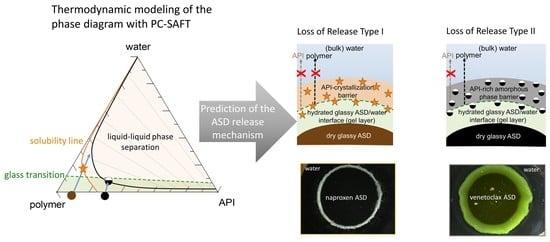
4.1. Modeling Approach for the ASD/Water Interface
As soon as the surface of an ASD encounters water, several interplay processes, including water sorption, phase separation, crystallization, and the viscosity of the formed (gel) layer, determine the ASD/water interfacial behavior and, subsequently, the dissolution performance of the formulation. The physicochemical properties of the APIs and the polymer, their interactions in the ASD, and, ultimately, their interactions with water determine the thermodynamics and kinetics of these processes. Previous works by Sadowski and coauthors demonstrated that phase transformations (crystallization and phase separation) associated with ASD/water can be thermodynamically modeled quantitatively [
11,
38,
39,
53,
58,
59,
60,
61]. The kinetics aspect is strongly influenced by the glass transition behavior of the system and the individual components [
12]. The interplay between phase transformations and glass transition during the ASD dissolution can be understood through the API/polymer/water ternary phase, coupled with the glass transition.
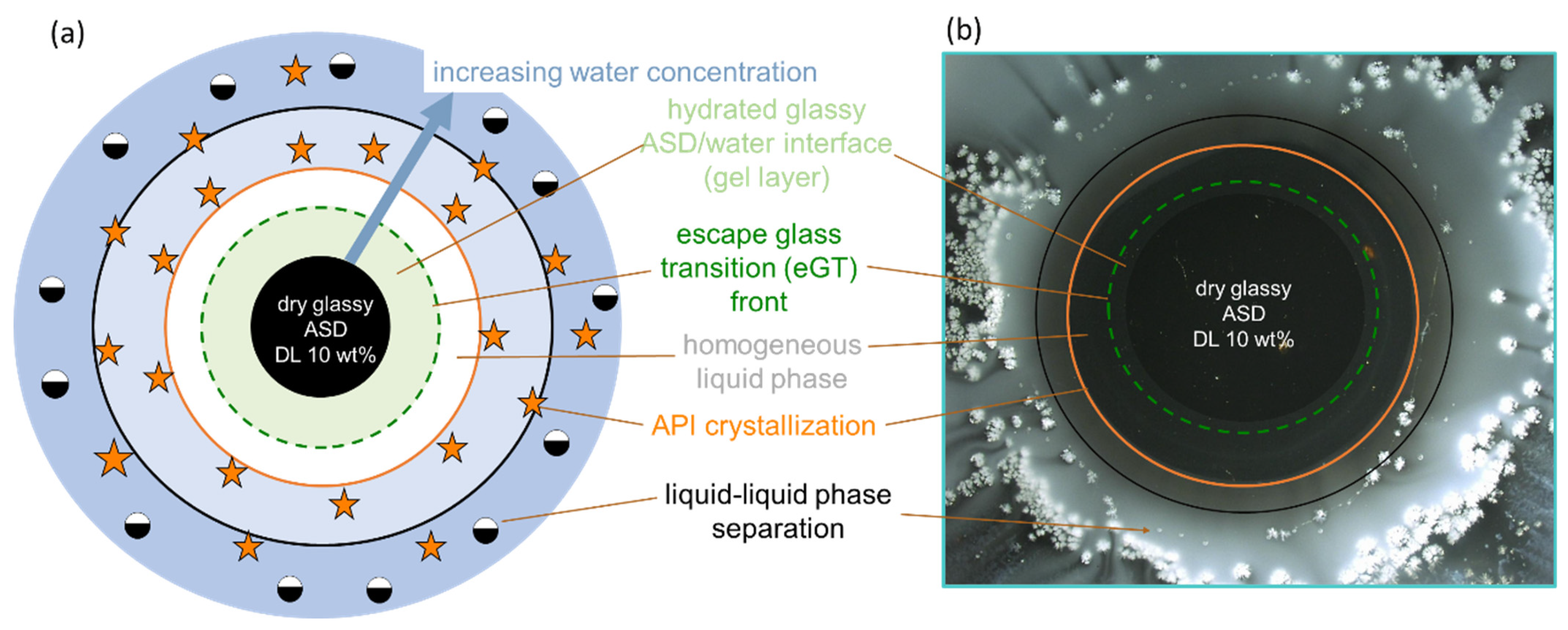
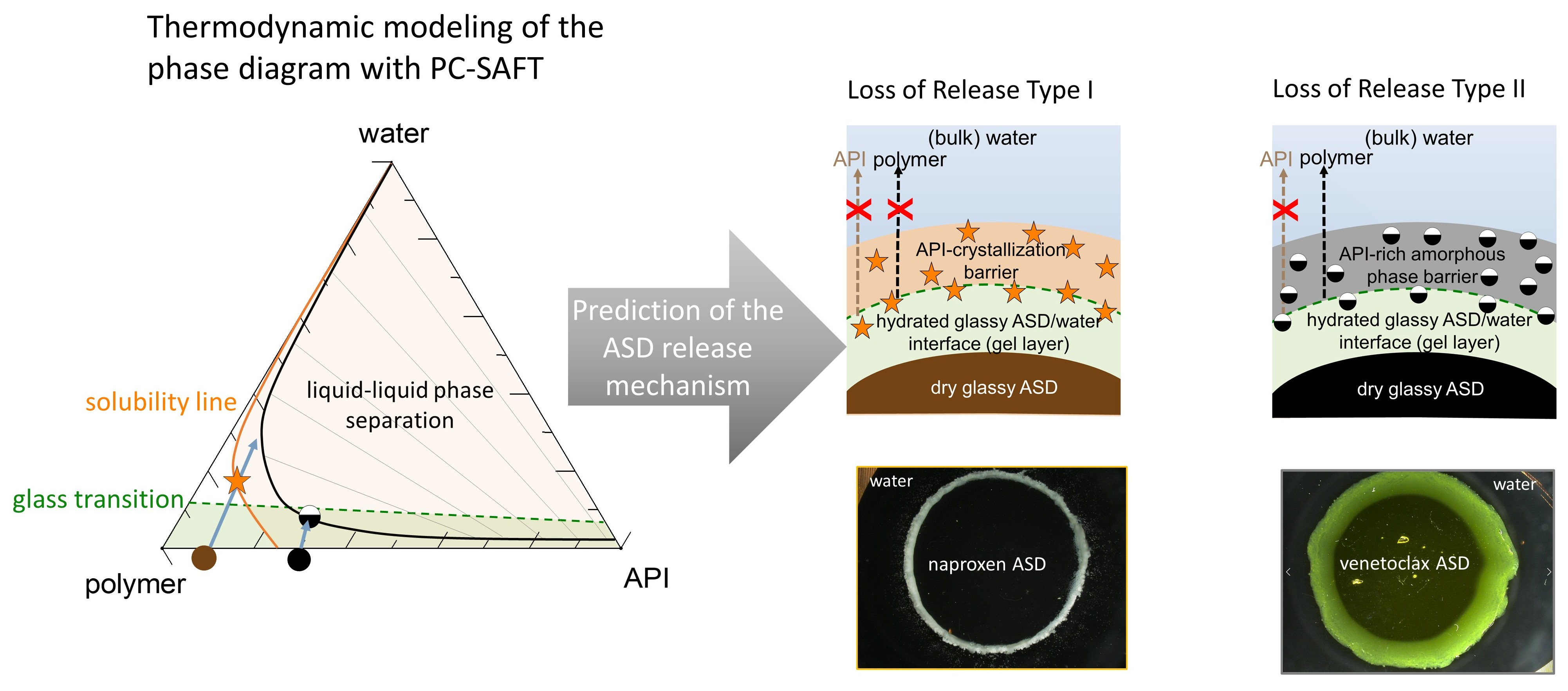
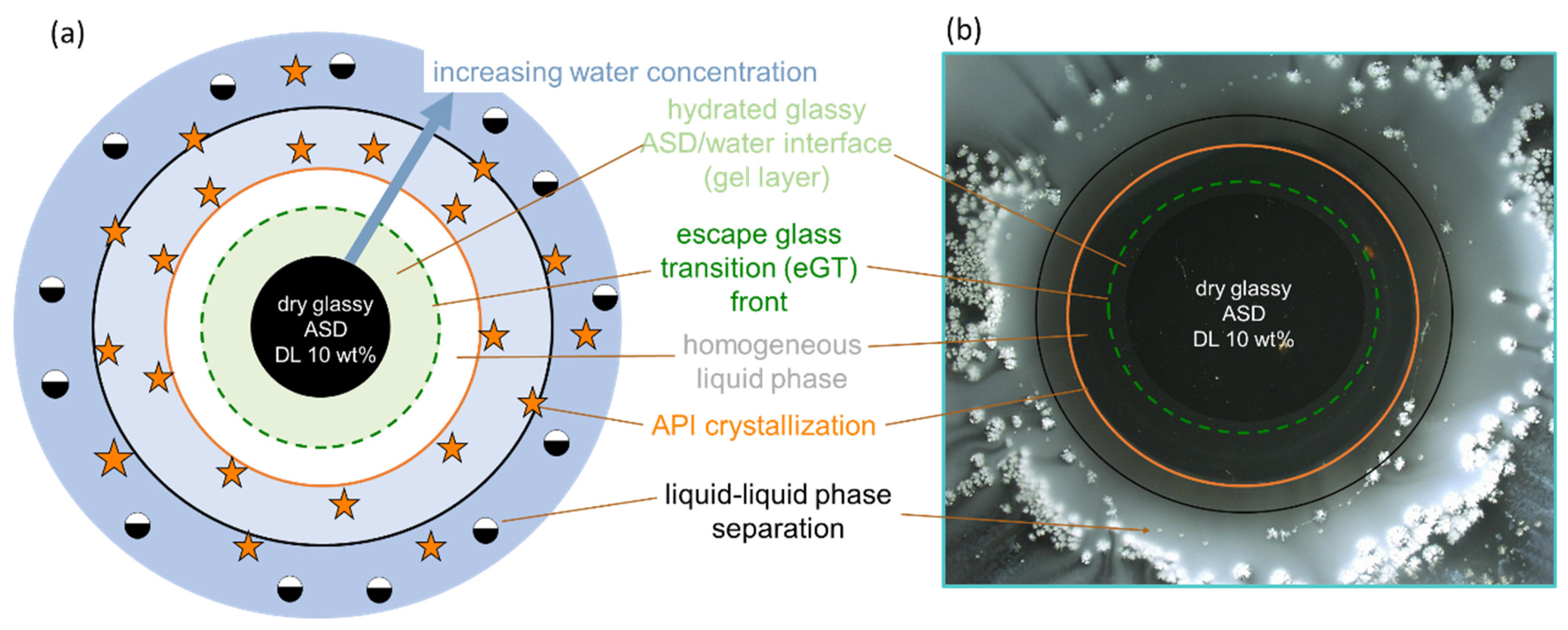
Figure 1 schematically shows the ternary phase diagram (
Figure 1a) of an API/polymer/water system and the hydration pathway of the ASD as it transitions into the bulk aqueous medium during dissolution, as exemplified for ASDs with 10 wt % (
Figure 1b) and 30 wt % of DL (
Figure 1c). The ternary phase depicts the regions of API crystallization or LLPS that form when the ASD is exposed to water. Depending on the concentration of the ternary mixture, the driving force(s) for equilibrium determines what phase changes occur, e.g., either crystallization in API-supersaturated regions or LLPS, or both. Furthermore, at concentrations below the glass transition (the green region in
Figure 1a), the mobility of the system is reduced, which can kinetically hinder phase changes.
