, R.; Yang, Z. A human monoclonal antibody to neutralize all four serotypes of dengue virus derived from patients at the convalescent phase of infection. Virology 2022, 576, 74–82. [Google Scholar] [CrossRef]
- Pinto, A.K.; Hassert, M.; Han, X.; Barker, D.; Carnelley, T.; Branche, E.; Steffen, T.L.; Stone, E.T.; Geerling, E.; Viramontes, K.M.; et al. The Ability of Zika virus Intravenous Immunoglobulin to Protect from or Enhance Zika Virus Disease. Front. Immunol. 2021, 12, 717425. [Google Scholar] [CrossRef]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef]
- Negron, C.; Fang, J.; McPherson, M.J.; Stine, W.B., Jr.; McCluskey, A.J. Separating clinical antibodies from repertoire antibodies, a path to in silico developability assessment. mAbs 2022, 14, 2080628. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.S.; Hoopes, E.M.; Falk, A.C.; Moore, D.J. A human IgM enriched immunoglobulin preparation, Pentaglobin, reverses autoimmune diabetes without immune suppression in NOD mice. Sci. Rep. 2022, 12, 11731. [Google Scholar] [CrossRef] [PubMed]
- Isa, M.B.; Martínez, L.C.; Ferreyra, L.J.; Giordano, M.O.; Barril, P.A.; Massachessi, G.; Nates, S.V. Measles Virus–Specific IgG4 Antibody Titer as a Serologic Marker of Post-vaccinal Immune Response. Viral Immunol. 2006, 19, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Siekman, S.L.; Pongracz, T.; Wang, W.; Nouta, J.; Kremsner, P.G.; da Silva-Neto, P.V.; Esen, M.; Kreidenweiss, A.; Held, J.; Trapé, Á.A.; et al. The IgG glycome of SARS-CoV-2 infected individuals reflects disease course and severity. Front. Immunol. 2022, 13, 993354. [Google Scholar] [CrossRef]
- Gardner, C.L.; Sun, C.; Luke, T.; Raviprakash, K.; Wu, H.; Jiao, J.-A.; Sullivan, E.; Reed, D.S.; Ryman, K.D.; Klimstra, W.B. Antibody Preparations from Human Transchromosomic Cows Exhibit Prophylactic and Therapeutic Efficacy against Venezuelan Equine Encephalitis Virus. J. Virol. 2017, 91, e00226-17. [Google Scholar] [CrossRef]
- Saied, A.A.; Nascimento, M.S.L.; Rangel, A.H.D.N.; Skowron, K.; Grudlewska-Buda, K.; Dhama, K.; Shah, J.; Abdeen, A.; El-Mayet, F.S.; Ahmed, H.; et al. Transchromosomic bovines-derived broadly neutralizing antibodies as potent biotherapeutics to counter important emerging viral pathogens with a special focus on SARS-CoV-2, MERS-CoV, Ebola, Zika, HIV-1, and influenza A virus. J. Med. Virol. 2022, 94, 4599–4610. [Google Scholar] [CrossRef]
- Cohn, E.J.; Strong, L.E.; Hughes, W.L.; Mulford, D.J.; Ashworth, J.N.; Melin, M.; Taylor, H.L. Preparation and Properties of Serum and Plasma Proteins. IV. A System for the Separation into Fractions of the Protein and Lipoprotein Components of Biological Tissues and Fluids. J. Am. Chem. Soc. 1946, 68, 459–475. [Google Scholar] [CrossRef]
- Oncley, J.L.; Melin, M.; Richert, D.A.; Cameron, J.W.; Gross, P.M. The Separation of the Antibodies, Isoagglutinins, Prothrombin, Plasminogen and β1-Lipoprotein into Subfractions of Human Plasma. J. Am. Chem. Soc. 1949, 71, 541–550. [Google Scholar] [CrossRef]
- Lebing, W.; Remington, K.M.; Schreiner, C.; Paul, H.-I. Properties of a new intravenous immunoglobulin (IGIV-C, 10%) produced by virus inactivation with caprylate and column chromatography. Vox Sang. 2003, 84, 193–201. [Google Scholar] [CrossRef]
- CytoGam Prescribing Information. Available online: https://www.accessdata.fda.gov/spl/data/2a40733c-106b-41cf-94f0-f10a03180ac8/2a40733c-106b-41cf-94f0-f10a03180ac8.xml (accessed on 3 May 2023).
- Vandeberg, P.; Cruz, M.; Diez, J.M.; Merritt, W.K.; Santos, B.; Trukawinski, S.; Wellhouse, A.; Jose, M.; Willis, T. Production of anti-SARS-CoV-2 hyperimmune globulin from convalescent plasma. Transfusion 2021, 61, 1705–1709. [Google Scholar] [CrossRef]
- Burnouf, T.; Gathof, B.; Bloch, E.M.; Bazin, R.; de Angelis, V.; Patidar, G.K.; Rastvorceva, R.M.G.; Oreh, A.; Goel, R.; Rahimi-Levene, N.; et al. Production and Quality Assurance of Human Polyclonal Hyperimmune Immunoglobulins against SARS-CoV-2. Transfus. Med. Rev. 2022, 36, 125–132. [Google Scholar] [CrossRef] [PubMed]
- HyperRAB Prescribing Information. Available online: https://www.accessdata.fda.gov/spl/data/f993778d-01fb-4670-af67-a0e08d6b258b/f993778d-01fb-4670-af67-a0e08d6b258b.xml (accessed on 3 May 2023).
- Imogam Prescribing Information. Available online: https://www.accessdata.fda.gov/spl/data/8026005f-7587-47fe-bb78-ec6247a3434b/8026005f-7587-47fe-bb78-ec6247a3434b.xml (accessed on 3 May 2023).
- Kedrab Prescribing Information. Available online: https://www.accessdata.fda.gov/spl/data/5e5c130a-693b-47f9-b44a-3d8f9cde3f98/5e5c130a-693b-47f9-b44a-3d8f9cde3f98.xml (accessed on 3 May 2023).
- VariZIG Prescribing Information. Available online: https://www.accessdata.fda.gov/spl/data/272379b7-f0e7-4560-8d79-3fd0024c3010/272379b7-f0e7-4560-8d79-3fd0024c3010.xml (accessed on 3 May 2023).
- Levin, M.J.; Duchon, J.M.; Swamy, G.K.; Gershon, A.A. Varicella zoster immune globulin (VARIZIG) administration up to 10 days after varicella exposure in pregnant women, immunocompromised participants, and infants: Varicella outcomes and safety results from a large, open-label, expanded-access program. PLoS ONE 2019, 14, e0217749. [Google Scholar] [CrossRef] [PubMed]
- Vaccinia Immune Globulin Prescribing Information. Available online: https://www.fda.gov/media/78174/download (accessed on 3 May 2023).
- Centers for Disease Control and Prevention. Household transmission of vaccinia virus from contact with a military smallpox vaccinee—Illinois and Indiana, 2007. MMWR Morb. Mortal. Wkly. Rep. 2007, 56, 478–481. [Google Scholar]
- Centers for Disease Control and Prevention. Progressive vaccinia in a military smallpox vaccinee—United States, 2009. MMWR Morb. Mortal. Wkly. Rep. 2009, 58, 532–536. [Google Scholar]
- Razonable, R.R.; Humar, A. Cytomegalovirus in solid organ transplant recipients—Guidelines of the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13512. [Google Scholar] [CrossRef]
- GamaSTAN Prescribing Information. Available online: https://www.accessdata.fda.gov/spl/data/38a323af-7c25-42d1-9c29-532ef61999b8/38a323af-7c25-42d1-9c29-532ef61999b8.xml (accessed on 3 May 2023).
- HyperHEP B Prescribing Information. Available online: https://www.accessdata.fda.gov/spl/data/391b2218-8a15-4e5e-8717-aa49efcc2210/391b2218-8a15-4e5e-8717-aa49efcc2210.xml (accessed on 3 May 2023).
- Nabi-HB Prescribing Information. Available online: https://www.accessdata.fda.gov/spl/data/ee1560c0-18e1-b617-e053-2a95a90aa1af/ee1560c0-18e1-b617-e053-2a95a90aa1af.xml (accessed on 3 May 2023).
- HepaGAM B Prescribing Information. Available online: https://www.accessdata.fda.gov/spl/data/56525de0-f47d-11eb-85b4-0800200c9a66/56525de0-f47d-11eb-85b4-0800200c9a66.xml (accessed on 3 May 2023).
- Te, H.; Doucette, K. Viral hepatitis: Guidelines by the American Society of Transplantation Infectious Disease Community of Practice. Clin. Transplant. 2019, 33, e13514. [Google Scholar] [CrossRef]
- FDA. Letter to Immune Globulin (Human) Licensed Manufacturers: Option to Lower Lot Release Specification for Required Measles Antibody Potency Testing. Available online: https://www.fda.gov/media/118428/download (accessed on 3 May 2023).
