


An Overview of the Development and Preclinical Evaluation of Antibody–Drug Conjugates for Non-Oncological Applications




Abstract
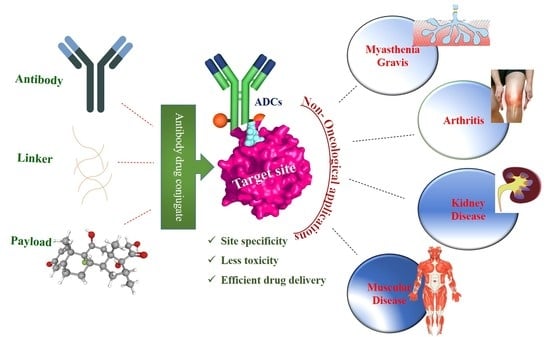
1. Introduction
- Antibody–drug conjugate mechanism of action:
- The fundamental mechanism after the entry of ADCs into the body is discussed below [38].
- Circulation: Antibody–drug solution administered by intravenous route enters the bloodstream.
- Binding: The mAb component of the ADC binds to the target antigen.
- Internalization: Internalization of ADC occurs via receptor-mediated endocytosis.
- Recycling: Early endosomes’ fraction of ADC binds to FcRn receptor.
- Release: Lysosomes fuse with late endosomes and release active moiety.
2. ADCs beyond the Cancer Treatment
2.1. Anti-Inflammatory
2.1.1. Anti-E Selectin Dexamethasone Conjugates
2.1.2. Anti-CD163 Dexamethasone Conjugates
2.1.3. Antibody–Curcumin Conjugates Anti-DR5
2.2. Antibody-Mediated siRNA Conjugates
2.2.1. Myasthenia Gravis (MG)
2.2.2. Antibody–siRNA Conjugate in Muscular Disorder
2.3. Antibody–Antibiotic Conjugate
2.4. Glomerular Nephritis
2.5. Rheumatoid Arthritis (RA)
2.6. Immuno-Suppression
2.7. Atherosclerosis
2.8. Systemic Sclerosis
3. Clinical Pharmacology Considerations for Antibody–Drug Conjugates
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tong, J.T.; Harris, P.W.; Brimble, M.A.; Kavianinia, I. An insight into FDA approved antibody-drug conjugates for cancer therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, S.; Ma, P.; Jiang, Y.; Cheng, K.; Yu, Y.; Jiang, N.; Miao, H.; Tang, Q.; Liu, F. Drug conjugate-based anticancer therapy-Current status and perspectives. Cancer Lett. 2022, 552, 215969. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, R.E.; Wang, F. Antibody-drug conjugates for non-oncological indications. Expert Opin. Biol. Ther. 2016, 16, 591–593. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Giles, F.J.; Estey, E.; O’Brien, S.; Keating, M.J.; Kantarjian, H.M. The role of gemtuzumab ozogamicin in acute leukaemia therapy. Br. J. Haematol. 2006, 132, 398–409. [Google Scholar] [CrossRef]
- Liberante, F.G.; Pouryahya, T.; McMullin, M.F.; Zhang, S.D.; Mills, K.I. Identification and validation of the dopamine agonist bromocriptine as a novel therapy for high-risk myelodysplastic syndromes and secondary acute myeloid leukemia. Oncotarget 2016, 7, 6609–6619. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Kantarjian, H.M.; Kornblau, S.M.; Thomas, D.A.; Garcia-Manero, G.; Waddelow, T.A.; David, C.L.; Phan, A.T.; Colburn, D.E.; Rashid, A. Mylotarg™(gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2001, 92, 406–413. [Google Scholar] [CrossRef]
- Wadleigh, M.; Richardson, P.G.; Zahrieh, D.; Lee, S.J.; Cutler, C.; Ho, V.; Alyea, E.P.; Antin, J.H.; Stone, R.M.; Soiffer, R.J. Prior gemtuzumab ozogamicin exposure significantly increases the risk of veno-occlusive disease in patients who undergo myeloablative allogeneic stem cell transplantation. Blood 2003, 102, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- The U.S. Food and Drug Administration. FDA Approves Mylotarg for Treatment of Acute Myeloid Leukemia. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-mylotarg-treatment-acute-myeloid-leukemia (accessed on 3 June 2023).