Considering the hypothetical 10 wt % DL ASD, on contact with water, the surface of the dry ASD immediately starts absorbing water along the hydration pathway (blue arrow). Simultaneously, the glass transition temperature (
Tg) at the ASD interface decreases, due to plasticization by the imbibed water molecules, thus forming a so-called gel layer at the surface of the ASD. The plasticization of the surface layer continues as more water is absorbed until the
Tg reaches the temperature of the dissolution medium. At this point, cooperative motion and the disentanglement of the polymer chains occur, initiating the dissolution of the ASD surface into the aqueous phase. We term the
Tg of the ASD surface at this point “escape glass transition” (eGT), as indicated by the dashed green line in
Figure 1a. Thus, the calculated eGT line represents the API/polymer/water compositions, wherein the
Tg of the system is equivalent to the temperature of the dissolution medium.
As the API and polymer continuously evolve from the interface into the aqueous phase along the hydration pathway, their concentration in the solution increases until the API’s maximum solubility is reached (orange line), beyond which the API may start to crystallize out of the supersaturated bulk solution. A further increase in API and polymer concentrations results in the solution entering the binodal region, wherein the system is prone to LLPS, leading to polymer-rich and API-rich phases along the tie lines shown in
Figure 1a. In
Figure 1b, the consequential impact of the hydration pathway on the 10 wt % DL ASD during dissolution is schematically depicted as an ASD disc in contact with water. Importantly, crystallization or LLPS is not expected to happen in the gel layer at the ASD/water interface since these phase transformations occur beyond the eGT. Similarly, following the hydration pathway of a hypothetical 30 wt % DL of ASD in
Figure 1a shows that the dry ASD is already in the supersaturated region to the right of the solubility line (orange line). Moreover, the hydration pathway encounters the LLPS region before reaching the eGT. Thus, contrary to the 10 wt % DL ASD, crystallization and/or LLPS is expected to occur in the gel layer at the ASD/water interface, as schematically depicted in the ASD disc in
Figure 1c.
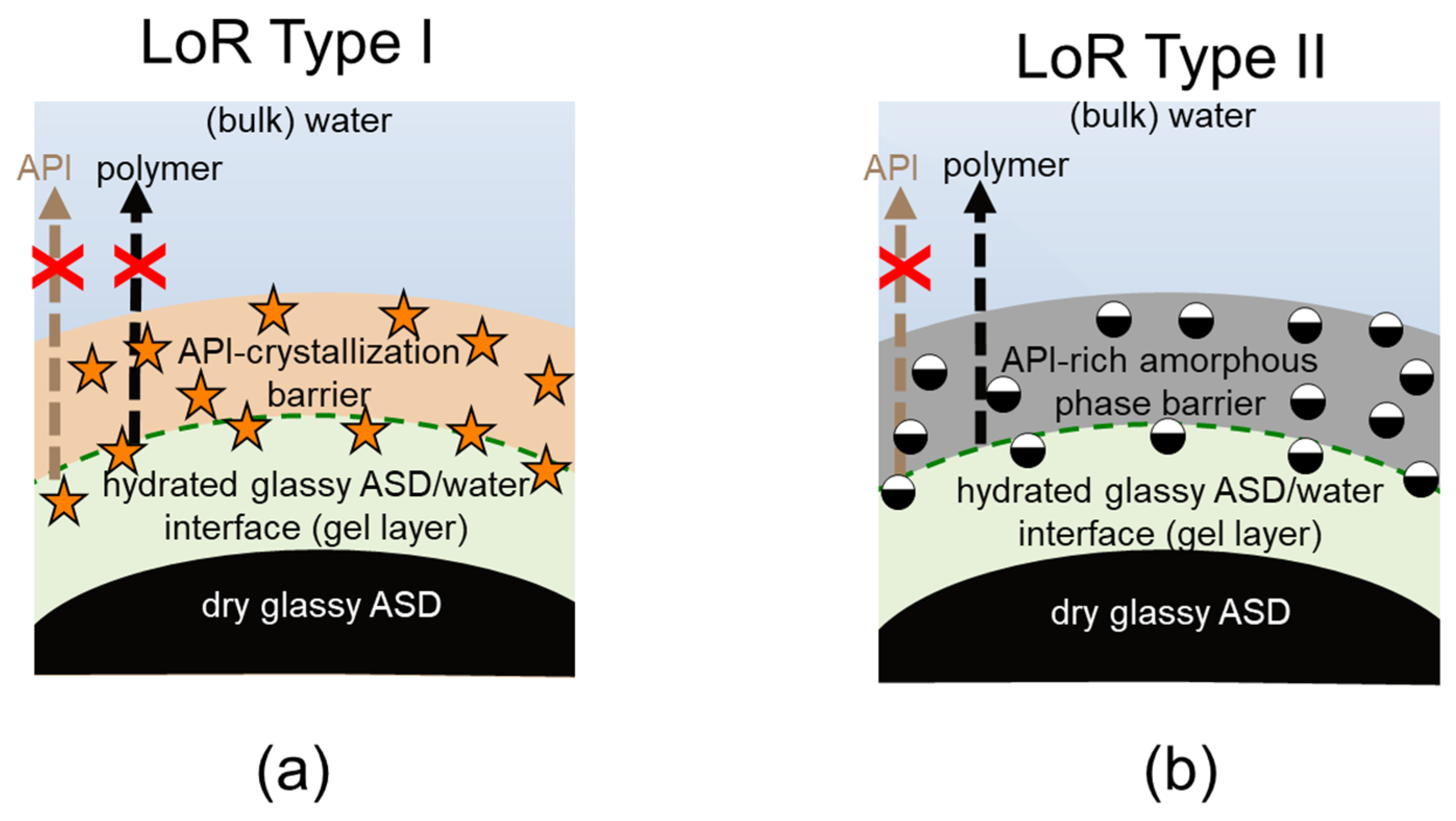
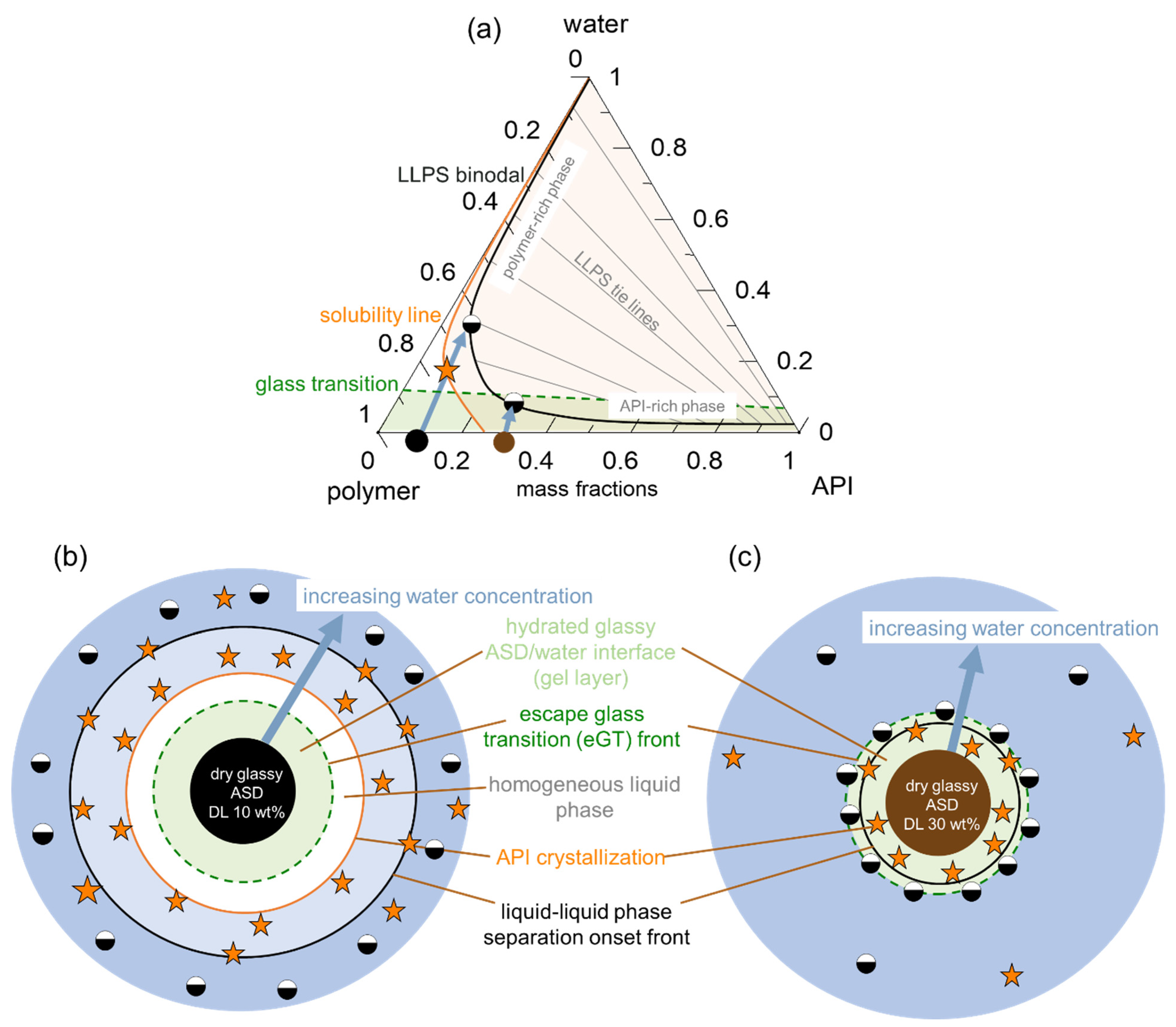
Obviously, the gel layer thus formed is not static and can be perturbed by hydrodynamics during the dissolution test of an ASD formulation. However, we postulate that it is critical to know the dominant thermodynamic driving forces at the ASD/water interface, especially in the first boundary layer below eGT, which is often referred to as the gel layer, in order to understand whether a phase change might or might not occur. Phase changes in the gel layer can lead to the passivation of the interface, resulting in the loss of release (LoR) of the API and polymer. The LoR mechanism can be categorized into two types, depending on whether the passivation is primarily driven by liquid-solid transformation (crystallization), LoR Type I, or liquid-liquid (amorphous) phase separation, LoR Type II, at the interface, as schematized in