- Stauft, C.B.; Tegenge, M.; Khurana, S.; Lee, Y.; Selvaraj, P.; Golding, H.; Wang, T.; Golding, B. Pharmacokinetics and Efficacy of Human Hyperimmune Intravenous Immunoglobulin Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 Infection in Adult Syrian Hamsters. Clin. Infect. Dis. 2021, 75, e459–e465. [Google Scholar] [CrossRef]
- Rao, A.K.; Petersen, B.W.; Whitehill, F.; Razeq, J.H.; Isaacs, S.N.; Merchlinsky, M.J.; Campos-Outcalt, D.; Morgan, R.L.; Damon, I.; Sánchez, P.J.; et al. Use of JYNNEOS (Smallpox and Monkeypox Vaccine, Live, Nonreplicating) for Preexposure Vaccination of Persons at Risk for Occupational Exposure to Orthopoxviruses: Recommendations of the Advisory Committee on Immunization Practices—United States, 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 734–742. [Google Scholar] [CrossRef]
- Mbaya, O.T.; Mukumbayi, P.; Mulangu, S. Review: Insights on Current FDA-Approved Monoclonal Antibodies against Ebola Virus Infection. Front. Immunol. 2021, 12, 721328. [Google Scholar] [CrossRef]
- Hammitt, L.L.; Dagan, R.; Yuan, Y.; Cots, M.B.; Bosheva, M.; Madhi, S.A.; Muller, W.J.; Zar, H.J.; Brooks, D.; Grenham, A.; et al. Nirsevimab for Prevention of RSV in Healthy Late-Preterm and Term Infants. N. Engl. J. Med. 2022, 386, 837–846. [Google Scholar] [CrossRef]
- de Melo, G.D.; Hellert, J.; Gupta, R.; Corti, D.; Bourhy, H. Monoclonal antibodies against rabies: Current uses in prophylaxis and in therapy. Curr. Opin. Virol. 2022, 53, 101204. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Crescioli, S.; Chenoweth, A.; Visweswaraiah, J.; Reichert, J.M. Antibodies to watch in 2023. mAbs 2022, 15, 2153410. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2021, 602, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradník, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.E.; Ginn, H.M.; Mentzer, A.J.; Tuekprakhon, A.; et al. SARS-CoV-2 Omicron-B.1.1.529 Leads to Widespread Escape from Neutralizing Antibody Responses. Cell 2022, 185, 467–484.e15. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef]
- Sheward, D.J.; Kim, C.; Fischbach, J.; Sato, K.; Muschiol, S.; Ehling, R.A.; Björkström, N.K.; Hedestam, G.B.K.; Reddy, S.T.; Albert, J.; et al. Omicron sublineage BA.2.75.2 exhibits extensive escape from neutralising antibodies. Lancet Infect. Dis. 2022, 22, 1538–1540. [Google Scholar] [CrossRef]
- Holland, T.L.; Ginde, A.A.; Paredes, R.; Murray, T.A.; Engen, N.; Grandits, G.; Vekstein, A.; Ivey, N.; Mourad, A.; Sandkovsky, U.; et al. Tixagevimab–cilgavimab for treatment of patients hospitalised with COVID-19: A randomised, double-blind, phase 3 trial. Lancet Respir. Med. 2022, 10, 972–984. [Google Scholar] [CrossRef]
- Imai, M.; Ito, M.; Kiso, M.; Yamayoshi, S.; Uraki, R.; Fukushi, S.; Watanabe, S.; Suzuki, T.; Maeda, K.; Sakai-Tagawa, Y.; et al. Efficacy of Antiviral Agents against Omicron Subvariants BQ.1.1 and XBB. N. Engl. J. Med. 2023, 388, 89–91. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 2023, 186, 279–286. [Google Scholar] [CrossRef]
- Coronavirus Disease 2019 (COVID-19) EUA Information. Available online: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs (accessed on 17 February 2023).
- Dibo, M.; Battocchio, E.C.; Dos Santos Souza, L.M.; Da Silva, M.D.V.; Banin-Hirata, B.K.; Sapla, M.M.; Marinello, P.; Rocha, S.P.D.; Faccin-Galhardi, L.C. Antibody Therapy for the Control of Viral Diseases: An Update. Curr. Pharm. Biotechnol. 2019, 20, 1108–1121. [Google Scholar] [CrossRef]
- Hastie, K.M.; Cross, R.W.; Harkins, S.S.; Zandonatti, M.A.; Koval, A.P.; Heinrich, M.L.; Rowland, M.M.; Robinson, J.E.; Geisbert, T.W.; Garry, R.F.; et al. Convergent Structures Illuminate Features for Germline Antibody Binding and Pan-Lassa Virus Neutralization. Cell 2019, 178, 1004–1015.e14. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Buck, T.; Zandonatti, M.; Yin, J.; Moon-Walker, A.; Fang, J.; Koval, A.; Heinrich, M.L.; Rowland, M.M.; Avalos, R.D.; et al. A cocktail of protective antibodies subverts the dense glycan shield of Lassa virus. Sci. Transl. Med. 2022, 14, eabq0991. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.K. Role of Fc-mediated antibody function in protective immunity against HIV-1. Immunology 2013, 142, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Asokan, M.; Dias, J.; Liu, C.; Maximova, A.; Ernste, K.; Pegu, A.; McKee, K.; Shi, W.; Chen, X.; Almasri, C.; et al. Fc-mediated effector function contributes to the in vivo antiviral effect of an HIV neutralizing antibody. Proc. Natl. Acad. Sci. USA 2020, 117, 18754–18763. [Google Scholar] [CrossRef] [PubMed]
- Vanderven, H.A.; Kent, S. The protective potential of Fc-mediated antibody functions against influenza virus and other viral pathogens. Immunol. Cell Biol. 2020, 98, 253–263. [Google Scholar] [CrossRef]
- Zhang, A.; Stacey, H.D.; D’Agostino, M.R.; Tugg, Y.; Marzok, A.; Miller, M.S. Beyond neutralization: Fc-dependent antibody effector functions in SARS-CoV-2 infection. Nat. Rev. Immunol. 2022, 1–16. [Google Scholar] [CrossRef]
- Cartwright, H.N.; Barbeau, D.J.; McElroy, A.K. Isotype-Specific Fc Effector Functions Enhance Antibody-Mediated Rift Valley Fever Virus Protection In Vivo. Msphere 2021, 6, e0055621. [Google Scholar] [CrossRef]
- Smatti, M.K.; Al Thani, A.A.; Yassine, H.M. Viral-Induced Enhanced Disease Illness. Front. Microbiol. 2018, 9, 2991. [Google Scholar] [CrossRef]
- Almagro, J.C.; Daniels-Wells, T.R.; Perez-Tapia, S.M.; Penichet, M.L. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front. Immunol. 2018, 8, 1751. [Google Scholar] [CrossRef]
- Liu, R.; Oldham, R.J.; Teal, E.; Beers, S.A.; Cragg, M.S. Fc-Engineering for Modulated Effector Functions—Improving Antibodies for Cancer Treatment. Antibodies 2020, 9, 64. [Google Scholar] [CrossRef]
- Ko, S.; Jo, M.; Jung, S.T. Recent Achievements and Challenges in Prolonging the Serum Half-Lives of Therapeutic IgG Antibodies Through Fc Engineering. Biodrugs 2021, 35, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Igawa, T.; Ishii, S.; Tachibana, T.; Maeda, A.; Higuchi, Y.; Shimaoka, S.; Moriyama, C.; Watanabe, T.; Takubo, R.; Doi, Y.; et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat. Biotechnol. 2010, 28, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Igawa, T.; Mimoto, F.; Hattori, K. pH-dependent antigen-binding antibodies as a novel therapeutic modality. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2014, 1844, 1943–1950. [Google Scholar] [CrossRef]
- Igawa, T.; Maeda, A.; Haraya, K.; Tachibana, T.; Iwayanagi, Y.; Mimoto, F.; Higuchi, Y.; Ishii, S.; Tamba, S.; Hironiwa, N.; et al. Engineered Monoclonal Antibody with Novel Antigen-Sweeping Activity In Vivo. PLoS ONE 2013, 8, e63236. [Google Scholar] [CrossRef] [PubMed]
- Reusch, D.; Tejada, M.L. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology 2015, 25, 1325–1334. [Google Scholar] [CrossRef]
- Golay, J.; Andrea, A.E.; Cattaneo, I. Role of Fc Core Fucosylation in the Effector Function of IgG1 Antibodies. Front. Immunol. 2022, 13, 929895. [Google Scholar] [CrossRef]
- Hatfield, G.; Tepliakova, L.; Gingras, G.; Stalker, A.; Li, X.; Aubin, Y.; Tam, R.Y. Specific location of galactosylation in an afucosylated antiviral monoclonal antibody affects its FcγRIIIA binding affinity. Front. Immunol. 2022, 13, 972168. [Google Scholar] [CrossRef]
- Tao, M.H.; Morrison, S.L. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 1989, 143, 2595–2601. [Google Scholar] [CrossRef]
- Bolt, S.; Routledge, E.; Lloyd, I.; Chatenoud, L.; Pope, H.; Gorman, S.D.; Clark, M.; Waldmann, H. The generation of a humanized, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur. J. Immunol. 1993, 23, 403–411. [Google Scholar] [CrossRef]
- Liu, D.; Shameem, M. Antiviral monoclonal antibody cocktails as a modern weapon in combating pandemics. Ther. Deliv. 2022, 13, 67–69. [Google Scholar] [CrossRef]
- Dacon, C.; Tucker, C.; Peng, L.; Lee, C.-C.D.; Lin, T.-H.; Yuan, M.; Cong, Y.; Wang, L.; Purser, L.; Williams, J.K.; et al. Broadly neutralizing antibodies target the coronavirus fusion peptide. Science 2022, 377, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Tarafdar, S.; Virata, M.L.; Yan, H.; Zhong, L.; Deng, L.; Xu, Y.; He, Y.; Struble, E.; Zhang, P. Multiple epitopes of hepatitis B virus surface antigen targeted by human plasma-derived immunoglobulins coincide with clinically observed escape mutations. J. Med. Virol. 2021, 94, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research OoPQ. Potency Assay Considerations for Monoclonal Antibodies and Other Therapeutic Proteins Targeting Viral Pathogens, Guidance for Industry (Draft). Available online: https://www.fda.gov/media/165746/download (accessed on 3 May 2023).