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19. [Google Scholar] [CrossRef]
- Waight, A.B.; Bargsten, K.; Doronina, S.; Steinmetz, M.O.; Sussman, D.; Prota, A.E. Structural basis of microtubule destabilization by potent auristatin anti-mitotics. PLoS ONE 2016, 11, e0160890. [Google Scholar] [CrossRef]
- Lambert, J.M.; Chari, R.V. Ado-trastuzumab Emtansine (T-DM1): An antibody–drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody–cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S. Antibody–Drug Conjugates for Immunology. J. Med. Chem. 2022, 65, 4496–4499. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Everts, M.; Kok, R.J.; Asgeirsdóttir, S.A.; Melgert, B.N.; Moolenaar, T.J.; Koning, G.A.; van Luyn, M.J.; Meijer, D.K.; Molema, G. Selective intracellular delivery of dexamethasone into activated endothelial cells using an E-selectin-directed immunoconjugate. J. Immunol. 2002, 168, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Theocharopoulos, C.; Lialios, P.-P.; Samarkos, M.; Gogas, H.; Ziogas, D.C. Antibody-drug conjugates: Functional principles and applications in oncology and beyond. Vaccines 2021, 9, 1111. [Google Scholar] [CrossRef]
- Graversen, J.H.; Svendsen, P.; Dagnæs-Hansen, F.; Dal, J.; Anton, G.; Etzerodt, A.; Petersen, M.D.; Christensen, P.A.; Møller, H.J.; Moestrup, S.K. Targeting the hemoglobin scavenger receptor CD163 in macrophages highly increases the anti-inflammatory potency of dexamethasone. Mol. Ther. 2012, 20, 1550–1558. [Google Scholar] [CrossRef]
- Brandish, P.E.; Palmieri, A.; Antonenko, S.; Beaumont, M.; Benso, L.; Cancilla, M.; Cheng, M.; Fayadat-Dilman, L.; Feng, G.; Figueroa, I.; et al. Development of Anti-CD74 Antibody–Drug Conjugates to Target Glucocorticoids to Immune Cells. Bioconjug. Chem. 2018, 29, 2357–2369. [Google Scholar] [CrossRef]
- Wang, R.E.; Liu, T.; Wang, Y.; Cao, Y.; Du, J.; Luo, X.; Deshmukh, V.; Kim, C.H.; Lawson, B.R.; Tremblay, M.S. An immunosuppressive antibody–drug conjugate. J. Am. Chem. Soc. 2015, 137, 3229–3232. [Google Scholar] [CrossRef]
- Lim, R.K.; Yu, S.; Cheng, B.; Li, S.; Kim, N.-J.; Cao, Y.; Chi, V.; Kim, J.Y.; Chatterjee, A.K.; Schultz, P.G. Targeted delivery of LXR agonist using a site-specific antibody–drug conjugate. Bioconjug. Chem. 2015, 26, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release 2016, 237, 1–13. [Google Scholar] [CrossRef]
- Ibtehaj, N.; Huda, R. High-dose BAFF receptor specific mAb-siRNA conjugate generates Fas-expressing B cells in lymph nodes and high-affinity serum autoantibody in a myasthenia mouse model. Clin. Immunol. 2017, 176, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Codina, A.; Nevskaya, T.; Pope, J. OP0172 Brentuximab Vedontin for Skin Involvement in Refractory Diffuse Cutaneous Systemic Sclerosis, Interim Results of a Phase IIB Open-Label Trial. BMJ 2021, 80, 103–104. [Google Scholar] [CrossRef]
- Lee, H.; Bhang, S.H.; Lee, J.H.; Kim, H.; Hahn, S.K. Tocilizumab–alendronate conjugate for treatment of rheumatoid arthritis. Bioconj. Chem. 2017, 28, 1084–1092. [Google Scholar] [CrossRef]
- Nagai, T.; Tanaka, M.; Tsuneyoshi, Y.; Matsushita, K.; Sunahara, N.; Matsuda, T.; Yoshida, H.; Komiya, S.; Onda, M.; Matsuyama, T. In vitro and in vivo efficacy of a recombinant immunotoxin against folate receptor β on the activation and proliferation of rheumatoid arthritis synovial cells. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2006, 54, 3126–3134. [Google Scholar] [CrossRef]