Figure 2.
For the LoR mechanism Type I, the formation of an inelastic crystalline passivation layer prevents the release of both the API and the polymer. On the other hand, for LoR mechanism Type II, the development of a hydrophobic API-rich amorphous phase barrier, due to LLPS, results in the preferential release of the complementary hydrophilic polymer-rich phase into the aqueous medium. Hence, the LoR Type II mechanism results in the so-called incongruent release of the API and polymer [
24]. It is also possible for both mechanisms to concurrently contribute to LoR, depending on the ternary phase diagram and crystallization propensity of the API.
To demonstrate the a priori applicability of the ternary phase diagram in predicting LoR, we chose two PVPVA64-based ASDs, formulated with either naproxen or venetoclax. For comparison purposes, having a common polymer for both formulations helps to normalize the polymer’s impact and other contributing factors to some extent in terms of the gel layer’s behavior, such as its viscosity and water-sorption kinetics.
4.2. LoR Type I: Naproxen/PVPVA64/Water System
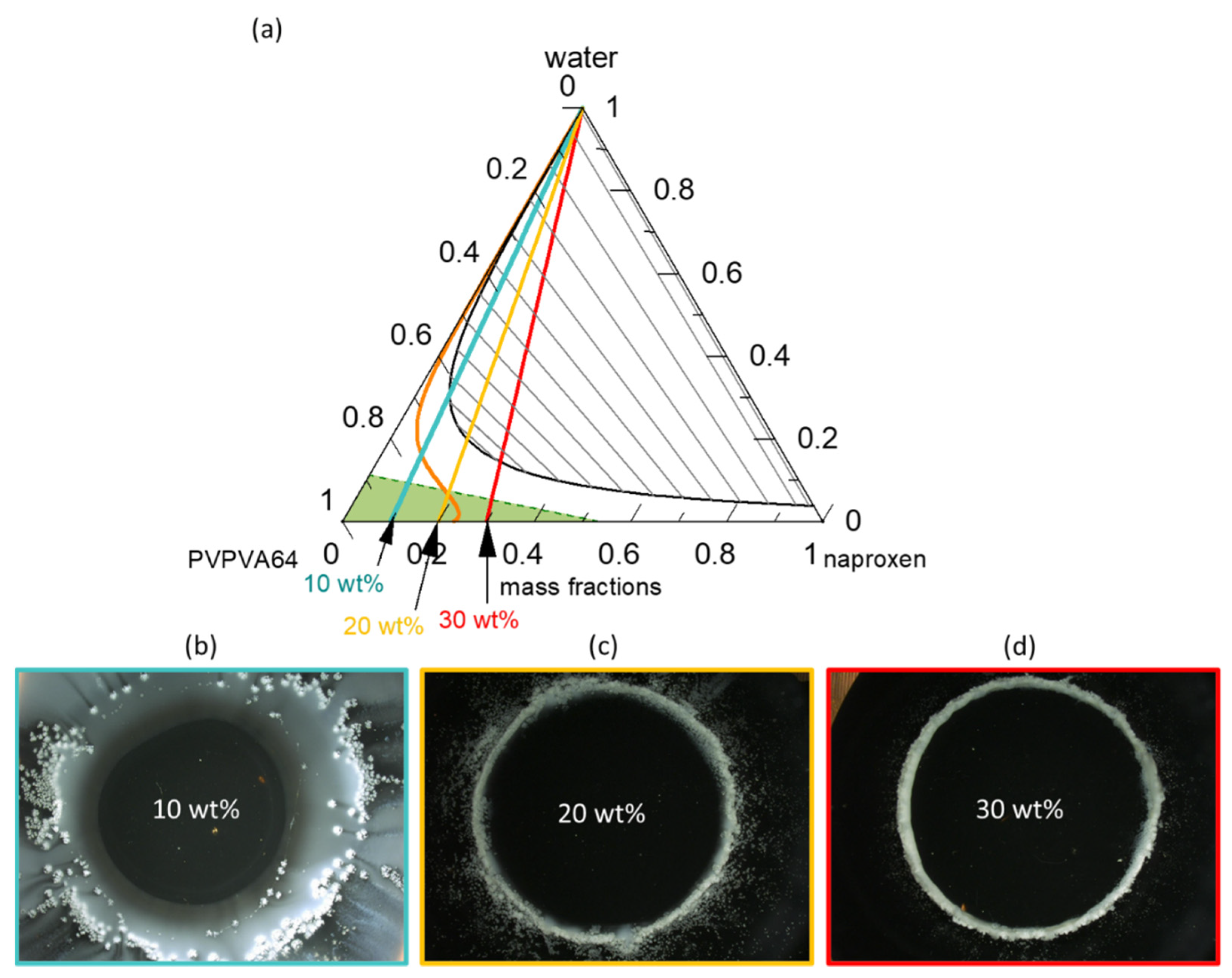
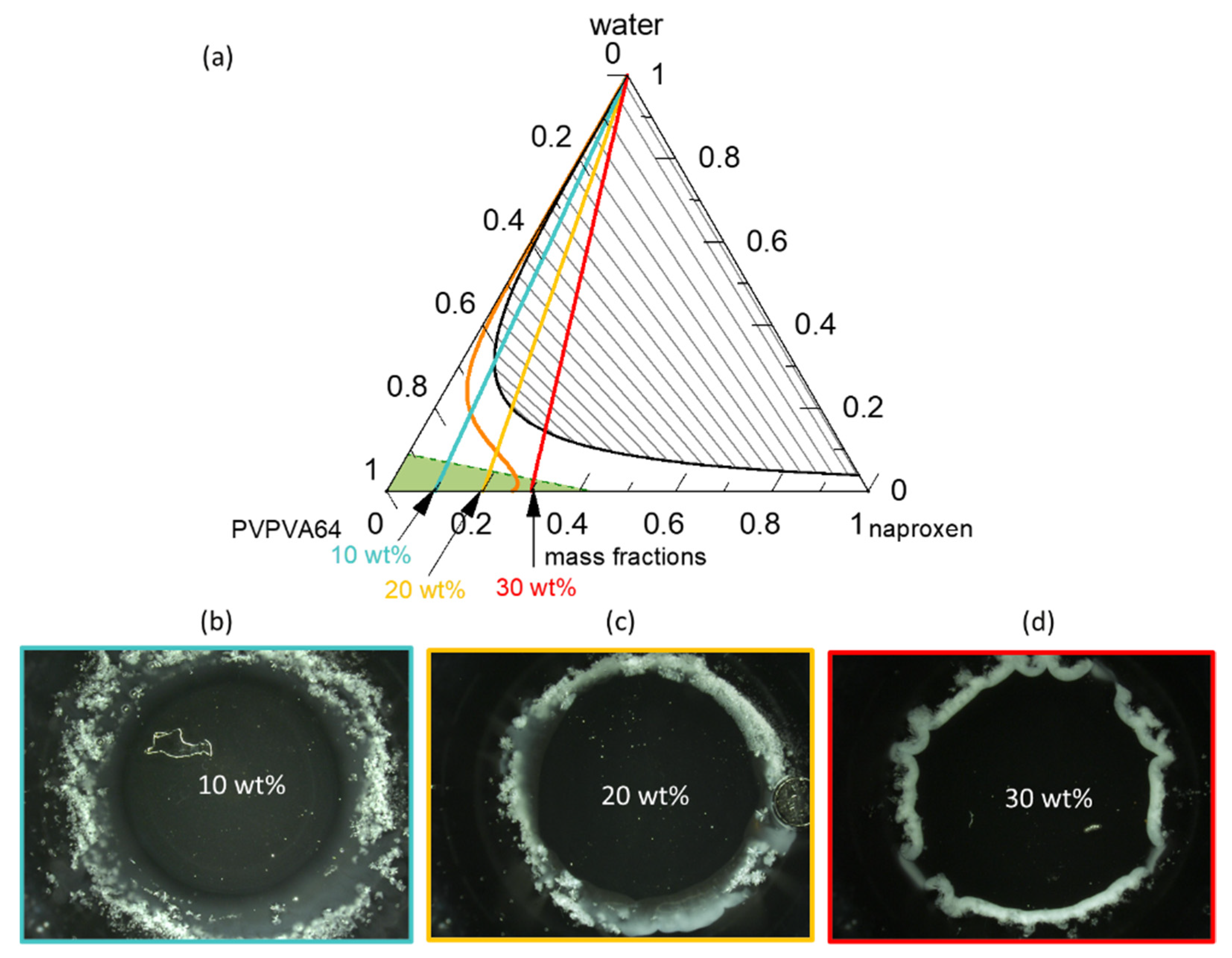
Figure 3a presents the modeled naproxen/PVPVA64/water ternary phase diagram at 37 °C;
Figure 3b–d shows the METT images of the ASDs at DLs of 10 wt %, 20 wt %, and 30 wt % when in contact with water.
The solid-liquid (API solubility line) and liquid-liquid equilibria lines were modeled using PC-SAFT and the eGT line was calculated using the Gordon-Taylor equation. The phase diagram shows that in the dry state, ASDs with DL up to 23 wt % are thermodynamically stable (intersection of solubility line with the PVPVA64/naproxen axis) and thus will not crystallize. From the phase diagram, it is clear that naproxen has a greater solubility in the PVPVA64 than in water since the solubility strongly decreases with increasing water concentration in the API/polymer/water mixture. The interplay between the solubility line and eGT line along the hydration pathway is decisive in predicting the crystallization of the API at the ASD/water interface during dissolution. As explained schematically in
Figure 1 for DLs below the intersection of the solubility line and the eGT line, crystallization is expected to occur in the aqueous phase away from the gel layer (e.g., in the dissolution medium). For DLs above this intersection, crystallization is expected in the gel layer, leading to the risk of LoR. Thus, based on the phase diagram, above 19 wt % DL ASD, there exists a risk of naproxen crystallization in the gel layer of the ASD because the hydration pathway intersects the solubility line before the eGT line. Moreover, the phase diagram predicts that regardless of the naproxen DL, LLPS will not occur in the gel layer but instead appear in the aqueous bulk medium since the LLPS phase transition is well above the eGT line. The readout API/water/polymer concentrations along the hydration pathway in the phase diagram for the investigated 10 wt %, 20 wt %, and 30 wt % DL naproxen ASDs are given in