- Jiang, X.-R.; Song, A.; Bergelson, S.; Arroll, T.; Parekh, B.; May, K.; Chung, S.; Strouse, R.; Mire-Sluis, A.; Schenerman, M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat. Rev. Drug Discov. 2011, 10, 101–111. [Google Scholar] [CrossRef]
- Schmidt, F.; Weisblum, Y.; Muecksch, F.; Hoffmann, H.-H.; Michailidis, E.; Lorenzi, J.C.; Mendoza, P.; Rutkowska, M.; Bednarski, E.; Gaebler, C.; et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J. Exp. Med. 2020, 217, e20201181. [Google Scholar] [CrossRef]
- Clapham, P.R. Vesicular Stomatitis Virus Pseudotypes of Retroviruses. Methods Mol. Biol. 2003, 8, 95–102. [Google Scholar] [CrossRef]
- Kim, Y.; Zheng, X.; Eschke, K.; Chaudhry, M.Z.; Bertoglio, F.; Tomić, A.; Krmpotić, A.; Hoffmann, M.; Bar-On, Y.; Boehme, J.; et al. MCMV-based vaccine vectors expressing full-length viral proteins provide long-term humoral immune protection upon a single-shot vaccination. Cell. Mol. Immunol. 2022, 19, 234–244. [Google Scholar] [CrossRef]
- Racine, T.; Kobinger, G.P.; Arts, E.J. Development of an HIV vaccine using a vesicular stomatitis virus vector expressing designer HIV-1 envelope glycoproteins to enhance humoral responses. AIDS Res. Ther. 2017, 14, 55. [Google Scholar] [CrossRef]
- Takada, A.; Feldmann, H.; Stroeher, U.; Bray, M.; Watanabe, S.; Ito, H.; McGregor, M.; Kawaoka, Y. Identification of Protective Epitopes on Ebola Virus Glycoprotein at the Single Amino Acid Level by Using Recombinant Vesicular Stomatitis Viruses. J. Virol. 2003, 77, 1069–1074. [Google Scholar] [CrossRef]
- Takada, A.; Robison, C.; Goto, H.; Sanchez, A.; Murti, K.G.; Whitt, M.A.; Kawaoka, Y. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. USA 1997, 94, 14764–14769. [Google Scholar] [CrossRef]
- Bannert, N.; Farzan, M.; Friend, D.S.; Ochi, H.; Price, K.S.; Sodroski, J.; Boyce, J.A. Human Mast Cell Progenitors Can Be Infected by Macrophagetropic Human Immunodeficiency Virus Type 1 and Retain Virus with Maturation In Vitro. J. Virol. 2001, 75, 10808–10814. [Google Scholar] [CrossRef]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr Is Required for Efficient Replication of Human Immunodeficiency Virus Type-1 in Mononuclear Phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O.; Englund, G.; Martin, M.A. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J. Virol. 1995, 69, 3949–3954. [Google Scholar] [CrossRef] [PubMed]
- Louder, M.K.; Sambor, A.; Chertova, E.; Hunte, T.; Barrett, S.; Ojong, F.; Sanders-Buell, E.; Zolla-Pazner, S.; McCutchan, F.E.; Roser, J.D.; et al. HIV-1 envelope pseudotyped viral vectors and infectious molecular clones expressing the same envelope glycoprotein have a similar neutralization phenotype, but culture in peripheral blood mononuclear cells is associated with decreased neutralization sensitivity. Virology 2005, 339, 226–238. [Google Scholar] [CrossRef]
- Lundquist, C.A.; Zhou, J.; Aiken, C. Nef Stimulates Human Immunodeficiency Virus Type 1 Replication in Primary T Cells by Enhancing Virion-Associated gp120 Levels: Coreceptor-Dependent Requirement for Nef in Viral Replication. J. Virol. 2004, 78, 6287–6296. [Google Scholar] [CrossRef] [PubMed]
- Sarzotti-Kelsoe, M.; Daniell, X.; Todd, C.A.; Bilska, M.; Martelli, A.; LaBranche, C.; Perez, L.G.; Ochsenbauer, C.; Kappes, J.C.; Rountree, W.; et al. Optimization and validation of a neutralizing antibody assay for HIV-1 in A3R5 cells. J. Immunol. Methods 2014, 409, 147–160. [Google Scholar] [CrossRef]
- Matsuura, Y.; Tania, H.; Suzukic, K.; Someyab, T.K.; Suzukib, R.; Aizakib, H.; Ishiib, K.; Moriishi, K.; Robison, C.S.; Whitt, M.A.; et al. Characterization of Pseudotype VSV Possessing HCV Envelope Proteins. Virology 2001, 286, 263–275. [Google Scholar] [CrossRef]
- Renelt, S.; Schult-Dietrich, P.; Baldauf, H.-M.; Stein, S.; Kann, G.; Bickel, M.; Kielland-Kaisen, U.; Bonig, H.; Marschalek, R.; Rieger, M.A.; et al. HIV-1 Infection of Long-Lived Hematopoietic Precursors In Vitro and In Vivo. Cells 2022, 11, 2968. [Google Scholar] [CrossRef]
- Riepler, L.; Rössler, A.; Falch, A.; Volland, A.; Borena, W.; Von Laer, D.; Kimpel, J. Comparison of Four SARS-CoV-2 Neutralization Assays. Vaccines 2020, 9, 13. [Google Scholar] [CrossRef]
- Chikere, K.; Webb, N.E.; Chou, T.; Borm, K.; Sterjovski, J.; Gorry, P.R.; Lee, B. Distinct HIV-1 entry phenotypes are associated with transmission, subtype specificity, and resistance to broadly neutralizing antibodies. Retrovirology 2014, 11, 48. [Google Scholar] [CrossRef]
- Mann, A.M.; Rusert, P.; Berlinger, L.; Kuster, H.; Günthard, H.F.; Trkola, A. HIV sensitivity to neutralization is determined by target and virus producer cell properties. Aids 2009, 23, 1659–1667. [Google Scholar] [CrossRef]
- Miyamoto, F.; Kawaji, K.; Oishi, S.; Fujii, N.; Kaku, M.; Kodama, E.N. Anti-HIV-1 activity determined by β-galactosidase activity in the multinuclear activation of an indicator assay is comparable with that by a conventional focus counting method. Antivir. Chem. Chemother. 2015, 24, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Spenlehauera, C.; Gordon, C.A.; Trkolac, A.; Moore, J.P. A Luciferase-Reporter Gene-Expressing T-Cell Line Facilitates Neutralization and Drug-Sensitivity Assays That Use Either R5 or X4 Strains of Human Immunodeficiency Virus Type 1. Virology 2001, 280, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Sarzotti-Kelsoe, M.; Bailer, R.T.; Turk, E.; Lin, C.-L.; Bilska, M.; Greene, K.M.; Gao, H.; Todd, C.A.; Ozaki, D.A.; Seaman, M.S.; et al. Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J. Immunol. Methods 2014, 409, 131–146. [Google Scholar] [CrossRef]
- Bentley, E.M.; Mather, S.T.; Temperton, N.J. The use of pseudotypes to study viruses, virus sero-epidemiology and vaccination. Vaccine 2015, 33, 2955–2962. [Google Scholar] [CrossRef] [PubMed]
- Comas-Garcia, M.; Colunga-Saucedo, M.; Rosales-Mendoza, S. The Role of Virus-Like Particles in Medical Biotechnology. Mol. Pharm. 2020, 17, 4407–4420. [Google Scholar] [CrossRef]
- Du, R.; Cui, Q.; Caffrey, M.; Rong, L. Ebola Virus Entry Inhibitors. Adv. Exp. Med. Biol. 2022, 1366, 155–170. [Google Scholar] [CrossRef]
- Kaku, Y.; Noguchi, A.; Marsh, G.A.; Barr, J.A.; Okutani, A.; Hotta, K.; Bazartseren, B.; Fukushi, S.; Broder, C.C.; Yamada, A.; et al. Second generation of pseudotype-based serum neutralization assay for Nipah virus antibodies: Sensitive and high-throughput analysis utilizing secreted alkaline phosphatase. J. Virol. Methods 2012, 179, 226–232. [Google Scholar] [CrossRef]
- Kaku, Y.; Noguchi, A.; Marsh, G.A.; McEachern, J.A.; Okutani, A.; Hotta, K.; Bazartseren, B.; Fukushi, S.; Broder, C.C.; Yamada, A.; et al. A neutralization test for specific detection of Nipah virus antibodies using pseudotyped vesicular stomatitis virus expressing green fluorescent protein. J. Virol. Methods 2009, 160, 7–13. [Google Scholar] [CrossRef]
- Khetawat, D.; Broder, C.C. A Functional Henipavirus Envelope Glycoprotein Pseudotyped Lentivirus Assay System. Virol. J. 2010, 7, 312. [Google Scholar] [CrossRef]
- Nooraei, S.; Bahrulolum, H.; Hoseini, Z.S.; Katalani, C.; Hajizade, A.; Easton, A.J.; Ahmadian, G. Virus-like particles: Preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnology 2021, 19, 59. [Google Scholar] [CrossRef]
- Rudometova, N.B.; Shcherbakov, D.N.; Rudometov, A.P.; Ilyichev, A.A.; Karpenko, L.I. Model systems of human immunodef iciency virus (HIV-1) for in vitro eff icacy assessment of candidate vaccines and drugs against HIV-1. Vavilov J. Genet. Breed. 2022, 26, 214–221. [Google Scholar] [CrossRef]
- Steeds, K.; Hall, Y.; Slack, G.S.; Longet, S.; Strecker, T.; Fehling, S.K.; Wright, E.; Bore, J.A.; Koundouno, F.R.; Konde, M.K.; et al. Pseudotyping of VSV with Ebola virus glycoprotein is superior to HIV-1 for the assessment of neutralising antibodies. Sci. Rep. 2020, 10, 14289. [Google Scholar] [CrossRef] [PubMed]
- Steffen, I.; Simmons, G. Pseudotyping Viral Vectors with Emerging Virus Envelope Proteins. Curr. Gene Ther. 2016, 16, 47–55. [Google Scholar] [CrossRef]
- Wang, B.; Meng, X.-J. Structural and molecular biology of hepatitis E virus. Comput. Struct. Biotechnol. J. 2021, 19, 1907–1916. [Google Scholar] [CrossRef]