- Mehta, G.; Scheinman, R.I.; Holers, V.M.; Banda, N.K. A new approach for the treatment of arthritis in mice with a novel conjugate of an anti-C5aR1 antibody and C5 small interfering RNA. J. Immunol. 2015, 194, 5446–5454. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Hiroshi Morisaki, J. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Hobson, A.D.; McPherson, M.J.; Hayes, M.E.; Goess, C.; Li, X.; Zhou, J.; Wang, Z.; Yu, Y.; Yang, J.; Sun, L.; et al. Discovery of ABBV-3373, an Anti-TNF Glucocorticoid Receptor Modulator Immunology Antibody Drug Conjugate. J. Med. Chem. 2022, 65, 15893–15934. [Google Scholar] [CrossRef]
- Hobson, A.D.; McPherson, M.J.; Waegell, W.; Goess, C.A.; Stoffel, R.H.; Li, X.; Zhou, J.; Wang, Z.; Yu, Y.; Hernandez, A., Jr.; et al. Design and Development of Glucocorticoid Receptor Modulators as Immunology Antibody–Drug Conjugate Payloads. J. Med. Chem. 2022, 65, 4500–4533. [Google Scholar] [CrossRef]
- Svendsen, P.; Graversen, J.H.; Etzerodt, A.; Hager, H.; Røge, R.; Grønbæk, H.; Christensen, E.I.; Møller, H.J.; Vilstrup, H.; Moestrup, S.K. Antibody-Directed Glucocorticoid Targeting to CD163 in M2-type Macrophages Attenuates Fructose-Induced Liver Inflammatory Changes. Mol. Methods Clin. Dev. 2017, 4, 50–61. [Google Scholar] [CrossRef]
- Han, A.; Olsen, O.; D’Souza, C.; Shan, J.; Zhao, F.; Yanolatos, J.; Hovhannisyan, Z.; Haxhinasto, S.; Delfino, F.; Olson, W. Development of Novel Glucocorticoids for Use in Antibody-Drug Conjugates for the Treatment of Inflammatory Diseases. J. Med. Chem. 2021, 64, 11958–11971. [Google Scholar] [CrossRef]
- Su, Z.; Xie, F.; Xu, X.; Liu, L.; Xiao, D.; Zhou, X.; Li, S. Development of a nitroreductase-dependent theranostic payload for antibody-drug conjugate. Bioorg. Chem. 2022, 129, 106190. [Google Scholar] [CrossRef] [PubMed]
- Stagg, N.J.; Katavolos, P.; Poon, K.A.; Zhong, S.; Ljumanovic, N.; Kamath, A.; Cai, H.; Carrasco-Triguero, M.; Halpern, W. Nonclinical toxicology development of a novel antibody antibiotic conjugate for treating invasive Staphylococcus Aureus infections. Toxicol. Appl. Pharmacol. 2022, 435, 115811. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Delaney, J.; Guillard, T.; Reffuveille, F.; Varin-Simon, J.; Li, K.; Wollacott, A.; Frapy, E.; Mong, S.; Tissire, H. Development of an antibody fused with an antimicrobial peptide targeting Pseudomonas aeruginosa: A new approach to prevent and treat bacterial infections. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hu, N.; Zhang, Y.; Yang, J.; Zhao, L.; Zhang, X.; Yang, Y.; Zhang, J.; Zou, Y.; Wei, K.; Zhao, C. Antibody-Antibiotic Conjugate Targeted Therapy for Orthopedic Implant-Associated Intracellular S. aureus Infections. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Kommineni, N.; Pandi, P.; Chella, N.; Domb, A.J.; Khan, W. Antibody drug conjugates: Development, characterization, and regulatory considerations. Polym. Adv. Technol. 2020, 31, 1177–1193. [Google Scholar] [CrossRef]
- Hasan, M.M.; Laws, M.; Jin, P.; Rahman, K.M. Factors influencing the choice of monoclonal antibodies for antibody–drug conjugates. Drug Discov. Today 2022, 27, 354–361. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Poetker, D.M.; Reh, D.D. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryngol. Clin. N. Am. 2010, 43, 753–768. [Google Scholar] [CrossRef]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef]
- Lin, Y.L.; Lin, C.Y.; Chi, C.W.; Huang, Y.T. Study on antifibrotic effects of curcumin in rat hepatic stellate cells. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2009, 23, 927–932. [Google Scholar] [CrossRef] [PubMed]