Table 5.
The water content at the solubility limit of the 10 wt % DL ASD is 16.3 wt %, which is more than the 9.2 wt % water content that is required to reach eGT; hence, API crystallization at high water concentrations outside the gel layer (i.e., in the dissolution bulk phase) is expected. For the 20 wt % DL ASD, the water content at the solubility limit and eGT are 6.1 wt % and 7.3 wt %, respectively, meaning that crystallization will occur in the gel layer but will be at the front, close to the aqueous bulk medium. However, for the 30 wt % DL ASD, the solubility limit is already exceeded in the dry state. Hence, crystallization will take place with the least amount of water absorbed at the interface, leading to the formation of a compact crystalline front. Thus, the consequential LoR Type I, due to crystallization in the gel layer, is expected to be more pronounced for the 30 wt % DL ASD than the 20 wt % DL ASD.
To verify the predicted gel layer behavior, the dissolution of 10 wt %, 20 wt %, and 30 wt % DL naproxen ASDs discs were monitored via METT experiments. According to the classification system presented by Baird et al. [
58], neat naproxen crystallizes extremely rapidly from the amorphous phase. Although the 30 wt % DL ASD in the initial dry state is thermodynamically unstable at 37 °C (
Figure 3a), naproxen does not spontaneously crystallize because the API molecules are kinetically stabilized in their glassy state in PVPVA64 (the green region in
Figure 3a) [
39].
Figure 3b–d presents the images of the ASD discs after 1 h of contact with water at 37 °C. Videos of the evolution of the ASD disc/water interface over the period are provided in the
Supplementary Materials. The METT experiment setup is a very effective technique for monitoring the gel layer since the hydrodynamic effects are minimal. As seen in
Figure 3b, naproxen crystallizes as a bright white domain well beyond the surface of the 10 wt % DL ASD disc, which appears as a dark circle in the image. Moreover, it can also be noticed that a concentric white solution phase has also developed far away from the interface. The observed effect is attributed to LLPS, which is predicted to occur beyond the gel layer. Thus, the evolution of the 10 wt % naproxen ASD/water interface agrees perfectly with the ternary phase diagram prediction shown in
Figure 3a. In contrast, for the naproxen ASDs with DLs of 20 wt % and 30 wt %, the white naproxen crystalline domains formed on the disc surface when in contact with the aqueous medium, as seen in
Figure 3c,d, respectively, indicating crystallization in the gel layer, as was predicted. Barely perceptible differences can be observed between the crystallized interfacial layer of the two formulations. Specifically, in the 30 wt % DL ASD, the crystals compact tightly at the surface of the disc, compared to the 20 wt % DL ASD disc. The METT images are, therefore, in very good agreement with the PC-SAFT-predicted phase behavior for ASD hydration pathways (
Figure 3a,
Table 6).
Figure 4 gives more detailed insights into the visual layer formation that occurred in the METT investigations (
Figure 3b).