- Ryu, W. Molecular Virology of Human Pathogenic Viruses, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2017; Chapter 19; pp. 263–275. [Google Scholar] [CrossRef]
- Gasmi, M.; Glynn, J.; Jin, M.-J.; Jolly, D.J.; Yee, J.-K.; Chen, S.-T. Requirements for Efficient Production and Transduction of Human Immunodeficiency Virus Type 1-Based Vectors. J. Virol. 1999, 73, 1828–1834. [Google Scholar] [CrossRef]
- Salmon, P.; Trono, D. Lentiviral Vectors for the Gene Therapy of Lympho-Hematological Disorders. Curr. Top. Microbiol. Immunol. 2002, 261, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Todd, C.A.; Greene, K.M.; Yu, X.; Ozaki, D.A.; Gao, H.; Huang, Y.; Wang, M.; Li, G.; Brown, R.; Wood, B.; et al. Development and implementation of an international proficiency testing program for a neutralizing antibody assay for HIV-1 in TZM-bl cells. J. Immunol. Methods 2012, 375, 57–67. [Google Scholar] [CrossRef]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of Resistant Human Immunodeficiency Virus Type 1 in Patients Receiving Fusion Inhibitor (T-20) Monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Seaman, M.S.; Janes, H.; Hawkins, N.; Grandpre, L.E.; Devoy, C.; Giri, A.; Coffey, R.T.; Harris, L.; Wood, B.; Daniels, M.G.; et al. Tiered Categorization of a Diverse Panel of HIV-1 Env Pseudoviruses for Assessment of Neutralizing Antibodies. J. Virol. 2010, 84, 1439–1452. [Google Scholar] [CrossRef]
- Hoenen, T. Minigenome Systems for Filoviruses. Methods Mol Biol. 2018, 1604, 237–245. [Google Scholar] [CrossRef]
- Cavrois, M.; De Noronha, C.; Greene, W.C. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 2002, 20, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bentsman, G.; Potash, M.J.; Volsky, D.J. Human immunodeficiency virus type 1 efficiently binds to human fetal astrocytes and induces neuroinflammatory responses independent of infection. BMC Neurosci. 2007, 8, 31. [Google Scholar] [CrossRef]
- Saeed, M.F.; Kolokoltsov, A.A.; Davey, R.A. Novel, rapid assay for measuring entry of diverse enveloped viruses, including HIV and rabies. J. Virol. Methods 2006, 135, 143–150. [Google Scholar] [CrossRef]
- Tobiume, M.; Lineberger, J.E.; Lundquist, C.A.; Miller, M.D.; Aiken, C. Nef Does Not Affect the Efficiency of Human Immunodeficiency Virus Type 1 Fusion with Target Cells. J. Virol. 2003, 77, 10645–10650. [Google Scholar] [CrossRef]
- Tscherne, D.M.; Manicassamy, B.; García-Sastre, A. An enzymatic virus-like particle assay for sensitive detection of virus entry. J. Virol. Methods 2010, 163, 336–343. [Google Scholar] [CrossRef]
- Wyma, D.J.; Jiang, J.; Shi, J.; Zhou, J.; Lineberger, J.E.; Miller, M.D.; Aiken, C. Coupling of Human Immunodeficiency Virus Type 1 Fusion to Virion Maturation: A Novel Role of the gp41 Cytoplasmic Tail. J. Virol. 2004, 78, 3429–3435. [Google Scholar] [CrossRef] [PubMed]
- Leroy, H.; Han, M.; Woottum, M.; Bracq, L.; Bouchet, J.; Xie, M.; Benichou, S. Virus-Mediated Cell-Cell Fusion. Int. J. Mol. Sci. 2020, 21, 9644. [Google Scholar] [CrossRef] [PubMed]
- Bossart, K.N.; Broder, C.C. Viral Glycoprotein-Mediated Cell Fusion Assays Using Vaccinia Virus Vectors. Methods Mol. Biol. 2004, 269, 309–331. [Google Scholar] [CrossRef]
- Moulard, M.; Phogat, S.K.; Shu, Y.; Labrijn, A.F.; Xiao, X.; Binley, J.M.; Zhang, M.-Y.; Sidorov, I.A.; Broder, C.C.; Robinson, J.; et al. Broadly cross-reactive HIV-1-neutralizing human monoclonal Fab selected for binding to gp120–CD4–CCR5 complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 6913–6918. [Google Scholar] [CrossRef]
- Saw, W.T.; Matsuda, Z.; Eisenberg, R.J.; Cohen, G.H.; Atanasiu, D. Using a split luciferase assay (SLA) to measure the kinetics of cell–cell fusion mediated by herpes simplex virus glycoproteins. Methods 2015, 90, 68–75. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, J.; Nguyen, L.N.T.; Adkins, J.L.; Schank, M.; Khanal, S.; Dang, X.; Cao, D.; Thakuri, B.K.C.; Lu, Z.; et al. Blockade of SARS-CoV-2 spike protein-mediated cell–cell fusion using COVID-19 convalescent plasma. Sci. Rep. 2021, 11, 5558. [Google Scholar] [CrossRef]
- Oguntuyo, K.Y.; Stevens, C.S.; Hung, C.T.; Ikegame, S.; Acklin, J.A.; Kowdle, S.S.; Carmichael, J.C.; Chiu, H.-P.; Azarm, K.D.; Haas, G.D.; et al. Quantifying Absolute Neutralization Titers against SARS-CoV-2 by a Standardized Virus Neutralization Assay Allows for Cross-Cohort Comparisons of COVID-19 Sera. MBio 2021, 12, e02492-20. [Google Scholar] [CrossRef]
- Collins, F.S.; Woodcock, J.; Graham, B.S.; Arvin, A.; Bieniasz, P.; Ho, D.; Alter, G.; Nussenzweig, M.; Burton, D.; Tavel, J.; et al. Therapeutic Neutralizing Monoclonal Antibodies: Report of a Summit Sponsored by Operation Warp Speed and the National Institutes of Health. Available online: https://www.nih.gov/sites/default/files/research-training/initiatives/activ/20200909-mAb-summit-pub.pdf (accessed on 17 May 2023).
- Li, Y.; O’Dell, S.; Walker, L.M.; Wu, X.; Guenaga, J.; Feng, Y.; Schmidt, S.D.; McKee, K.; Louder, M.K.; Ledgerwood, J.E.; et al. Mechanism of Neutralization by the Broadly Neutralizing HIV-1 Monoclonal Antibody VRC01. J. Virol. 2011, 85, 8954–8967. [Google Scholar] [CrossRef]
- Lorenzi, J.C.C.; Mendoza, P.; Cohen, Y.Z.; Nogueira, L.; Lavine, C.; Sapiente, J.; Wiatr, M.; Mugo, N.R.; Mujugira, A.; Delany, S.; et al. Neutralizing Activity of Broadly Neutralizing Anti-HIV-1 Antibodies against Primary African Isolates. J. Virol. 2021, 95, e01909-20. [Google Scholar] [CrossRef]
- Sanders, D.A. No false start for novel pseudotyped vectors. Curr. Opin. Biotechnol. 2002, 13, 437–442. [Google Scholar] [CrossRef]
- Li, Q.; Liu, Q.; Huang, W.; Li, X.; Wang, Y. Current status on the development of pseudoviruses for enveloped viruses. Rev. Med. Virol. 2017, 28, e1963. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N.; et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726. [Google Scholar] [CrossRef]
- Cho, A.; Muecksch, F.; Schaefer-Babajew, D.; Wang, Z.; Finkin, S.; Gaebler, C.; Ramos, V.; Cipolla, M.; Mendoza, P.; Agudelo, M.; et al. Anti-SARS-CoV-2 receptor-binding domain antibody evolution after mRNA vaccination. Nature 2021, 600, 517–522. [Google Scholar] [CrossRef]
- Dong, J.; Zost, S.J.; Greaney, A.J.; Starr, T.N.; Dingens, A.S.; Chen, E.C.; Chen, R.E.; Case, J.B.; Sutton, R.E.; Gilchuk, P.; et al. Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail. Nat. Microbiol. 2021, 6, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Lusvarghi, S.; Pollett, S.D.; Neerukonda, S.N.; Wang, W.; Wang, R.; Vassell, R.; Epsi, N.J.; Fries, A.C.; Agan, B.K.; Lindholm, D.A.; et al. SARS-CoV-2 BA.1 variant is neutralized by vaccine booster–elicited serum but evades most convalescent serum and therapeutic antibodies. Sci. Transl. Med. 2022, 14, eabn8543. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef]
- Yamasoba, D.; Kimura, I.; Nasser, H.; Morioka, Y.; Nao, N.; Ito, J.; Uriu, K.; Tsuda, M.; Zahradnik, J.; Shirakawa, K.; et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell 2022, 185, 2103–2115.e19. [Google Scholar] [CrossRef]
- Farrell, A.G.; Dadonaite, B.; Greaney, A.J.; Eguia, R.; Loes, A.N.; Franko, N.M.; Logue, J.; Carreño, J.M.; Abbad, A.; Chu, H.Y.; et al. Receptor-Binding Domain (RBD) Antibodies Contribute More to SARS-CoV-2 Neutralization When Target Cells Express High Levels of ACE2. Viruses 2022, 14, 2061. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Soriaga, L.B.; Montiel-Ruiz, M.; Benigni, F.; Noack, J.; Park, Y.-J.; Bianchi, S.; Walls, A.C.; Bowen, J.E.; Zhou, J.; et al. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature 2021, 598, 342–347. [Google Scholar] [CrossRef]
- Van Blargan, L.A.; Errico, J.M.; Halfmann, P.J.; Zost, S.J.; Crowe, J.E.; Purcell, L.A.; Kawaoka, Y.; Corti, D.; Fremont, D.H.; Diamond, M.S. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 2022, 28, 490–495. [Google Scholar] [CrossRef] [PubMed]
- FDA. Integrated Review Application Number 761172. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761172Orig1s000IntegratedR.pdf (accessed on 17 February 2023).
- FDA. Multi-Discipline Review, Application Number 761169. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761169Orig1s000MultidisciplineR.pdf (accessed on 17 February 2023).