- You, D.G.; Kim, C.H.; Kwon, S.; Um, W.; Oh, B.H.; An, J.Y.; Jeon, J.; Park, J.H. An anti-DR5 antibody-curcumin conjugate for the enhanced clearance of activated hepatic stellate cells. Int. J. Biol. Macromol. 2021, 192, 1231–1239. [Google Scholar]
- Gandhi, C.R. Oxidative stress and hepatic stellate cells: A paradoxical relationship. Trends Cell Mol. Biol. 2012, 7, 1. [Google Scholar]
- Choi, Y.J.; Kim, D.H.; Kim, S.J.; Kim, J.; Jeong, S.-I.; Chung, C.H.; Yu, K.-Y.; Kim, S.-Y. Decursin attenuates hepatic fibrogenesis through interrupting TGF-beta-mediated NAD (P) H oxidase activation and Smad signaling in vivo and in vitro. Life Sci. 2014, 108, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q. Site-Specific Antibody Conjugation with Payloads beyond Cytotoxins. Molecules 2023, 28, 917. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Li, R.; Pei, X.; Chai, M.; Sun, L.; Huang, Y.; Wang, J.; Barth, S.; Yu, F.; He, H. Antibody-siRNA conjugates (ARC): Emerging siRNA drug formulation. Med. Drug Discov. 2022, 15, 100128. [Google Scholar] [CrossRef]
- Lefvert, A.K.; Zhao, Y.; Ramanujam, R.; Yu, S.; Pirskanen, R.; Hammarström, L. PTPN22 R620W promotes production of anti-AChR autoantibodies and IL-2 in myasthenia gravis. J. Neuroimmunol. 2008, 197, 110–113. [Google Scholar] [CrossRef]
- Dabi, A.; Solieman, N.; Kurukumbi, M.; Kalyanam, J. Myasthenia gravis: A review. Autoimmune Dis. 2012, 2012, 874680. [Google Scholar]
- Mariathasan, S.; Tan, M.-W. Antibody–antibiotic conjugates: A novel therapeutic platform against bacterial infections. Trends Mol. Med. 2017, 23, 135–149. [Google Scholar] [CrossRef]
- Cavaco, M.; Castanho, M.A.; Neves, V. The use of antibody-antibiotic conjugates to fight bacterial infections. Front. Microbiol. 2022, 13, 666. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–drug conjugates: The last decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Peck, M.; Rothenberg, M.E.; Deng, R.; Lewin-Koh, N.; She, G.; Kamath, A.V.; Carrasco-Triguero, M.; Saad, O.; Castro, A.; Teufel, L. A phase 1, randomized, single-ascending-dose study to investigate the safety, tolerability, and pharmacokinetics of DSTA4637S, an anti-Staphylococcus aureus thiomab antibody-antibiotic conjugate, in healthy volunteers. Antimicrob. Agents Chemother. 2019, 63, e02588-18. [Google Scholar] [CrossRef]
- Kajihara, K.K.; Pantua, H.; Hernandez-Barry, H.; Hazen, M.; Deshmukh, K.; Chiang, N.; Ohri, R.; Castellanos, E.R.; Martin, L.; Matsumoto, M.L. Potent killing of Pseudomonas aeruginosa by an antibody-antibiotic conjugate. Mbio 2021, 12, e00202–e00221. [Google Scholar] [CrossRef]
- Kvirkvelia, N.; McMenamin, M.; Gutierrez, V.I.; Lasareishvili, B.; Madaio, M.P. Human anti-α3 (IV) NC1 antibody drug conjugates target glomeruli to resolve nephritis. Am. J. Physiol.-Ren. Physiol. 2015, 309, F680–F684. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef]
- Shen, H.; Xiu, Z.; Tian, Y.; Xia, L.; Lu, J. AB0065 Elevated Serum and Synovial Fluid Levels of Tumor Necrosis Factor-Like Ligand 1A and Decoy Receptor 3 in Rheumatoid Arthritis: Induce Interleukin-17 Production. Ann. Rheum. Dis. 2014, 73, 825. [Google Scholar] [CrossRef]
- Mullard, A. Maturing antibody-drug conjugate pipeline hits 30. Nat. Rev. Drug Discov. 2013, 12, 329–333. [Google Scholar] [CrossRef]
- Bohdziewicz, A.; Pawlik, K.K.; Maciejewska, M.; Sikora, M.; Alda-Malicka, R.; Czuwara, J.; Rudnicka, L. Future treatment options in systemic sclerosis—Potential targets and ongoing clinical trials. J. Clin. Med. 2022, 11, 1310. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Brentuximab Vedotin for Systemic Sclerosis (BRAVOS). Available online: https://clinicaltrials.gov/ct2/show/NCT03222492 (accessed on 6 June 2023).