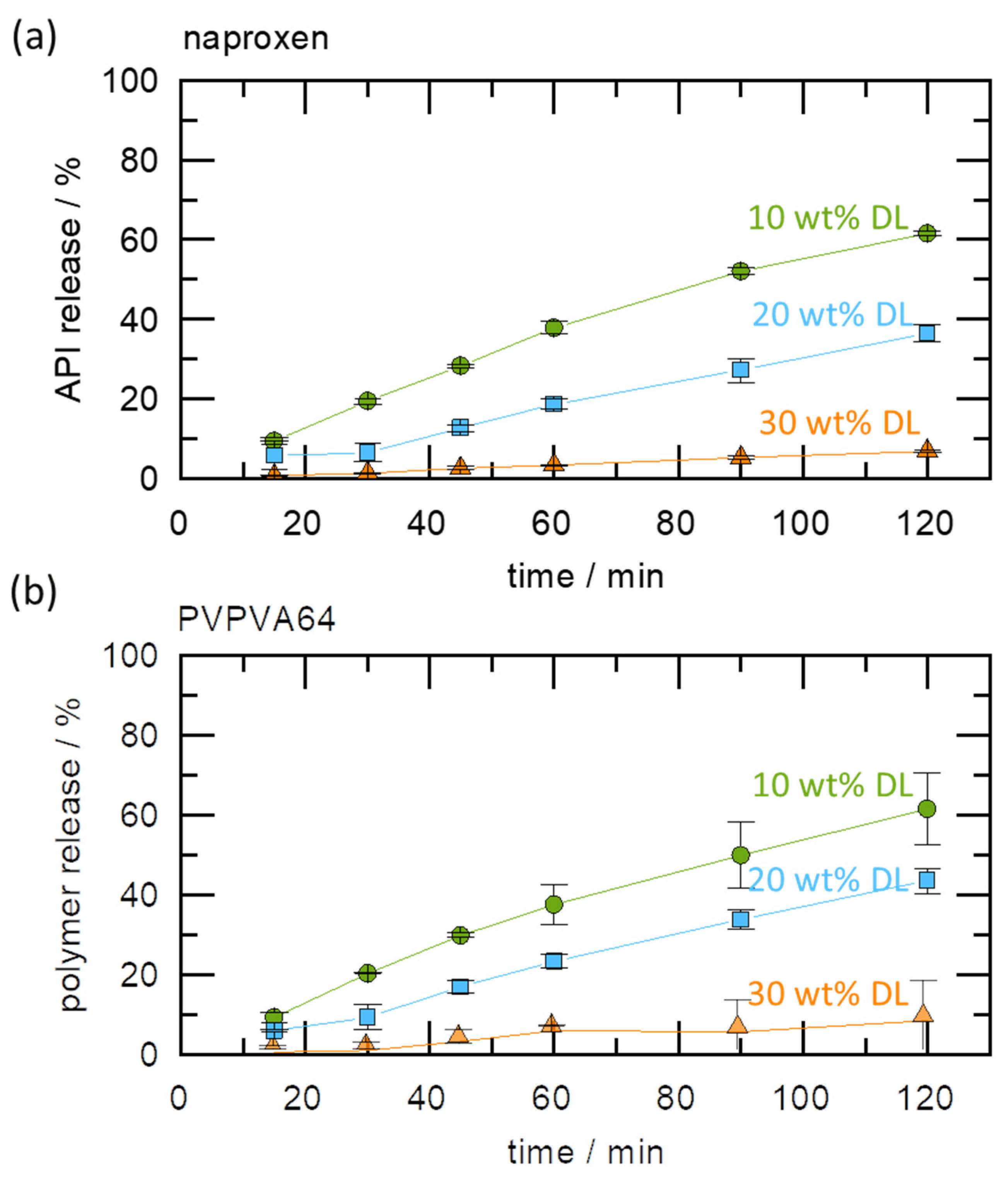
The impact of the gel layer crystallization on dissolution was further investigated by monitoring the release profiles of naproxen and PVPVA64 from the ASDs at 37 °C, using a USP 2 apparatus. The results are displayed in
Figure 5a,b.
It can be observed that APIs and polymer release levels are highest for 10 wt % DLASD and lowest for 30 wt % DL ASD. While the former released 60% of both the API and polymer in 2 h, the latter released only 10% in the same period. As pointed out earlier, the Type I LoR mechanism triggers the formation of a solid crystalline barrier at the ASD/water interface, as seen in the METT image, which prevents the free release of both the API and the polymer. For the 20 wt % DLASD, although there is an LoR, the API and polymer release levels are significantly higher compared to the 30 wt % DL ASD. This can be explained by the fact that the crystallization barrier at the ASD/water interface is expected to be less compact and is more easily disrupted by the hydrodynamics of the dissolution experiments than for the 30 wt % DL ASD. It is worth noting that from the three ASDs, both the API and the polymer released at similar rates, the so-called congruent release, even when LoR occurred. This supports our hypothesis that API and polymer release rates are nearly equally affected when LoR is triggered by the LoR Type I mechanism. Thus, the results of the dissolution and METT experiments support and validate the quantitatively predicted release behavior, based on the modeled ternary phase diagram at 37 °C (
Figure 3).
4.3. LoR Type II: Venetoclax/PVPVA64/Water System
For an API that does not readily crystallize from the amorphous phase, as with venetoclax, the solubility line in the ternary phase diagram is less decisive for LoR prediction. In such a system, the interplay between the LLPS boundary and eGT dictates the LoR prediction that is based on the phase diagram (