- Hsieh, Y.-T.; Aggarwal, P.; Cirelli, D.; Gu, L.; Surowy, T.; Mozier, N.M. Characterization of FcγRIIIA effector cells used in in vitro ADCC bioassay: Comparison of primary NK cells with engineered NK-92 and Jurkat T cells. J. Immunol. Methods 2017, 441, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Parekh, B.S.; Berger, E.; Sibley, S.; Cahya, S.; Xiao, L.; LaCerte, M.A.; Vaillancourt, P.; Wooden, S.; Gately, D. Development and validation of an antibody-dependent cell-mediated cytotoxicity-reporter gene assay. mAbs 2012, 4, 310–318. [Google Scholar] [CrossRef]
- de Taeye, S.W.; Rispens, T.; Vidarsson, G. The Ligands for Human IgG and Their Effector Functions. Antibodies 2019, 8, 30. [Google Scholar] [CrossRef]
- Goncalvez, A.P.; Engle, R.E.; St Claire, M.; Purcell, R.H.; Lai, C.-J. Monoclonal antibody-mediated enhancement of dengue virus infection in vitro and in vivo and strategies for prevention. Proc. Natl. Acad. Sci. USA 2007, 104, 9422–9427. [Google Scholar] [CrossRef]
- Huang, X.; Yue, Y.; Li, D.; Zhao, Y.; Qiu, L.; Chen, J.; Pan, Y.; Xi, J.; Wang, X.; Sun, Q.; et al. Antibody-dependent enhancement of dengue virus infection inhibits RLR-mediated Type-I IFN-independent signalling through upregulation of cellular autophagy. Sci. Rep. 2016, 6, 22303. [Google Scholar] [CrossRef]
- Littaua, R.; Kurane, I.; Ennis, F.A. Human IgG Fc receptor II mediates antibody-dependent enhancement of dengue virus infection. 1990, 144, 3183–3186. J. Immunol. [CrossRef]
- Stettler, K.; Beltramello, M.; Espinosa, D.A.; Graham, V.; Cassotta, A.; Bianchi, S.; Vanzetta, F.; Minola, A.; Jaconi, S.; Mele, F.; et al. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 2016, 353, 823–826. [Google Scholar] [CrossRef]
- Shafer, R.W.; Chou, S. Mechanisms of Resistance to Antiviral Agents. Man. Clin. Microbiol. 2015, 111, 1894–1912. [Google Scholar] [CrossRef]
- Vere Hodge, A.; Field, H.J. General Mechanisms of Antiviral Resistance. Genet. Evol. Infect. Dis. 2011, 339–362. [Google Scholar] [CrossRef]
- FDA. Antiviral Product Development—Conducting and Submitting Virology Studies to the Agency, Guidance for Industry. Available online: https://www.fda.gov/media/71223/download (accessed on 2 May 2023).
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef]
- Copin, R.; Baum, A.; Wloga, E.; Pascal, K.E.; Giordano, S.; Fulton, B.O.; Zhou, A.; Negron, N.; Lanza, K.; Chan, N.; et al. The monoclonal antibody combination REGEN-COV protects against SARS-CoV-2 mutational escape in preclinical and human studies. Cell 2021, 184, 3949–3961.e11. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Ibalizumab Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761065lbl.pdf (accessed on 2 May 2023).
- Ruiz, S.I.; Zumbrun, E.E.; Nalca, A. Animal Models of Human Viral Diseases. Anim. Model. Study Hum. Dis. 2017, 853–901. [Google Scholar]
- Beddingfield, B.J.; Maness, N.J.; Fears, A.C.; Rappaport, J.; Aye, P.P.; Russell-Lodrigue, K.; Doyle-Meyers, L.A.; Blair, R.V.; Carias, A.M.; Madden, P.J.; et al. Effective Prophylaxis of COVID-19 in Rhesus Macaques Using a Combination of Two Parenterally-Administered SARS-CoV-2 Neutralizing Antibodies. Front. Cell. Infect. Microbiol. 2021, 11, 753444. [Google Scholar] [CrossRef]
- Haagmans, B.L.; Noack, D.; Okba, N.M.A.; Li, W.; Wang, C.; Bestebroer, T.; de Vries, R.; Herfst, S.; de Meulder, D.; Verveer, E.; et al. SARS-CoV-2 Neutralizing Human Antibodies Protect against Lower Respiratory Tract Disease in a Hamster Model. J. Infect. Dis. 2021, 223, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; Barker, D.; Lew, J.; Manoharan, V.; van Kessel, J.; Haupt, R.; Toth, D.; Frieman, M.; Falzarano, D.; Kodihalli, S. Efficacy of COVID-HIGIV in animal models of SARS-CoV-2 infection. Sci. Rep. 2022, 12, 16956. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ryu, D.-K.; Lee, J.; Kim, Y.-I.; Seo, J.-M.; Kim, Y.-G.; Jeong, J.-H.; Kim, M.; Kim, J.-I.; Kim, P.; et al. A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat. Commun. 2021, 12, 288. [Google Scholar] [CrossRef]
- Maisonnasse, P.; Aldon, Y.; Marc, A.; Marlin, R.; Dereuddre-Bosquet, N.; Kuzmina, N.A.; Freyn, A.W.; Snitselaar, J.L.; Gonçalves, A.; Caniels, T.G.; et al. COVA1-18 neutralizing antibody protects against SARS-CoV-2 in three preclinical models. Nat. Commun. 2021, 12, 6097. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.S.; Gilchuk, P.; Yu, J.; Bailey, A.L.; Chen, R.E.; Chong, Z.; Zost, S.J.; Jang, H.; Huang, Y.; Allen, J.D.; et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell 2021, 184, 1804–1820.e16. [Google Scholar] [CrossRef]
- Yadav, P.D.; Mendiratta, S.K.; Mohandas, S.; Singh, A.K.; Abraham, P.; Shete, A.; Bandyopadhyay, S.; Kumar, S.; Parikh, A.; Kalita, P.; et al. ZRC3308 Monoclonal Antibody Cocktail Shows Protective Efficacy in Syrian Hamsters against SARS-CoV-2 Infection. Viruses 2021, 13, 2424. [Google Scholar] [CrossRef] [PubMed]
- Zost, S.J.; Gilchuk, P.; Case, J.B.; Binshtein, E.; Chen, R.E.; Nkolola, J.P.; Schäfer, A.; Reidy, J.X.; Trivette, A.; Nargi, R.S.; et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 2020, 584, 443–449. [Google Scholar] [CrossRef]
- FDA. S6(R1) Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals, Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/s6r1-preclinical-safety-evaluation-biotechnology-derived-pharmaceuticals (accessed on 17 February 2023).
- Mahmood, I.; Tegenge, M.A. Prediction of tissue concentrations of monoclonal antibodies in mice from plasma concentrations. Regul. Toxicol. Pharmacol. 2018, 97, 57–62. [Google Scholar] [CrossRef]
- Keeler, S.P.; Fox, J.M. Requirement of Fc-Fc Gamma Receptor Interaction for Antibody-Based Protection against Emerging Virus Infections. Viruses 2021, 13, 1037. [Google Scholar] [CrossRef]
- Schmaljohn, A.L.; Orlandi, C.; Lewis, G.K. Deciphering Fc-mediated Antiviral Antibody Functions in Animal Models. Front. Immunol. 2019, 10, 1602. [Google Scholar] [CrossRef]
- Robbie, G.J.; Criste, R.; Dall’Acqua, W.F.; Jensen, K.; Patel, N.K.; Losonsky, G.A.; Griffin, M.P. A Novel Investigational Fc-Modified Humanized Monoclonal Antibody, Motavizumab-YTE, Has an Extended Half-Life in Healthy Adults. Antimicrob. Agents Chemother. 2013, 57, 6147–6153. [Google Scholar] [CrossRef]
- Smith, P.; Di Lillo, D.J.; Bournazos, S.; Li, F.; Ravetch, J.V. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc. Natl. Acad. Sci. USA 2012, 109, 6181–6186. [Google Scholar] [CrossRef] [PubMed]
- Proetzel, G.; Roopenian, D.C. Humanized FcRn mouse models for evaluating pharmacokinetics of human IgG antibodies. Methods 2013, 65, 148–153. [Google Scholar] [CrossRef]
- FDA. Product Development under the Animal Rule, Guidance for Industry. Available online: https://www.fda.gov/media/88625/download (accessed on 17 February 2023).
- FDA. Animal Rule Approvals. Available online: https://www.fda.gov/drugs/nda-and-bla-approvals/animal-rule-approvals (accessed on 17 February 2023).
- Sharp, J.C.; Fletcher, W.B. Experience of anti-vaccinia immunoglobulin in the United Kingdom. Lancet 1973, 301, 656–659. [Google Scholar] [CrossRef]
- Bahmanyar, M.; Fayaz, A.; Nour-Salehi, S.; Mohammadi, M.; Koprowski, H. Successful protection of humans exposed to rabies infection. Postexposure treatment with the new human diploid cell rabies vaccine and antirabies serum. JAMA 1976, 236, 2751–2754. [Google Scholar] [CrossRef] [PubMed]
- Nelson, N.P.; Weng, M.K.; Hofmeister, M.G.; Moore, K.L.; Doshani, M.; Kamili, S.; Koneru, A.; Haber, P.; Hagan, L.; Romero, J.R.; et al. Prevention of Hepatitis A Virus Infection in the United States: Recommendations of the Advisory Committee on Immunization Practices, 2020. MMWR. Recomm. Rep. 2020, 69, 1–38. [Google Scholar] [CrossRef]
- Fisher, R.W.; Reed, J.L.; Snoy, P.J.; Mikolajczyk, M.G.; Bray, M.; Scott, D.E.; Kennedy, M.C. Postexposure Prevention of Progressive Vaccinia in SCID Mice Treated with Vaccinia Immune Globulin. Clin. Vaccine Immunol. 2011, 18, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.; Wright, M.E. Progressive Vaccinia. Clin. Infect. Dis. 2003, 36, 766–774. [Google Scholar] [CrossRef]
- FDA. REGEN-COV (Casirivimab and Imdevimab). Available online: https://www.fda.gov/media/145611/download (accessed on 17 February 2023).
- FDA. Bamlanivimab and Etesevimab. Available online: https://www.fda.gov/media/145802/download (accessed on 17 February 2023).
- FDA. EVUSHELD™ (Tixagevimab Co-Packaged with Cilgavimab). Available online: https://www.fda.gov/media/154701/download (accessed on 17 February 2023).
