
Combining Molecular Dynamics Simulations and Biophysical Characterization to Investigate Protein-Specific Excipient Effects on Reteplase during Freeze Drying

 , ,
, ,
Abstract
:
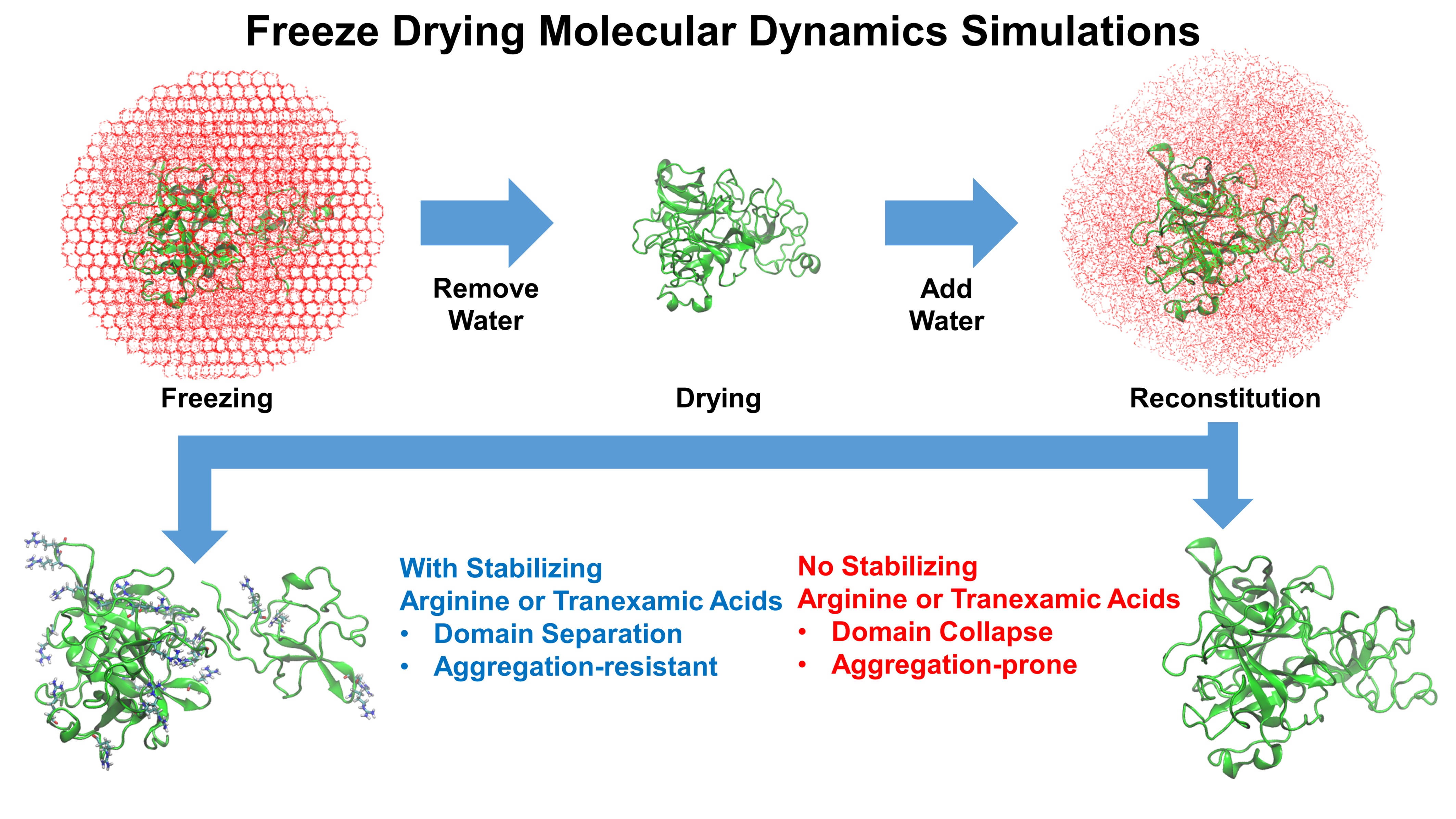
1. Introduction
2. Materials and Methods