Figure 6).
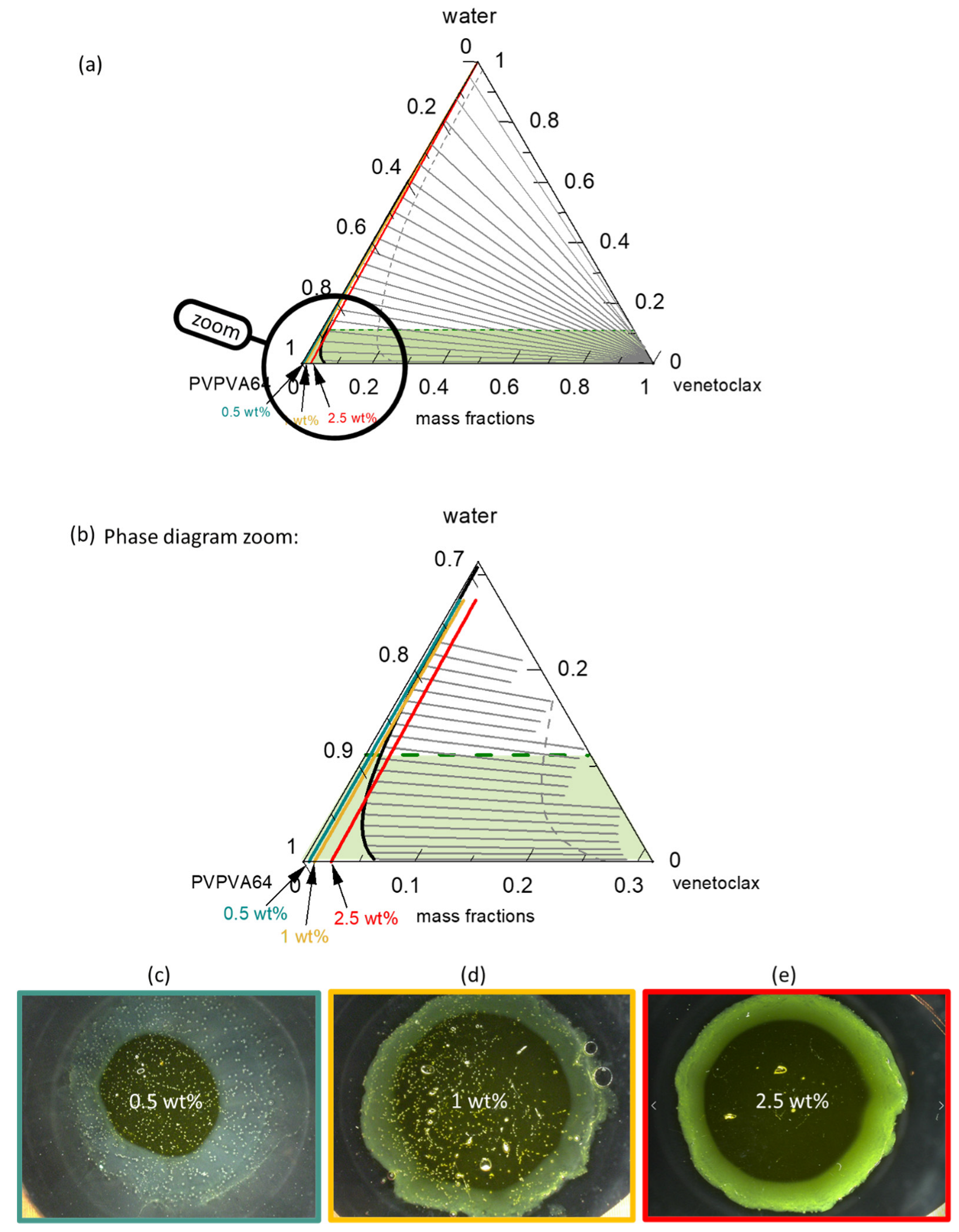
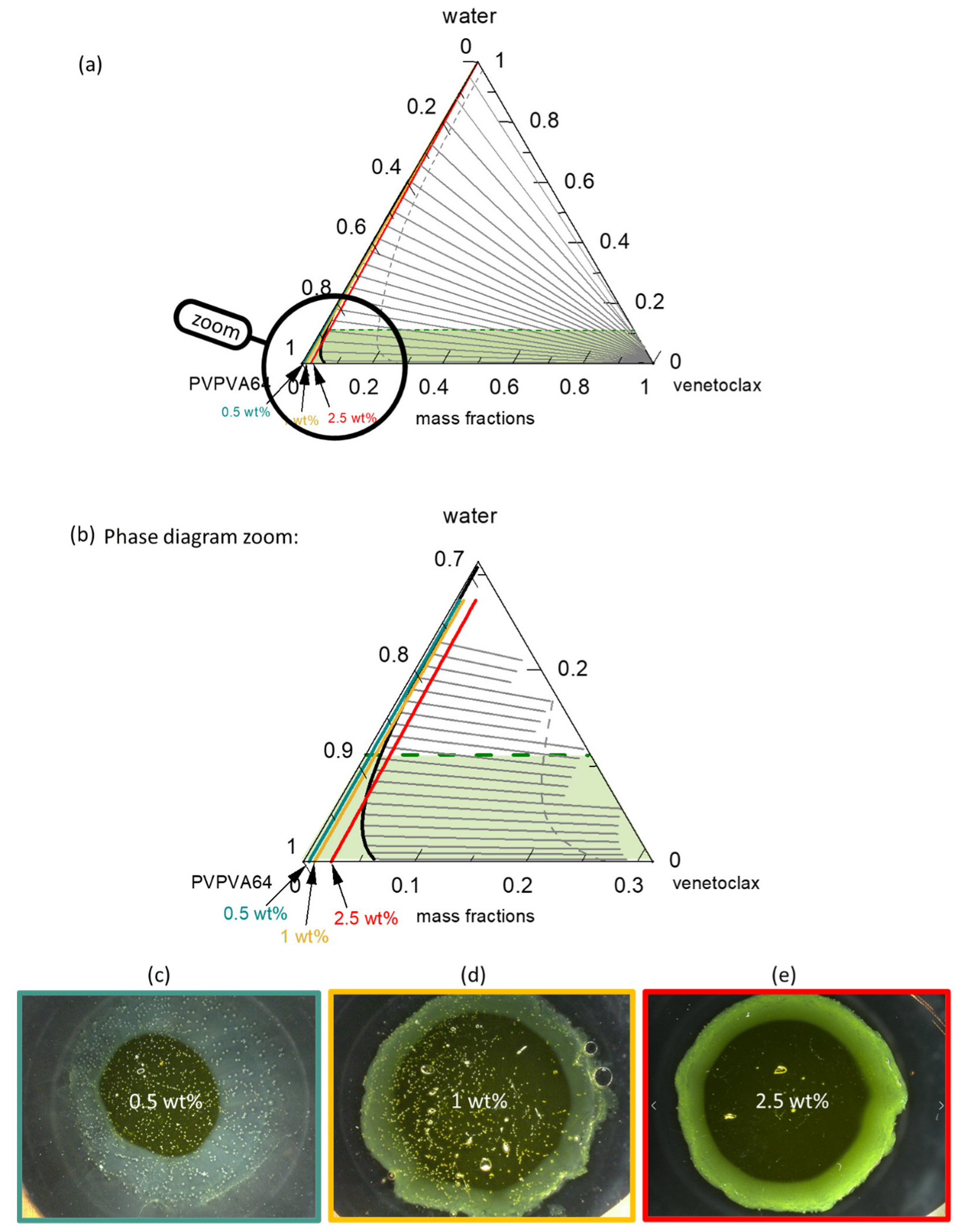
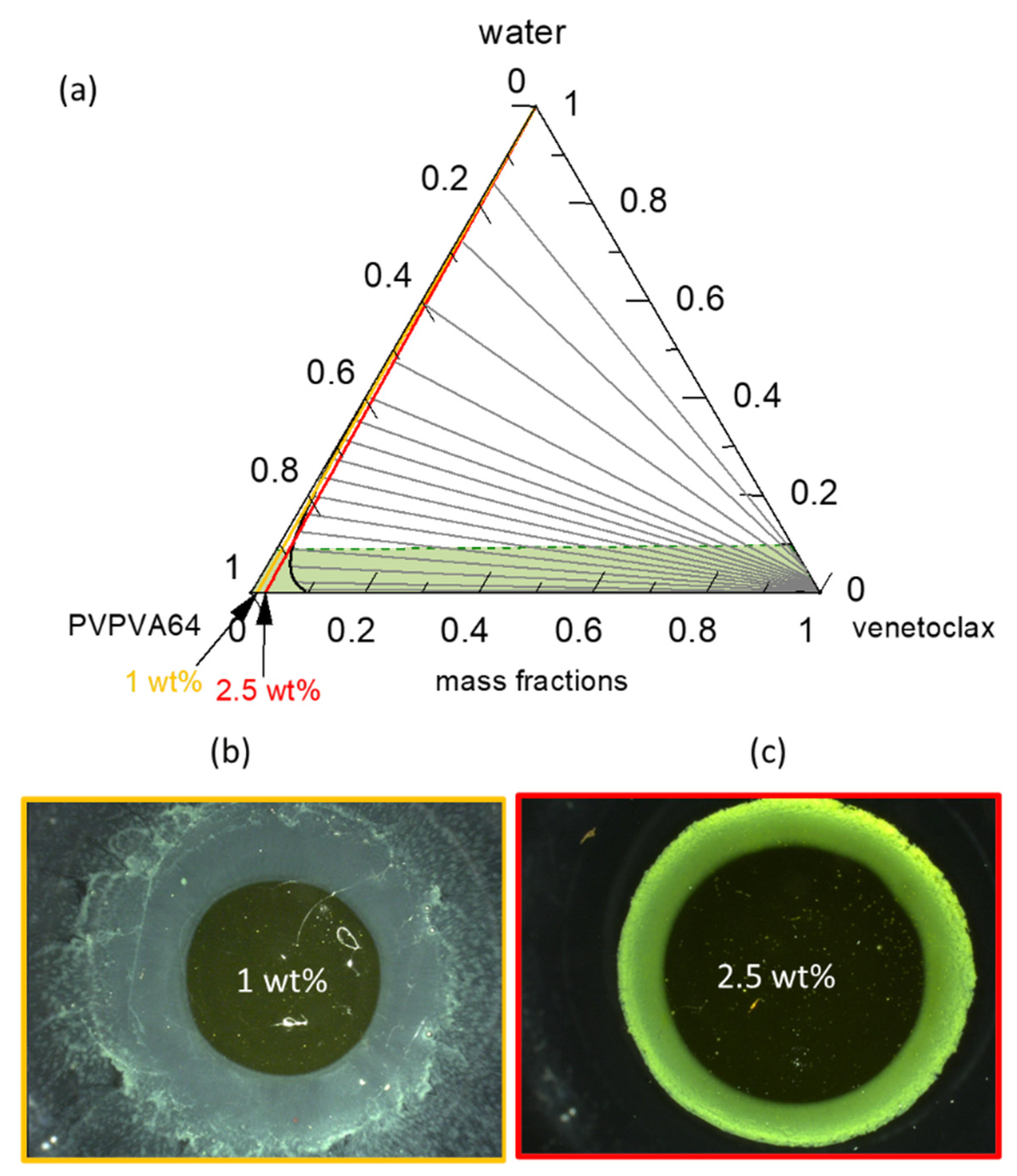
Figure 6a presents the venetoclax/PVPVA64/water ternary phase diagram at 37 °C with the hydration pathway for 0.5 wt %, 1 wt %, and 2.5 wt % DL ASDs. The API has extremely low solubility in both the polymer and water, such that the solubility line (orange line) runs indistinguishably parallel on the PVPVA64/water axis, implying that all three ASD DLs are already supersaturated in the dry state. Moreover, the system exhibits a very large LLPS region. The predicted large miscibility gap for the ternary system covers almost the entire phase diagram, resulting in an API-rich phase containing almost only venetoclax, along with the polymer-rich phase containing a low venetoclax amount, as indicated by the predicted tie lines. Already, the dry ASD with a DL higher than 5 wt % is thermodynamically predicted to be in the LLPS region and, therefore, is prone to phase separation. Nonetheless, it must be recognized that the system has significantly high
Tg (
Figure S1 in the Supplementary Materials) and is, thus, kinetically well-stabilized, which can hinder phase separation in the dry state. Zooming into the phase diagram (
Figure 6b) provides insight into the hydration pathway of the different DLs of the ASD. The exact compositions along the hydration pathway in the ternary phase diagram are given in
Table 6.
Table 6.
Venetoclax/PVPVA64/water concentrations along the hydration pathway for 0.5 wt %, 1 wt %, and 2.5 wt % DL venetoclax ASDs at 37 °C (
Figure 6a), with the calculated corresponding
Tg.
Table 6.
Venetoclax/PVPVA64/water concentrations along the hydration pathway for 0.5 wt %, 1 wt %, and 2.5 wt % DL venetoclax ASDs at 37 °C (
Figure 6a), with the calculated corresponding
Tg.