- ClinicalTrials.gov. Brentuximab Vedotin in Early Diffuse Cutaneous Systemic Sclerosis. Available online: https://clinicaltrials.gov/ct2/show/NCT03198689 (accessed on 6 June 2023).
- ClinicalTrials.gov. Open Label Extension Study of Brentuximab Vedotin in Early dcSSc. Available online: https://clinicaltrials.gov/ct2/show/NCT05149768?term=NCT05149768&draw=2&rank=1 (accessed on 6 June 2023).
- ClinicalTrials.gov. Brentuximab Vedotin in Treating Patients with Steroid-Resistant Acute Graft-Versus-Host Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT01616680?term=NCT01616680&draw=2&rank=1 (accessed on 6 June 2023).
- The U.S. Food & Drug Administraion. Clinical Pharmacology Considerations for Antibody-Drug Conjugates Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-pharmacology-considerations-antibody-drug-conjugates-guidance-industry (accessed on 4 June 2023).



{kind=link}
{kind=link}
{kind=link}
| Indications | ADCs | Antibody | Linkers | Payloads | Testing Status | Ref. |
|---|---|---|---|---|---|---|
| Inflammation | Anti-E Selectin Dex (Dexa–AbhEsel) | Murine anti-E-selectin mAb (H18/7) | Succinate linker | Dexamethasone | In vitro pre-clinical | [17] |
| Chronic models of inflammation | Anti-CD163 Dex (Cymac-001) | Murine anti-CD163 mAb (Ed-2) | Hemisuccinate linker | Dexamethasone | In vivo pre-clinical | [19] |
| Autoimmune models | Anti-CD74 fluticasone propionate (Anti-CD74-flu449) | Human anti-CD74 mAb | Pyrophosphate acetal linker | Fluticasone propionate | In vivo pre-clinical | [20] |
| Autoimmune and inflammatory models | Anti-CXCR4 dasatinib | Humanized anti-CXCR4 mAb (HLCX) | Tetra-poly-ethylene glycol linker | Dasatinib | In vitro pre-clinical | [21] |
| Atherosclerosis | Anti-CD11a LXR agonist | Humanized anti-CD11 mAb | PEG4-Phe-Lys | Amino acid para-acetylphenylalanine | In vitro pre-clinical | [22] |
| Muscular diseases | Anti-CD71 siRNA | Murine anti-CD71 | mAb Maleimide linker | siRNA | In vivo pre-clinical | [23] |
| Myasthenia gravis | Anti-TNFRSF13c siRNA | anti-TNFRSF13c mA | Protamine linker | siRNA | In vivo pre-clinical | [24] |
| Systemic sclerosis | Anti-CD30 Vedotin (ADCETRIS) | Chimeric anti-CD30 mAb (cAC10, SGN-30) | Val-Cit linker | MMAE (Monomethyl auristatin E) | Phase II clinical trial (NCT03198689), (NCT03222492) | [25] |
| Rheumatoid arthritis | Anti-IL-6 alendronate | Humanized anti-IL-6 mAb (tocilizumab) | PDPH-PEG-NHS | Alendronate (ALD) | In vivo pre-clinical | [26] |
| Rheumatoid arthritis | Anti-FRβ Pseudomonas exotoxin A (PE38) | Murine anti-FRβ mAb | NA | Pseudomonas exotoxin A (PE38) | In vivo pre-clinical | [27] |
| Rheumatoid arthritis | Anti–C5aR1 C5 siRNA | Murine anti-C5aR1 mAb | Protamine linker | C5 siRNA | In vivo pre-clinical | [28] |