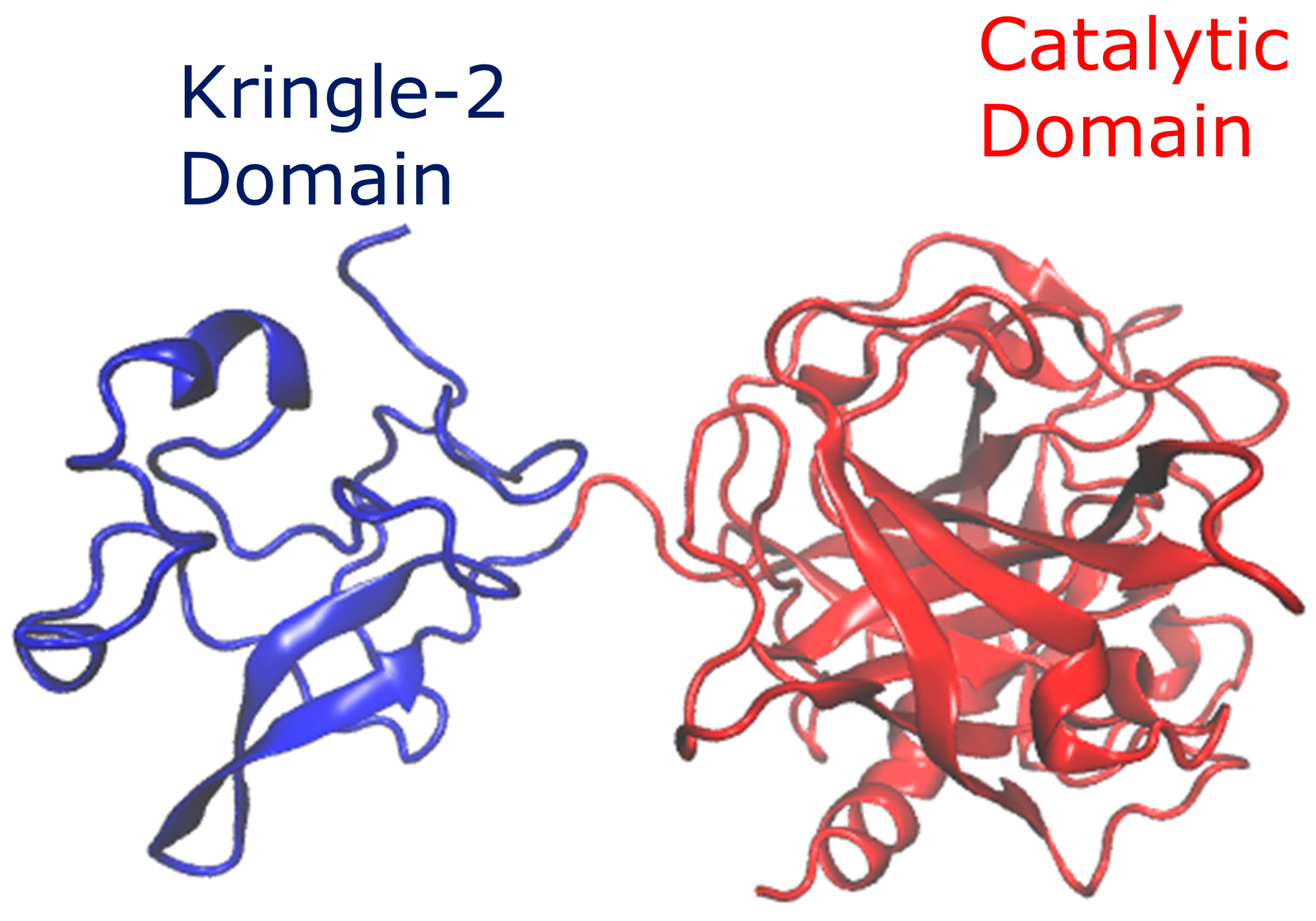
2.1. Structure
2.2. Coarse-Grained Simulations
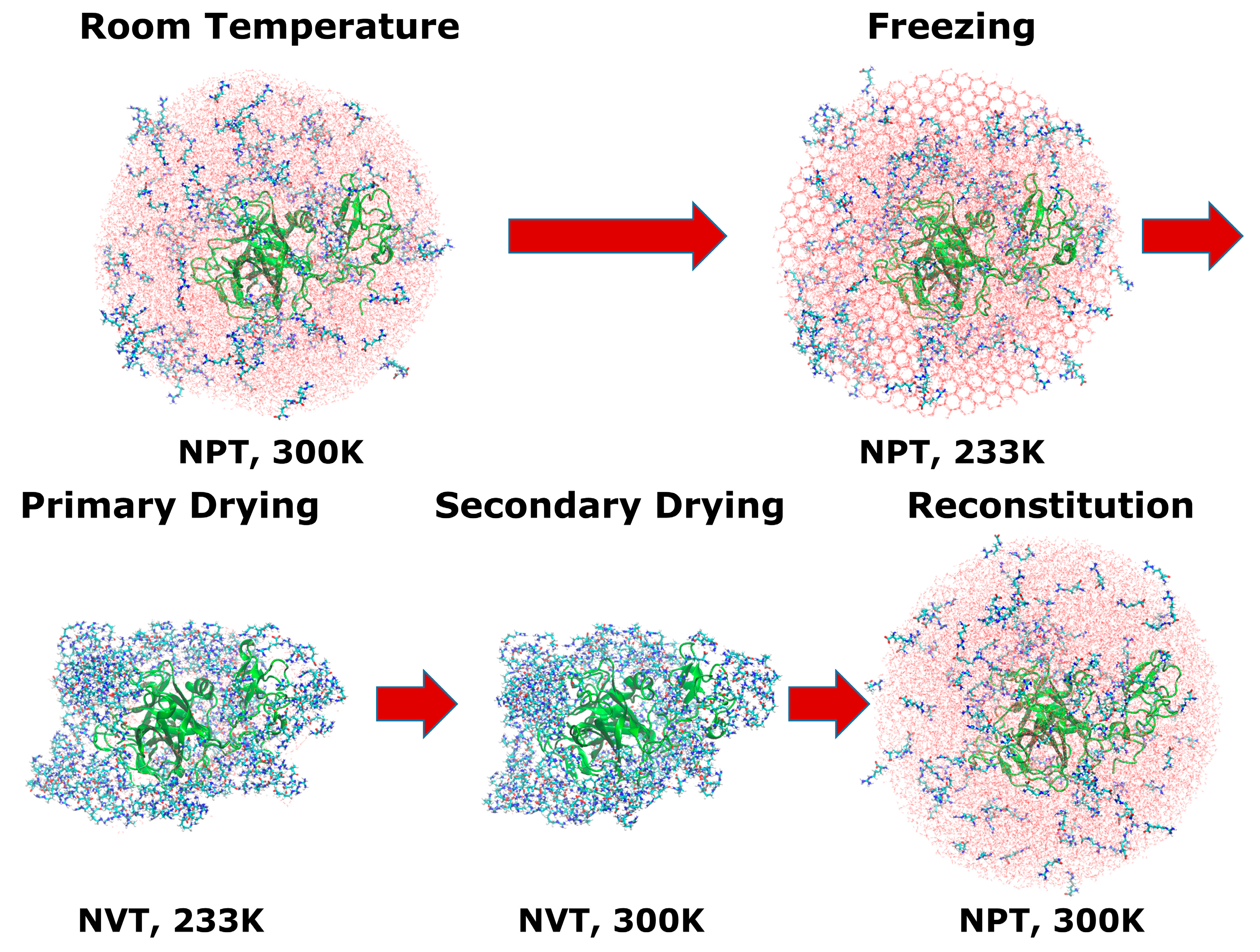