| | Water | Venetoclax | PVPVA64 | Tg/°C |
|---|
| | 0.5 wt % DL ASD |
| ASD (dry) | 0 wt % | 0.5 wt % | 99.5 wt % | 111.0 |
| eGT | 11.2 wt % | 0.4 wt % | 88.4 wt % | 37.0 |
| Solubility limit | APIs supersaturated in the dry state |
| Polymer-rich phase at binodal line | 20.5 wt % | 0.6 wt % | 78.9 wt % | −4.4 |
| API-rich phase at binodal line | 0.6 wt % | 99.4 wt % | 0.0 wt % | 113.9 |
| | 1 wt % DL ASD |
| ASD (dry) | 0 wt % | 1.0 wt % | 99.0 wt % | 111.1 |
| eGT | 11.2 w % | 0.9 wt % | 87.9 wt % | 37.0 |
| Solubility limit | APIs supersaturated in the dry state |
| Polymer-rich phase at binodal line | 15.6 wt % | 0.9 wt % | 83.5 wt % | 15.8 |
| API-rich phase at binodal line | 0.4 wt % | 99.6 wt % | 0.0 wt % | 115.8 |
| | 2.5 wt % DL ASD |
| ASD (dry) | 0 wt % | 2.50 wt % | 97.50 wt % | 111.2 |
| Solubility limit | APIs supersaturated in the dry state |
| eGT | LLPS occurs before eGT | 37.0 |
| Polymer-rich phase at binodal line | 6.1 wt % | 2.47 wt % | 91.4 wt % | 66.5 |
| API-rich phase at binodal line | 0.2 wt % | 99.8 wt % | 0.0 wt % | 118.3 |
The ASD with 0.5 wt % of venetoclax will most likely release into the aqueous medium before LLPS because its hydration pathway meets the binodal line far above eGT. This is evidenced by the high amount of water, 20 wt %, at the binodal line compared to 11 wt % of water content at eGT (
Table 6). Thus, phase separation in the gel layer prior to the dissolution of the ASD is unlikely, and a simultaneous API and polymer release is expected. For the 1 wt % DL ASD, the hydration pathway encounters the binodal line at 15 wt % water content, which is very close to the 12 wt % water content at eGT. Thus, LLPS is likely to occur close to the gel layer. The risk of LLPS in the gel layer becomes more obvious with an increasing DL from 1 wt % to 2.5 wt %. For the 2.5 wt % DL ASD, the hydration pathway fully encounters the binodal line before the eGT line, suggesting that LLPS will occur in the gel layer. When LLPS occurs, the PC-SAFT calculated API-rich phase consists of nearly only API with very small amount of water, while the polymer-rich phase consists mostly of polymer and water (
Table 6). Hence, the resulting
Tg value of the LLPS phases differs from the initial homogeneous phase. For instance, the calculated
Tg of the formed polymer-rich phase for the 1 wt % DL at the binodal line is 15.8 °C, suggesting high mobility of the polymer-rich phase since the dissolution medium temperature of 37 °C is much higher than the
Tg value. Thus, the high mobility in the polymer-rich phase will allow its release into the aqueous media. In contrast, due to the high
Tg value of the API-rich phase (
Tg = 115.8 °C) and lesser mobility, most of the API will enrich near the gel layer. Situations where LLPS leads to an API-rich phase with a
Tg value higher than the dissolution medium temperature are often referred to as glass liquid phase separation (GLPS) [
62]. For the 2.5 wt % DL ASD, according to the LLPS tie lines in
Figure 6a, at eGT, the thermodynamic endpoint composition of the polymer-rich phase is estimated to be 12 wt % water and 87 wt % polymer, with a
Tg of 33 °C, while that of the API-rich phase is approximately 0.3 wt % of water and 99 wt % of API, with a
Tg of 117 °C. Due to the high
Tg value of the API-rich phase, most of the APIs can be trapped within the gel layer in the glassy state because the
Tg of this phase is significantly higher than the dissolution medium temperature, 37 °C. According to the lever rule, it is estimated that the API-rich phase constitutes approximately 5% of the total phases at this point. However, considering that when the polymer-rich phase is releasing preferentially, the API-to-polymer ratio will increase; thus, the hydration pathway would curve towards the right over the dissolution process. Thus, the enrichment of venetoclax at the ASD surface over time, coupled with its high
Tg glassy attribute, will cause passivation of the ASD surface, leading to LoR Type II.
Figure 6c–e shows the METT images of the 0.5 wt %, 1 wt %, and 2.5 wt % DL venetoclax ASDs discs after exposure to water. The multiple small spherical structures seen across the images, especially in the 0.5 wt % and 1 wt % DL ASDs, are trapped air bubbles. Videos of the experimental evolution of the METT ASD disc/water interface are provided in the
Supplementary Materials. The images show that the 0.5 wt % DL formulation does not build an LLPS passivating gel layer, unlike the 1 wt % and 2.5 wt % DL formulations. It also appears that the passivated gel layer of the 1 wt % DL formulation is fluid and not rigid, as in the case of the 2.5 wt % DL formulation, because the LLPS occurs closely above eGT. It can also be observed that a light yellowish dispersion is formed in the solution phase away from the interface for the 0.5 wt % DL ASD, which is attributable to the LLPS-driven drug-rich phase droplet formation occurring in the aqueous phase. Thus, applying the ternary phase diagram and concepts presented here, the debate that is often found in the literature as to whether API-rich phase droplet formation occurs in the gel layer or in the solution can be predicted through rigorous thermodynamic modeling [
33,
35,
63]. Overall, the observations from the METT experiments agree very well with the predicted phase behavior of the gel layer, based on the ternary phase diagram, and exactly match the predicted hydration pathways of the ASDs (
Table 6), with compositions and
Tgs explaining the mobility and occurrence of the resulting phases.
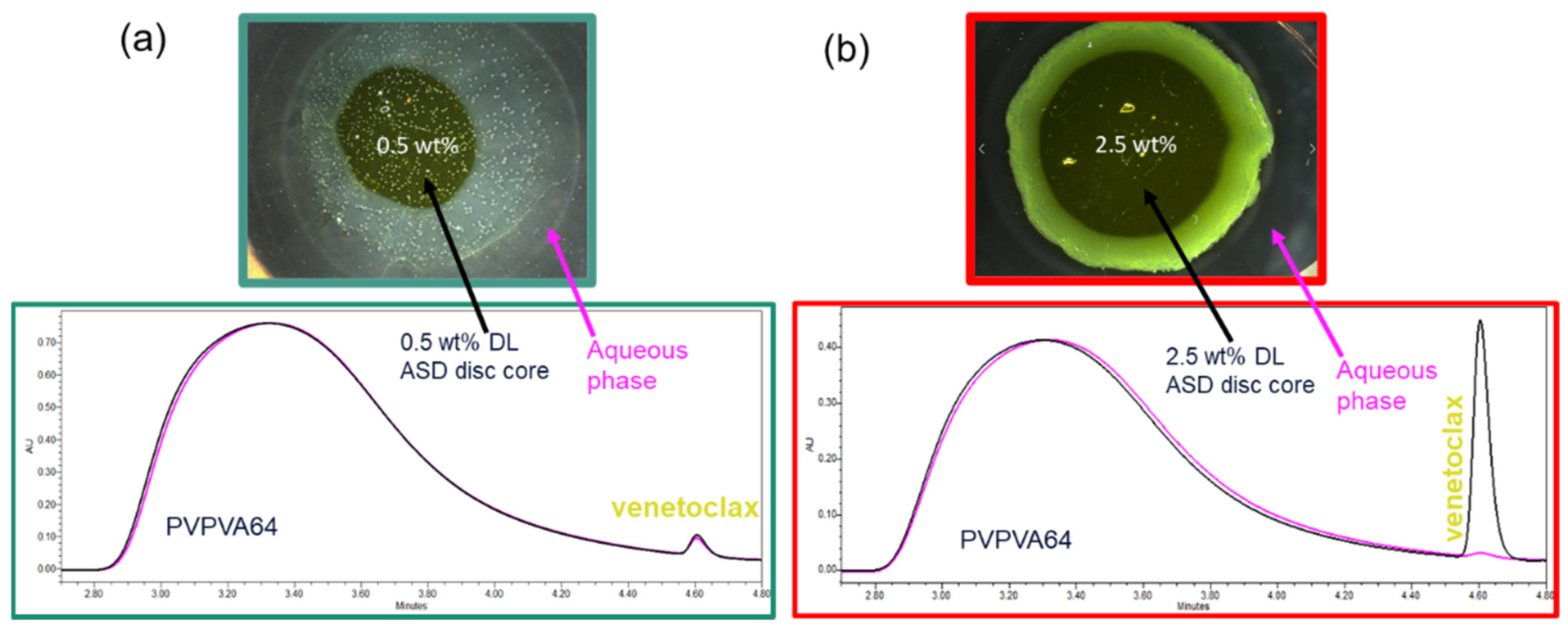
Since the LoR of venetoclax ASDs occurs at such a low DL, dissolution experiments with the USP 2 apparatus could not be conducted. Nevertheless, the relative amounts of API and polymer released were monitored via the METT experiments. The composition of venetoclax and PVPVA64 in the aqueous phase at the end of the METT experiments was determined via SEC and was then compared with the ASD disc core composition (
Figure 7). The
supporting information provides all the chromatogram data (
Figure S2 in the Supplementary Materials).