- Ejemel, M.; Smith, T.G.; Greenberg, L.; Carson, W.C.; Lowe, D.; Yang, Y.; Jackson, F.R.; Morgan, C.N.; Martin, B.E.; Kling, C.; et al. A cocktail of human monoclonal antibodies broadly neutralizes North American rabies virus variants as a promising candidate for rabies post-exposure prophylaxis. Sci. Rep. 2022, 12, 9403. [Google Scholar] [CrossRef]
- Koszalka, P.; Subbarao, K.; Baz, M. Preclinical and clinical developments for combination treatment of influenza. PLoS Pathog. 2022, 18, e1010481. [Google Scholar] [CrossRef]
- Maertens, J.; Logan, A.C.; Jang, J.; Long, G.; Tang, J.-L.; Hwang, W.Y.K.; Koh, L.P.; Chemaly, R.; Gerbitz, A.; Winkler, J.; et al. Phase 2 Study of Anti-Human Cytomegalovirus Monoclonal Antibodies for Prophylaxis in Hematopoietic Cell Transplantation. Antimicrob. Agents Chemother. 2020, 64, e02467-19. [Google Scholar] [CrossRef] [PubMed]
- Mahomed, S.; Garrett, N.; Baxter, C.; Karim, Q.A.; Karim, S.S.A. Clinical Trials of Broadly Neutralizing Monoclonal Antibodies for Human Immunodeficiency Virus Prevention: A Review. J. Infect. Dis. 2020, 223, 370–380. [Google Scholar] [CrossRef] [PubMed]
- FDA. Codevelopment of Two or More New Investigational Drugs for Use in Combination, Guidance for Industry. Available online: https://www.fda.gov/media/80100/download (accessed on 17 February 2023).
- Verrier, F.; Nádas, A.; Gorny, M.K.; Zolla-Pazner, S. Additive Effects Characterize the Interaction of Antibodies Involved in Neutralization of the Primary Dualtropic Human Immunodeficiency Virus Type 1 Isolate 89.6. J. Virol. 2001, 75, 9177–9186. [Google Scholar] [CrossRef]
- Keck, Z.-Y.; Girard-Blanc, C.; Wang, W.; Lau, P.; Zuiani, A.; Rey, F.A.; Krey, T.; Diamond, M.S.; Foung, S.K.H. Antibody Response to Hypervariable Region 1 Interferes with Broadly Neutralizing Antibodies to Hepatitis C Virus. J. Virol. 2016, 90, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Mankowski, M.C.; Kinchen, V.J.; Wasilewski, L.N.; Flyak, A.I.; Ray, S.C.; Crowe, J.E.; Bailey, J.R. Synergistic anti-HCV broadly neutralizing human monoclonal antibodies with independent mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, E82–E91. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Katinger, H.; Posner, M.R.; Cavacini, L.; Zolla-Pazner, S.; Gorny, M.K.; Sodroski, J.; Chou, T.-C.; Baba, T.W.; Ruprecht, R.M. Synergistic Neutralization of Simian-Human Immunodeficiency Virus SHIV-vpu + by Triple and Quadruple Combinations of Human Monoclonal Antibodies and High-Titer Anti-Human Immunodeficiency Virus Type 1 Immunoglobulins. J. Virol. 1998, 72, 3235–3240. [Google Scholar] [CrossRef]
- Miglietta, R.; Pastori, C.; Venuti, A.; Ochsenbauer, C.; Lopalco, L. Synergy in monoclonal antibody neutralization of HIV-1 pseudoviruses and infectious molecular clones. J. Transl. Med. 2014, 12, 346. [Google Scholar] [CrossRef]
- Ter Meulen, J.; Van Den Brink, E.N.; Poon, L.L.M.; Marissen, W.E.; Leung, C.S.W.; Cox, F.; Cheung, C.Y.; Bakker, A.Q.; Bogaards, J.A.; van Deventer, E.; et al. Human Monoclonal Antibody Combination against SARS Coronavirus: Synergy and Coverage of Escape Mutants. PLoS Med. 2006, 3, e237. [Google Scholar] [CrossRef]
- Zhong, L.; Haynes, L.; Struble, E.B.; Tamin, A.; Virata-Theimer, M.L.; Zhang, P. Antibody-mediated synergy and interference in the neutralization of SARS-CoV at an epitope cluster on the spike protein. Biochem. Biophys. Res. Commun. 2009, 390, 1056–1060. [Google Scholar] [CrossRef]
- Patel, H.D.; Nikitin, P.; Gesner, T.; Lin, J.J.; Barkan, D.T.; Ciferri, C.; Carfi, A.; Yazdi, T.A.; Skewes-Cox, P.; Wiedmann, B.; et al. In Vitro Characterization of Human Cytomegalovirus-Targeting Therapeutic Monoclonal Antibodies LJP538 and LJP539. Antimicrob. Agents Chemother. 2016, 60, 4961–4971. [Google Scholar] [CrossRef]
- Gottlieb, R.L.; Nirula, A.; Chen, P.; Boscia, J.; Heller, B.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. Effect of Bamlanivimab as Monotherapy or in Combination with Etesevimab on Viral Load in Patients with Mild to Moderate COVID-19: A Randomized Clinical Trial. JAMA 2021, 325, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.A.; Shapiro, M.B.; Haigwood, N.L.; Hessell, A.J. Advancing HIV Broadly Neutralizing Antibodies: From Discovery to the Clinic. Front. Public Health 2021, 9, 690017. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.; Peacock, T.P.; Harvey, W.T.; Hughes, J.; Wright, D.W.; Willett, B.J.; Thomson, E.; Gupta, R.K.; Peacock, S.J.; Robertson, D.L.; et al. SARS-CoV-2 variant evasion of monoclonal antibodies based on in vitro studies. Nat. Rev. Microbiol. 2023, 21, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Julg, B.; Stephenson, K.E.; Wagh, K.; Tan, S.C.; Zash, R.; Walsh, S.; Ansel, J.; Kanjilal, D.; Nkolola, J.; Walker-Sperling, V.E.K.; et al. Safety and antiviral activity of triple combination broadly neutralizing monoclonal antibody therapy against HIV-1: A phase 1 clinical trial. Nat. Med. 2022, 28, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.E.Z.; Seah, S.G.K.; Chye, D.H.; Massey, S.; Torres, M.; Lim, A.P.C.; Wong, S.K.K.; Neo, J.J.Y.; Wong, P.S.; Lim, J.H.; et al. The Fc-mediated effector functions of a potent SARS-CoV-2 neutralizing antibody, SC31, isolated from an early convalescent COVID-19 patient, are essential for the optimal therapeutic efficacy of the antibody. PLoS ONE 2021, 16, e0253487. [Google Scholar] [CrossRef]
- Schäfer, A.; Muecksch, F.; Lorenzi, J.C.; Leist, S.R.; Cipolla, M.; Bournazos, S.; Schmidt, F.; Maison, R.M.; Gazumyan, A.; Martinez, D.R.; et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J. Exp. Med. 2021, 218, e20201993. [Google Scholar] [CrossRef]
- Yamin, R.; Jones, A.T.; Hoffmann, H.-H.; Schäfer, A.; Kao, K.S.; Francis, R.L.; Sheahan, T.P.; Baric, R.S.; Rice, C.M.; Ravetch, J.V.; et al. Fc-engineered antibody therapeutics with improved anti-SARS-CoV-2 efficacy. Nature 2021, 599, 465–470. [Google Scholar] [CrossRef]
- Chigutsa, E.; Jordie, E.; Riggs, M.; Nirula, A.; Elmokadem, A.; Knab, T.; Chien, J.Y. A Quantitative Modeling and Simulation Framework to Support Candidate and Dose Selection of Anti-SARS-CoV-2 Monoclonal Antibodies to Advance Bamlanivimab into a First-in-Human Clinical Trial. Clin. Pharmacol. Ther. 2021, 111, 595–604. [Google Scholar] [CrossRef]
- Chigutsa, E.; O’brien, L.; Ferguson-Sells, L.; Long, A.; Chien, J. Population Pharmacokinetics and Pharmacodynamics of the Neutralizing Antibodies Bamlanivimab and Etesevimab in Patients with Mild to Moderate COVID-19 Infection. Clin. Pharmacol. Ther. 2021, 110, 1302–1310. [Google Scholar] [CrossRef]
- Magyarics, Z.; Leslie, F.; Bartko, J.; Rouha, H.; Luperchio, S.; Schörgenhofer, C.; Schwameis, M.; Derhaschnig, U.; Lagler, H.; Stiebellehner, L.; et al. Randomized, Double-Blind, Placebo-Controlled, Single-Ascending-Dose Study of the Penetration of a Monoclonal Antibody Combination (ASN100) Targeting Staphylococcus aureus Cytotoxins in the Lung Epithelial Lining Fluid of Healthy Volunteers. Antimicrob. Agents Chemother. 2019, 63, e00350-19. [Google Scholar] [CrossRef]
- Nehls, J.; Businger, R.; Hoffmann, M.; Brinkmann, C.; Fehrenbacher, B.; Schaller, M.; Maurer, B.; Schönfeld, C.; Kramer, D.; Hailfinger, S.; et al. Release of Immunomodulatory Ebola Virus Glycoprotein-Containing Microvesicles Is Suppressed by Tetherin in a Species-Specific Manner. Cell Rep. 2019, 26, 1841–1853.e6. [Google Scholar] [CrossRef] [PubMed]
- Rydell, G.E.; Prakash, K.; Norder, H.; Lindh, M. Hepatitis B surface antigen on subviral particles reduces the neutralizing effect of anti-HBs antibodies on hepatitis B viral particles in vitro. Virology 2017, 509, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Deming, D.J.; Patel, N.; McCarthy, M.P.; Mishra, L.; Shapiro, A.M.; Suzich, J.A. Potential for Palivizumab Interference with Commercially Available Antibody–antigen Based Respiratory Syncytial Virus Diagnostic Assays. Pediatr. Infect. Dis. J. 2013, 32, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- General Chapter: USP. General Tests and Assays, Biological Tests and Assays, <85> Bacterial Endotoxins Test. The United States Pharmacopeia—National Formulary, Rockville, MD. Available online: https://doi.usp.org/USPNF/USPNF_M98830_02_01.html (accessed on 17 May 2023).
- Vir Biothechnology, Inc. Press Release, 11 June 2022. Available online: https://investors.vir.bio/news-releases/news-release-details/vir-biotechnology-presents-new-data-evaluating-potential-vir-0 (accessed on 17 February 2023).