2.3. Freeze-Drying Simulations
2.4. Materials
2.5. Sample Dialysis and Preparation
2.6. Freeze-Thaw Cycles and Lyophilization
2.7. Differential Scanning Fluorimetry (nanoDSF)
2.8. Circular Dichroism (CD) Spectroscopy
2.9. High-Performance Size Exclusion Chromatography (HP-SEC)
3. Results and Discussion
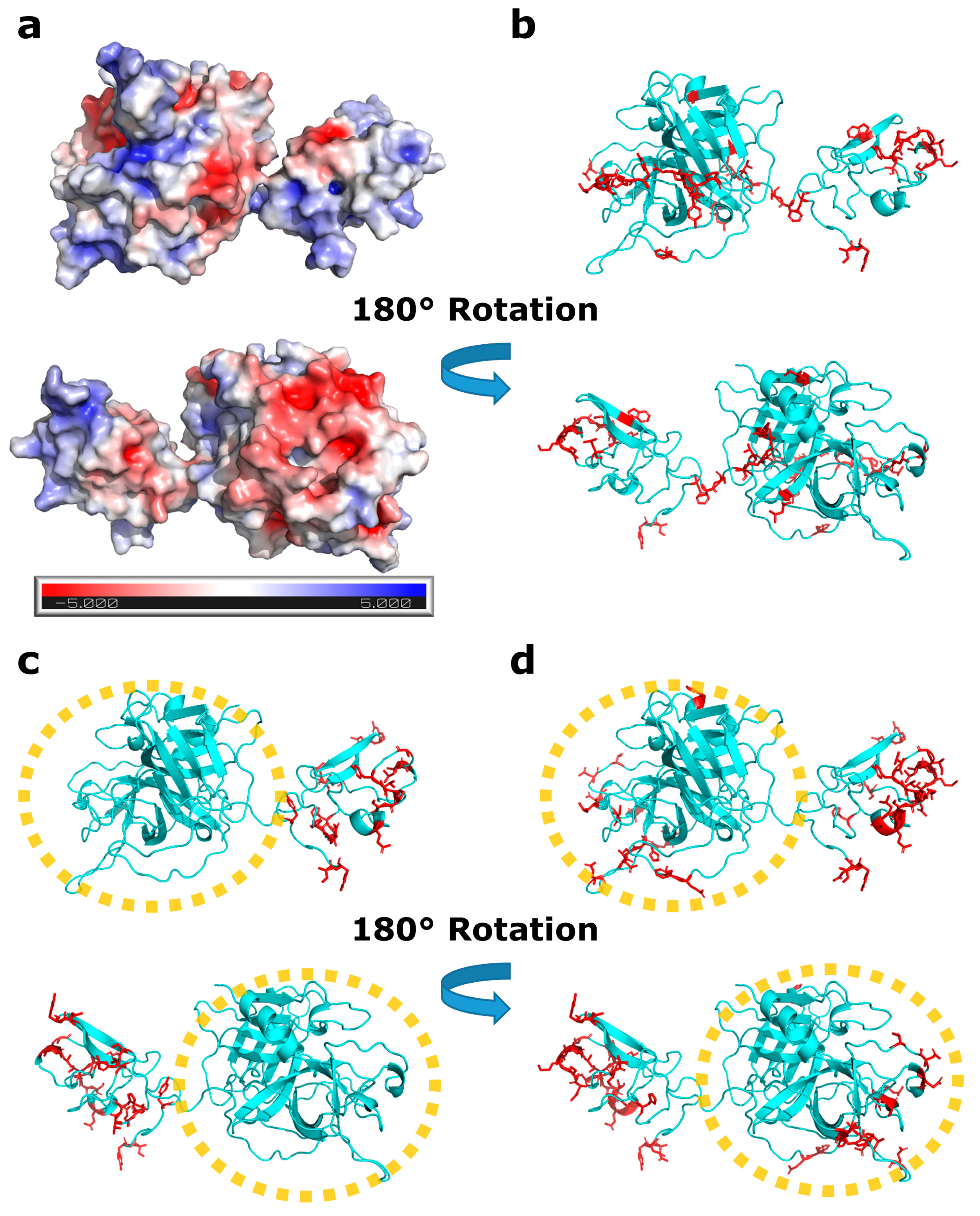
3.1. Protein–Protein Interaction Study of Reteplase