As shown in
Figure 7a, the chromatogram of the aqueous phase of the 0.5 wt % DL ASD is very similar to the chromatogram of the ASD core, suggesting that both the API and polymer were congruently released into the aqueous phase. In contrast, the chromatogram of the aqueous phase of the 2.5 wt % DL of ASD (
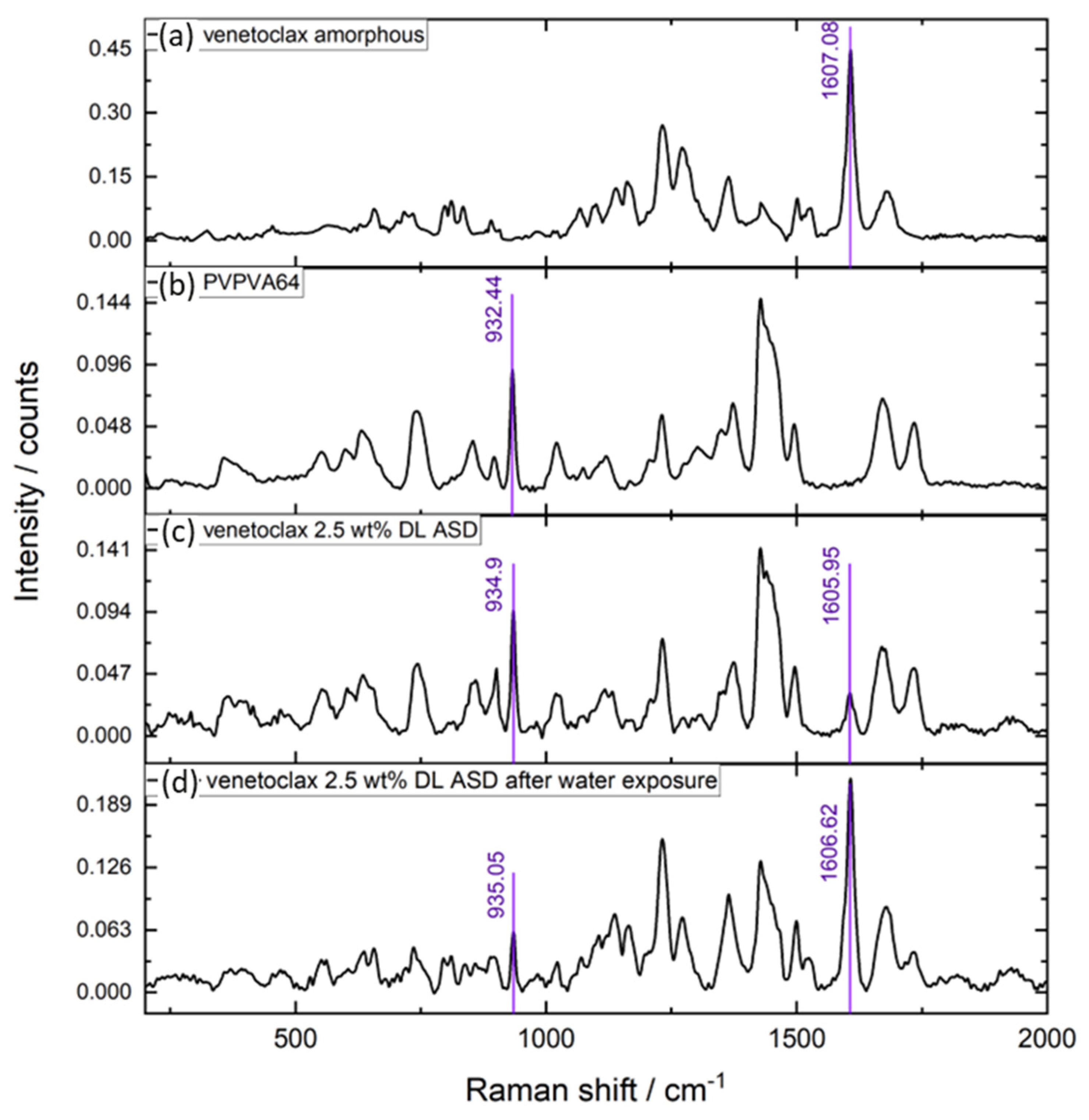
Figure 7b), compared to the chromatogram of the ASD core, shows only a PVPVA64 peak and an inconspicuous venetoclax peak, thus indicating that the polymer was preferentially released into the aqueous phase while the API was trapped in the gel layer. The enrichment of the API in the gel layer was also directly confirmed via Raman spectroscopy after exposing the 2.5 wt % DL to water, which is shown in
Figure 8.
The Raman data show very strong intensification of the API band at ~1607 cm
−1 after exposure of the ASD to water, while concurrently, the polymer band at ~935 cm
−1 strongly diminished.
Table 7 summarizes the relative amount of API and polymer determined in the aqueous phase for the three formulations after the METT experiments, which confirms that the API and polymer released congruently for the 0.5 wt % and 1 wt % DL ASDs, while at 2.5 wt % DL, LoR occurred, leading to almost only PVPVA64 in the aqueous phase, as predicted using the ternary phase diagram. Thus, for the venetoclax/PVPVA64 ASD system, the limit of congruent (LoC) API and polymer release is 1 wt % DL.
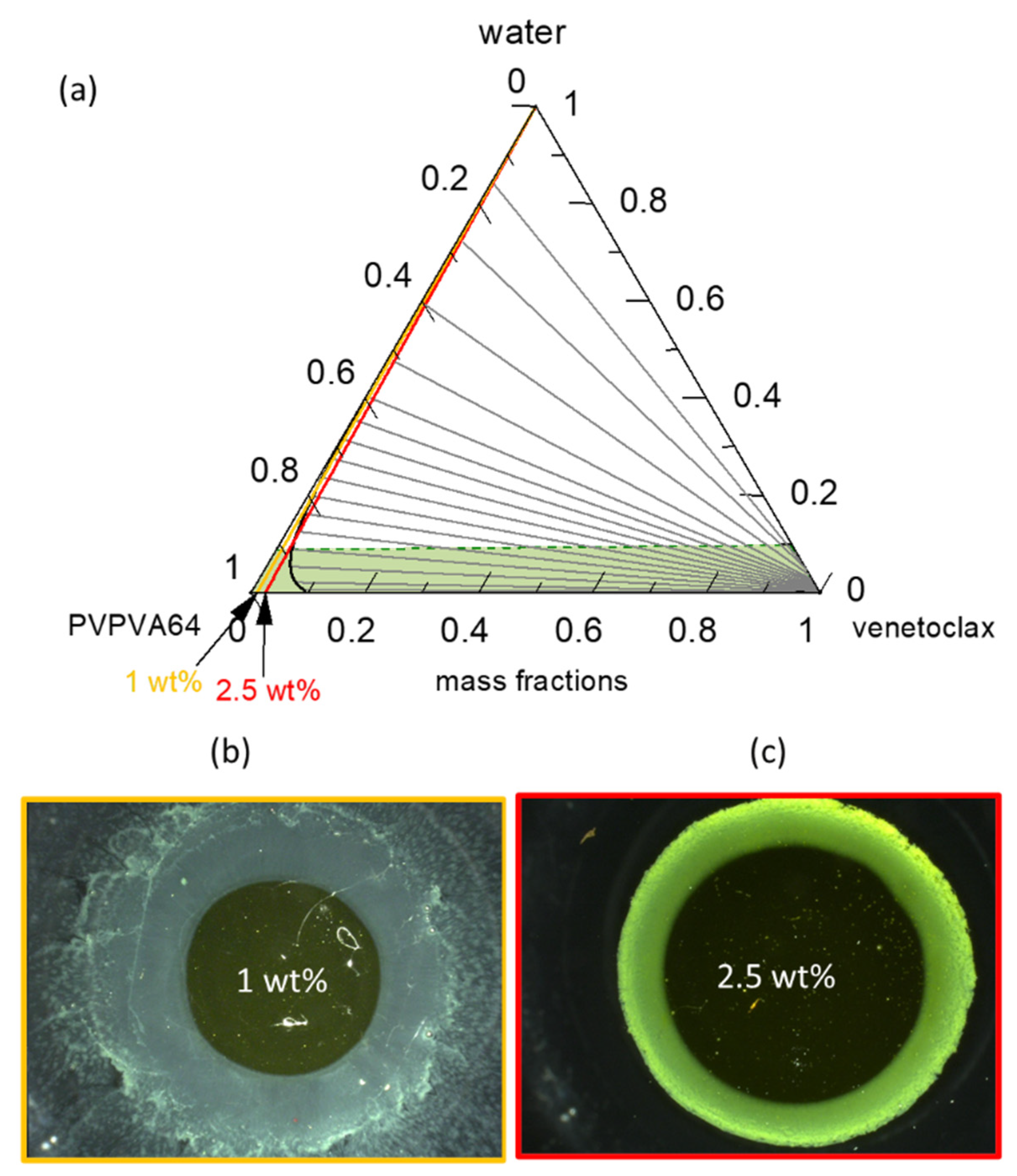
Several attempts have been made in the literature to explain the thermodynamic origin of the typically observed DL-dependent LoR of ASDs. Using a hypothetical ternary phase diagram, Han et al. [
35] proposed that LLPS and the eventual passivation of the gel layer occur when spinodal decomposition is triggered at a high drug load below an arbitrarily defined percolation threshold. They argued that this condition leads to the formation of a morphological continuous API-rich phase that passivates the gel layer, while the polymer-rich phase is preferentially released. Recently, the Taylor group [
25], also relying on a hypothetical ternary phase diagram, proposed that LoC is defined by the plait point (critical point) wherein the binodal and spinodal lines converge, and assumed that phase separation primarily occurs via spinodal decomposition. To verify these hypotheses, we introduced the PC-SAFT-calculated spinodal line (gray dashed line) in the phase diagram in
Figure 6. Based on the hypotheses and assumptions detailed above, phase separation in the gel layer should not be feasible for any of the DL values investigated for the venetoclax/PVPVA64 ASDs since none of the hydration paths runs through the spinodal decomposition region (on the right side of the gray dashed line). However, our experimental data and observations do not support these hypotheses. In our opinion, firstly, these hypotheses are not based on data-driven rigorous thermodynamic modeling and ignore the impact of glass transition, which primarily drives the kinetics of phase separation. Secondly, since dissolution at the ASD/water interface is dynamic, the binodal line is more relevant because it defines compositions at which the phase separation is thermodynamically favorable.



 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}