- Vir Biothechnology, Inc. Press Release, 25 June 22. Available online: https://investors.vir.bio/news-releases/news-release-details/vir-biotechnology-announces-new-clinical-data-its-broad (accessed on 22 February 2022).
- Xiao, F.; Fofana, I.; Thumann, C.; Mailly, L.; Alles, R.; Robinet, E.; Meyer, N.; Schaeffer, M.; Habersetzer, F.; Doffoël, M.; et al. Synergy of entry inhibitors with direct-acting antivirals uncovers novel combinations for prevention and treatment of hepatitis C. Gut 2015, 64, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Caskey, M.; Klein, F.; Lorenzi, J.C.C.; Seaman, M.S.; West, A.P., Jr.; Buckley, N.; Kremer, G.; Nogueira, L.; Braunschweig, M.; Scheid, J.F.; et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 2015, 522, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Mouquet, H.; Scharf, L.; Euler, Z.; Liu, Y.; Eden, C.; Scheid, J.F.; Halper-Stromberg, A.; Gnanapragasam, P.N.P.; Spencer, D.I.R.; Seaman, M.S.; et al. Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc. Natl. Acad. Sci. USA 2012, 109, E3268–E3277. [Google Scholar] [CrossRef] [PubMed]
- Clinicalinfo. Available online: https://clinicalinfo.hiv.gov/en/drugs (accessed on 3 May 2023).
- Kiso, M.; Yamayoshi, S.; Kawaoka, Y. Triple combination therapy of favipiravir plus two monoclonal antibodies eradicates influenza virus from nude mice. Commun. Biol. 2020, 3, 219. [Google Scholar] [CrossRef]
- Cross, R.W.; Bornholdt, Z.A.; Prasad, A.N.; Borisevich, V.; Agans, K.N.; Deer, D.J.; Abelson, D.M.; Kim, D.H.; Shestowsky, W.S.; Campbell, L.A.; et al. Combination therapy protects macaques against advanced Marburg virus disease. Nat. Commun. 2021, 12, 1891. [Google Scholar] [CrossRef]
- Cross, R.W.; Bornholdt, Z.A.; Prasad, A.N.; Woolsey, C.; Borisevich, V.; Agans, K.N.; Deer, D.J.; Abelson, D.M.; Kim, D.H.; Shestowsky, W.S.; et al. Combination therapy with remdesivir and monoclonal antibodies protects nonhuman primates against advanced Sudan virus disease. J. Clin. Investig. 2022, 7, e159090. [Google Scholar] [CrossRef]
- Nakamura, G.; Chai, N.; Park, S.; Chiang, N.; Lin, Z.; Chiu, H.; Fong, R.; Yan, D.; Kim, J.; Zhang, J.; et al. An In Vivo Human-Plasmablast Enrichment Technique Allows Rapid Identification of Therapeutic Influenza a Antibodies. Cell Host Microbe 2013, 14, 93–103. [Google Scholar] [CrossRef]
- Paules, C.I.; Lakdawala, S.; McAuliffe, J.M.; Paskel, M.; Vogel, L.; Kallewaard, N.L.; Zhu, Q.; Subbarao, K. The Hemagglutinin A Stem Antibody MEDI8852 Prevents and Controls Disease and Limits Transmission of Pandemic Influenza Viruses. J. Infect. Dis. 2017, 216, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Tharakaraman, K.; Subramanian, V.; Viswanathan, K.; Sloan, S.; Yen, H.-L.; Barnard, D.L.; Leung, Y.H.C.; Szretter, K.J.; Koch, T.J.; Delaney, J.C.; et al. A broadly neutralizing human monoclonal antibody is effective against H7N9. Proc. Natl. Acad. Sci. USA 2015, 112, 10890–10895. [Google Scholar] [CrossRef] [PubMed]
- Yi, K.S.; Choi, J.-A.; Kim, P.; Ryu, D.-K.; Yang, E.; Son, D.; Shin, J.; Park, H.; Lee, S.; Lee, H.; et al. Broader neutralization of CT-P27 against influenza A subtypes by combining two human monoclonal antibodies. PLoS ONE 2020, 15, e0236172. [Google Scholar] [CrossRef] [PubMed]
- Lindorfer, M.A.; Taylor, R.P. FcγR-Mediated Trogocytosis 2.0: Revisiting History Gives Rise to a Unifying Hypothesis. Antibodies 2022, 11, 45. [Google Scholar] [CrossRef]
- Massanella, M.; Puigdomènech, I.; Cabrera, C.; Fernandez-Figueras, M.T.; Aucher, A.; Gaibelet, G.; Hudrisier, D.; García, E.; Bofill, M.; Clotet, B.; et al. Antigp41 antibodies fail to block early events of virological synapses but inhibit HIV spread between T cells. Aids 2009, 23, 183–188. [Google Scholar] [CrossRef]
- Richardson, S.I.; Crowther, C.; Mkhize, N.N.; Morris, L. Measuring the ability of HIV-specific antibodies to mediate trogocytosis. J. Immunol. Methods 2018, 463, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Kramski, M.; Parsons, M.S.; Stratov, I.; Kent, S.J. HIV-specific antibody immunity mediated through NK cells and monocytes. Curr. HIV Res. 2013, 11, 388–406. [Google Scholar] [CrossRef] [PubMed]
- Shiakolas, A.R.; Kramer, K.J.; Wrapp, D.; Richardson, S.I.; Schäfer, A.; Wall, S.; Wang, N.; Janowska, K.; Pilewski, K.A.; Venkat, R.; et al. Cross-reactive coronavirus antibodies with diverse epitope specificities and Fc effector functions. Cell Rep. Med. 2021, 2, 100313. [Google Scholar] [CrossRef]
- Low, L.A.; Sutherland, M.; Lumelsky, N.; Selimovic, S.; Lundberg, M.S.; Tagle, D.A. Organs-on-a-Chip. Adv. Exp. Med. Biol. 2020, 1230, 27–42. [Google Scholar] [CrossRef]
- Washburn, N.; Schwab, I.; Ortiz, D.; Bhatnagar, N.; Lansing, J.C.; Medeiros, A.; Tyler, S.; Mekala, D.; Cochran, E.; Sarvaiya, H.; et al. Controlled tetra-Fc sialylation of IVIg results in a drug candidate with consistent enhanced anti-inflammatory activity. Proc. Natl. Acad. Sci. USA 2015, 112, E1297–E1306. [Google Scholar] [CrossRef]
- Keating, S.M.; Mizrahi, R.A.; Adams, M.S.; Asensio, M.A.; Benzie, E.; Carter, K.P.; Chiang, Y.; Edgar, R.C.; Gautam, B.K.; Gras, A.; et al. Generation of recombinant hyperimmune globulins from diverse B-cell repertoires. Nat. Biotechnol. 2021, 39, 989–999. [Google Scholar] [CrossRef]
- Frost, G.I. Recombinant human hyaluronidase (rHuPH20): An enabling platform for subcutaneous drug and fluid administration. Expert Opin. Drug Deliv. 2007, 4, 427–440. [Google Scholar] [CrossRef]