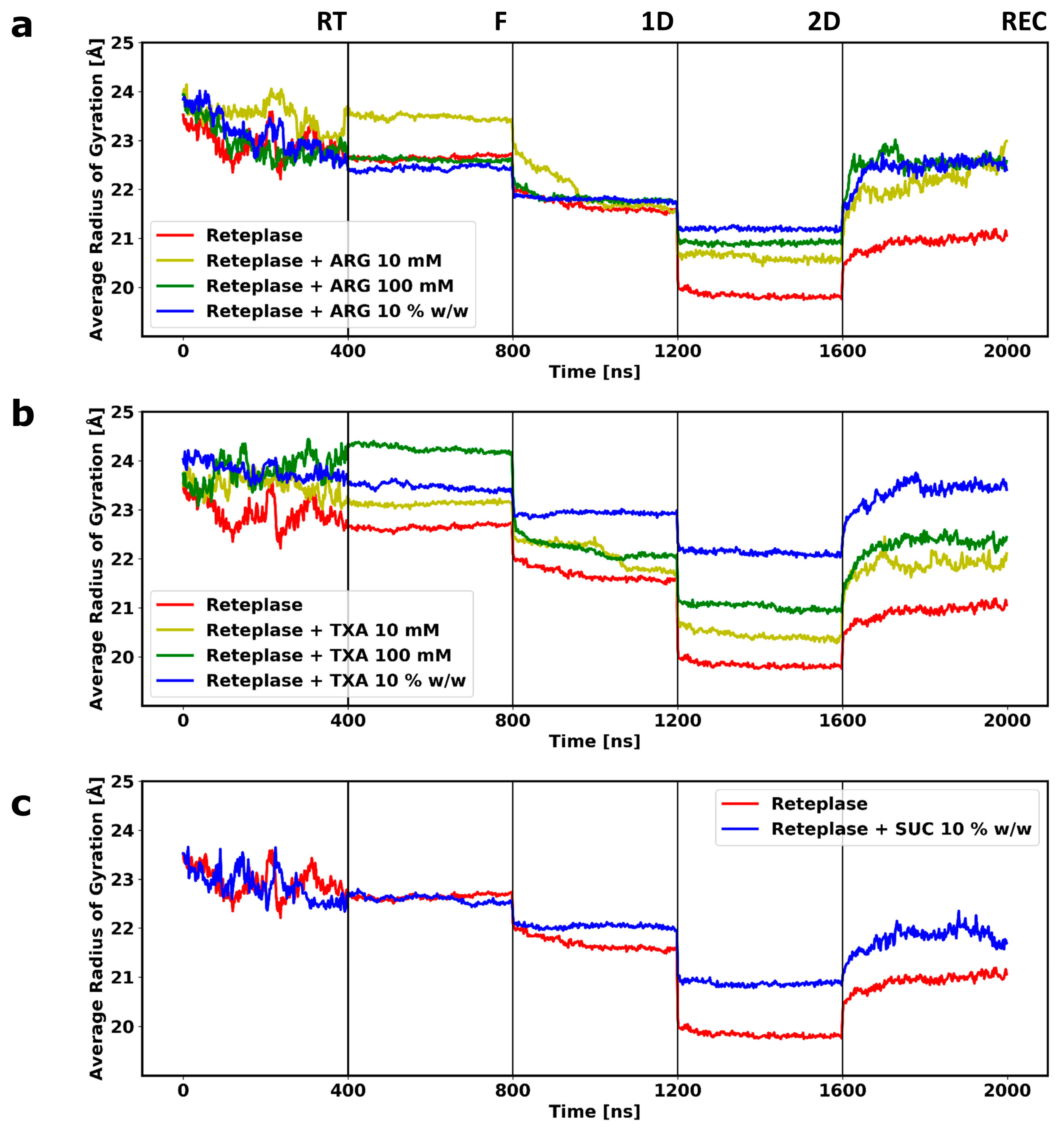
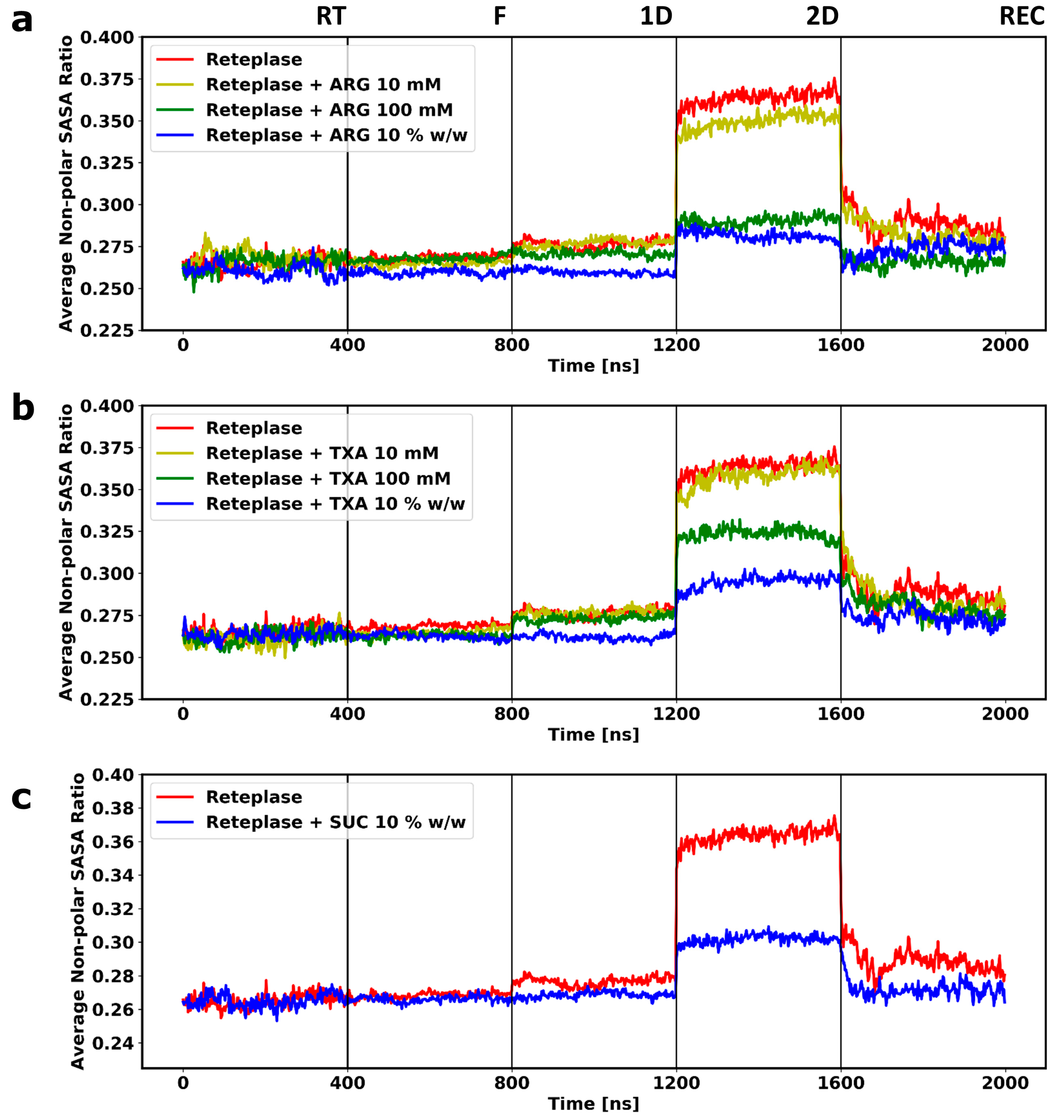
3.2. Molecular Dynamics Simulations of the Freeze-Drying Process