| S. aureus bacteremia | Anti-S. aureus Antibiotic (DSTA4637S) | Human anti-β-Nacetylglucosamine cell-wall teichoic acid (β-GlcNAc- WTA) mAb | MC-Val-Cit-PAB-OH | Antibiotic (rifalogue) | Phase I clinical trial (NCT03162250) | [29] |
| Arthritis | Anti-mTNF GRM (ABBV-3373) | Antibody: alpha TNF | MP-Ala-Ala | Glucocorticoid Receptor Modulator (GRM) | In vitro pre-clinical | [30] |
| Arthritis | α-TNF-GRM ADC | Antibody: alpha TNF | M-Gly-Ala-Ala | GRM | In vitro pre-clinical | [31] |
| Inflammation | Anti-TNFα glucocorticoid | Anti-TNF mAb | Dipeptide-based (Ala-Ala) protease-cleavable | Dexamethasone | In vitro pre-clinical | [15] |
| Non-alcoholic fatty liver disease | Anti-CD163-IgG- Dex | Anti-CD163 mAb | Hemisuccinate linker | Dexamethasone | In vivo pre-clinical | [32] |
| Bowel disease, ulcerative colitis, and Crohn’s disease. | anti-CD70 mAb–Budesonide | Anti-CD70 mAb | Carbamate linkage | Budesonide | In vitro and vivo pre-clinical study | [33] |
| Theragnostic agent | Trastuzumab-7- nitro-3-hydroxyethyl- coumarin- Monomethyl auristatin E | Trastuzumab | Carbamate linkage | L-233 | In vitro pre-clinical study | [34] |
| Staphylococcus Aureus infections | THIOMAB™ antibody antibiotic conjugate/ DSTA4637A | Human IgG1 anti-S. aureus THIOMAB™ monoclonal antibody | valine-citrulline linker | Antibiotic dmDNA31 | In vitro and In vivo study | [35] |
| Lung infection | VSX-D297 antimicrobial antibody conjugate | VSX | Enzymatically coupling with Sortase A | Antimicrobial peptides | In vitro studies | [36] |
| Orthopedic implant-associated intracellular S. aureus infections | M0662-MC-Val-Cit-PAB-Vancomycin | Human monoclonal antibody (M0662) against the surface antigen Staphylococcal protein A (SpA) of S. aureus | Mc-Val-Cit-PAB | Vancomycin | In vitro studies | [37] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pal, L.B.; Bule, P.; Khan, W.; Chella, N. An Overview of the Development and Preclinical Evaluation of Antibody–Drug Conjugates for Non-Oncological Applications. Pharmaceutics 2023, 15, 1807. https://doi.org/10.3390/pharmaceutics15071807
Pal LB, Bule P, Khan W, Chella N. An Overview of the Development and Preclinical Evaluation of Antibody–Drug Conjugates for Non-Oncological Applications. Pharmaceutics. 2023; 15(7):1807. https://doi.org/10.3390/pharmaceutics15071807
Chicago/Turabian StylePal, Lal Bahadur, Prajakta Bule, Wahid Khan, and Naveen Chella. 2023. "An Overview of the Development and Preclinical Evaluation of Antibody–Drug Conjugates for Non-Oncological Applications" Pharmaceutics 15, no. 7: 1807. https://doi.org/10.3390/pharmaceutics15071807
APA StylePal, L. B., Bule, P., Khan, W., & Chella, N. (2023). An Overview of the Development and Preclinical Evaluation of Antibody–Drug Conjugates for Non-Oncological Applications. Pharmaceutics, 15(7), 1807. https://doi.org/10.3390/pharmaceutics15071807