- Pitiot, A.; Heuzé-Vourc’h, N.; Sécher, T. Alternative Routes of Administration for Therapeutic Antibodies—State of the Art. Antibodies 2022, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Mahomed, S.; Garrett, N.; Karim, Q.A.; Zuma, N.Y.; Capparelli, E.; Baxter, C.; Gengiah, T.; Archary, D.; Samsunder, N.; Rose, N.D.; et al. Assessing the safety and pharmacokinetics of the anti-HIV monoclonal antibody CAP256V2LS alone and in combination with VRC07-523LS and PGT121 in South African women: Study protocol for the first-in-human CAPRISA 012B phase I clinical trial. BMJ Open 2020, 10, e042247. [Google Scholar] [CrossRef]
- Nyakatura, E.K.; Soare, A.Y.; Lai, J.R. Bispecific antibodies for viral immunotherapy. Hum. Vaccines Immunother. 2017, 13, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Sroga, P.; Safronetz, D.; Stein, D.R. Nanobodies: A new approach for the diagnosis and treatment of viral infectious diseases. Futur. Virol. 2020, 15, 195–205. [Google Scholar] [CrossRef]
- Walser, M.; Mayor, J.; Rothenberger, S. Designed Ankyrin Repeat Proteins: A New Class of Viral Entry Inhibitors. Viruses 2022, 14, 2242. [Google Scholar] [CrossRef] [PubMed]
- Wensel, D.; Sun, Y.; Davis, J.; Li, Z.; Zhang, S.; McDonagh, T.; Fabrizio, D.; Cockett, M.; Krystal, M. A Novel gp41-Binding Adnectin with Potent Anti-HIV Activity Is Highly Synergistic when Linked to a CD4-Binding Adnectin. J. Virol. 2018, 92, e00421-18. [Google Scholar] [CrossRef] [PubMed]
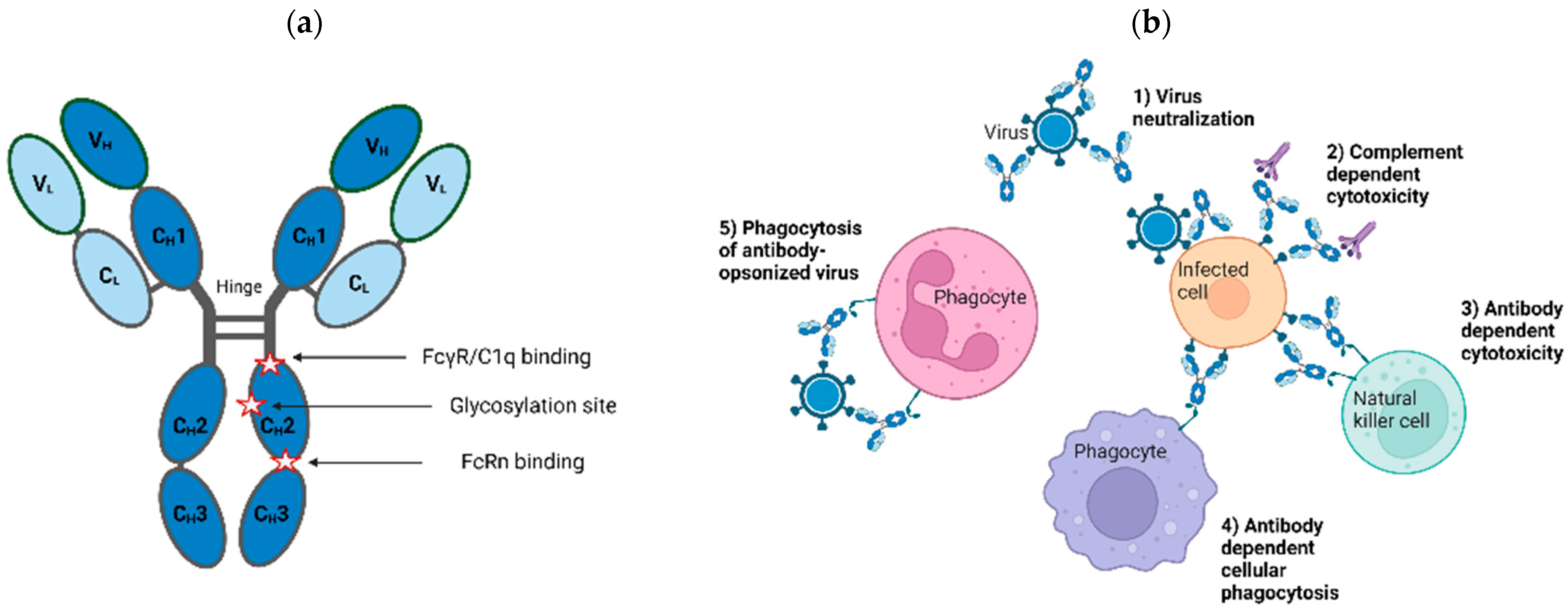

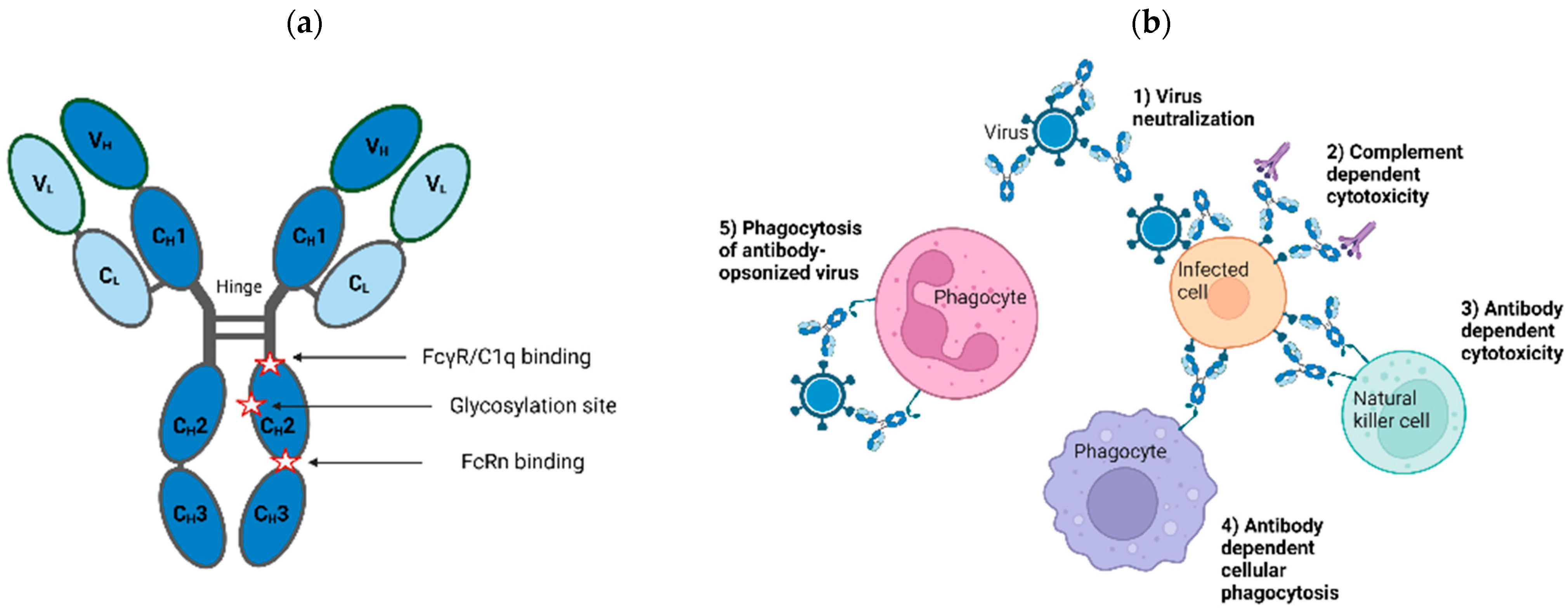{kind=link}
| Target | Trade Name(s) | Donors | Indications | Used With |
|---|---|---|---|---|
| Rabies [44,45,46] | HyperRAB, Imogam, KedRab | Vaccinated | Post-exposure prophylaxis | Rabies vaccine (required, see prescribing information for rabies IG) |
| Varicella [47] | VARIZIG | Donations selected from high-titer donors after natural infection * | Post-exposure prophylaxis in patients at risk for severe infection | Concomitant use of acyclovir reported to occur in clinical practice [48] |
| Vaccinia [49] | Vaccinia Immune Globulin (Human) | Vaccinated | Treatment of severe complications after smallpox vaccination | Investigational antiviral drugs and/or cidofovir [50,51] |
| Cytomegalovirus (CMV) [41] | CytoGam | Donations selected from source plasma | Prevention of CMV disease in patients receiving organ transplants from CMV donors | Ganciclovir recommended in prescribing information; other drugs recommended in practice guidelines [52] |
| Hepatitis A (HAV) [53] | GamaSTAN | Regular donors | Pre- and post-exposure prophylaxis | None |
| Hepatitis B (HBVIG/IGIV) [54,55,56] | HyperHEP B, Nabi-HB | Vaccinated | Post-exposure prophylaxis | None |
| HepaGam-B | Vaccinated | Post-exposure prophylaxis Prevention of HBV recurrence in HBsAg+ liver transplant recipients | Concomitant treatment with other drugs recommended in practice guidelines [57] | |
| Measles [53] † | GamaSTAN | Regular donors | Prevention or attenuation of measles in susceptible individuals | None |
| Rubella [28] | GamaSTAN | Regular donors | To modify rubella in exposed pregnant women who will not be undergoing a therapeutic abortion | None |
| Non-Proprietary Name | Trade Name | Target | Indication |
|---|---|---|---|
| palivizumab | Synagis | RSV F protein | For the prevention of serious lower respiratory tract disease resulting from RSV in pediatric patients (specific conditions and age limitations) |
| ibalizumab | Trogarzo | CD4 (post-attachment HIV-1 inhibitor) | In combination with other antiretroviral(s), for the treatment of HIV-1 infection in heavily treatment-experienced adults with multidrug-resistant HIV-1 infection failing their current antiretroviral regimen |
| atoltivimab, maftivimab, odesivimab-ebgn | Inmazeb | Ebola virus glycoprotein | For the treatment of infection resulting from Zaire ebolavirus in adult and pediatric patients, including neonates born to a mother who is RT-PCR positive for Zaire ebolavirus infection |
| ansuvimab-zykl | Ebanga | Ebola virus glycoprotein | For the treatment of infection resulting from Zaire ebolavirus in adult and pediatric patients, including neonates born to a mother who is RT-PCR positive for Zaire ebolavirus infection |
| Specific Polyclonal Antibodies | Monoclonal Antibodies | |
|---|---|---|
| Advantages |
|
|
| Disadvantages |
|
|
| Virus | mAb(s) (Target) | Antiviral(s) | Stage | References |
|---|---|---|---|---|
| HBV/HDV | VIR-3434 (HBsAg) | VIR-2218 ± NrtI ± pegIFN | Phase 2 | NCT04856085 *, also see [232,233] |
| HCV | various (HCV receptors) † | Various † | Pre-clinical | [234] |
| HIV-1 | Teropavimab + zinlirvimab (gp120) | Various ‡ | Phase 1–2 | NCT04811040 *, also see [235,236,237] |
| HIV-1 | Ibalizumab (CD4) | Approved antiretrovirals | Approved | [178] |
| IAV | Various (HA) § | Oseltamivir | Phase 2 | [206] |
| IAV | CR9114 + F3A19 (HA) | Favipiravir | Pre-clinical | [238] |
| MARV | MR186-YTE (GP) | Remdesivir | Pre-clinical | [239] |
| SUDV | ADI-15878 + ADI-23774 (GP) | Remdesivir | Pre-clinical | [240] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Struble, E.B.; Rawson, J.M.O.; Stantchev, T.; Scott, D.; Shapiro, M.A. Uses and Challenges of Antiviral Polyclonal and Monoclonal Antibody Therapies. Pharmaceutics 2023, 15, 1538. https://doi.org/10.3390/pharmaceutics15051538
Struble EB, Rawson JMO, Stantchev T, Scott D, Shapiro MA. Uses and Challenges of Antiviral Polyclonal and Monoclonal Antibody Therapies. Pharmaceutics. 2023; 15(5):1538. https://doi.org/10.3390/pharmaceutics15051538
Chicago/Turabian StyleStruble, Evi B., Jonathan M. O. Rawson, Tzanko Stantchev, Dorothy Scott, and Marjorie A. Shapiro. 2023. "Uses and Challenges of Antiviral Polyclonal and Monoclonal Antibody Therapies" Pharmaceutics 15, no. 5: 1538. https://doi.org/10.3390/pharmaceutics15051538
APA StyleStruble, E. B., Rawson, J. M. O., Stantchev, T., Scott, D., & Shapiro, M. A. (2023). Uses and Challenges of Antiviral Polyclonal and Monoclonal Antibody Therapies. Pharmaceutics, 15(5), 1538. https://doi.org/10.3390/pharmaceutics15051538