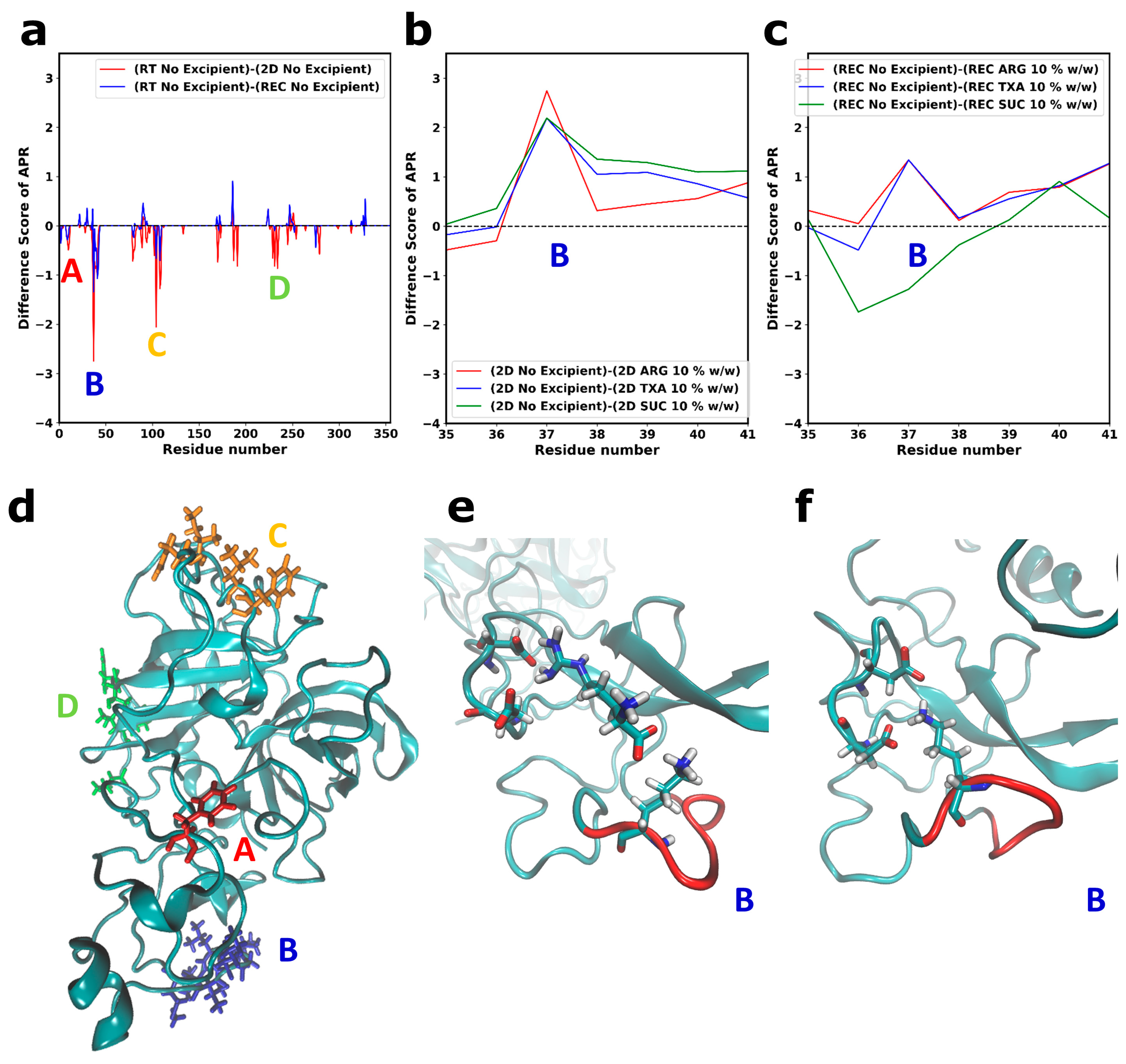
3.3. Local Structural Changes and Effects of Excipients
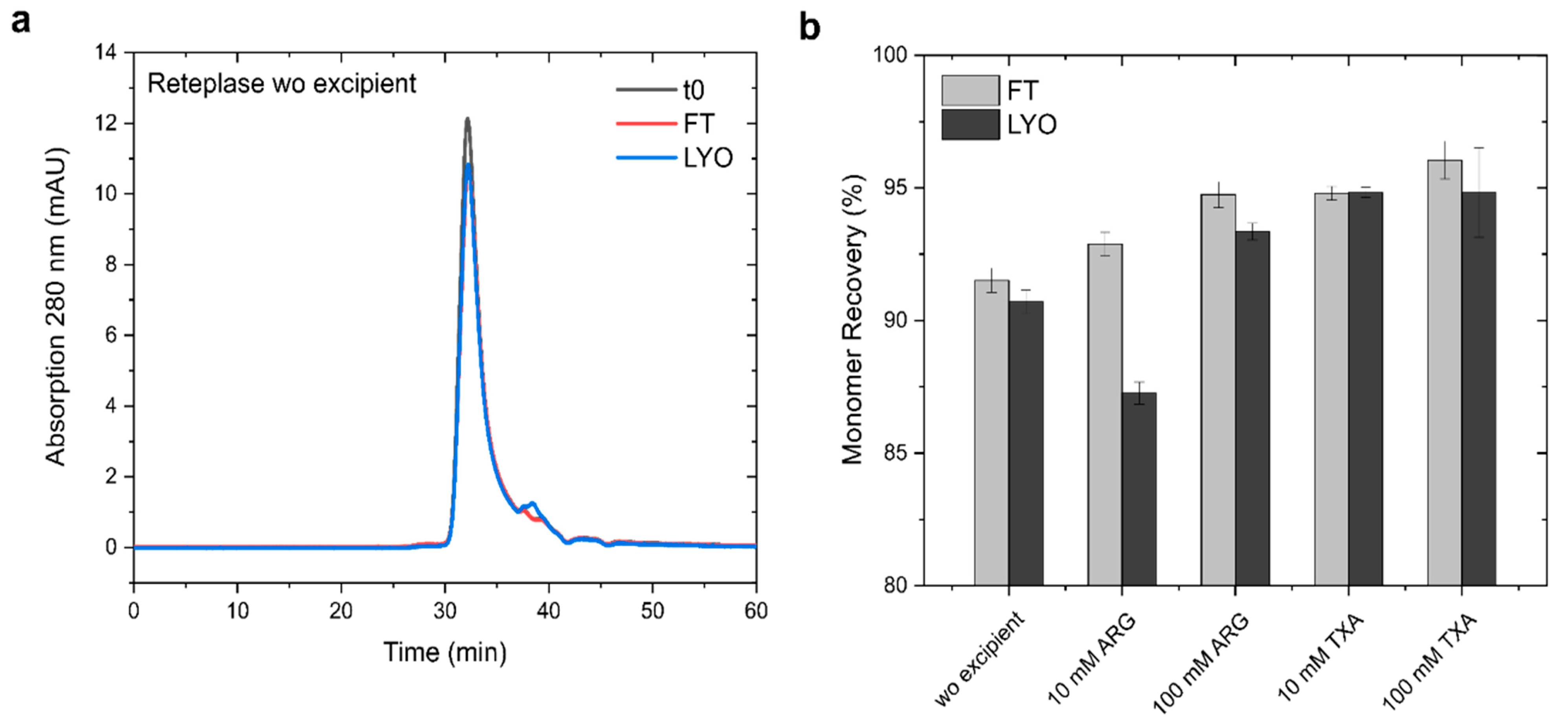
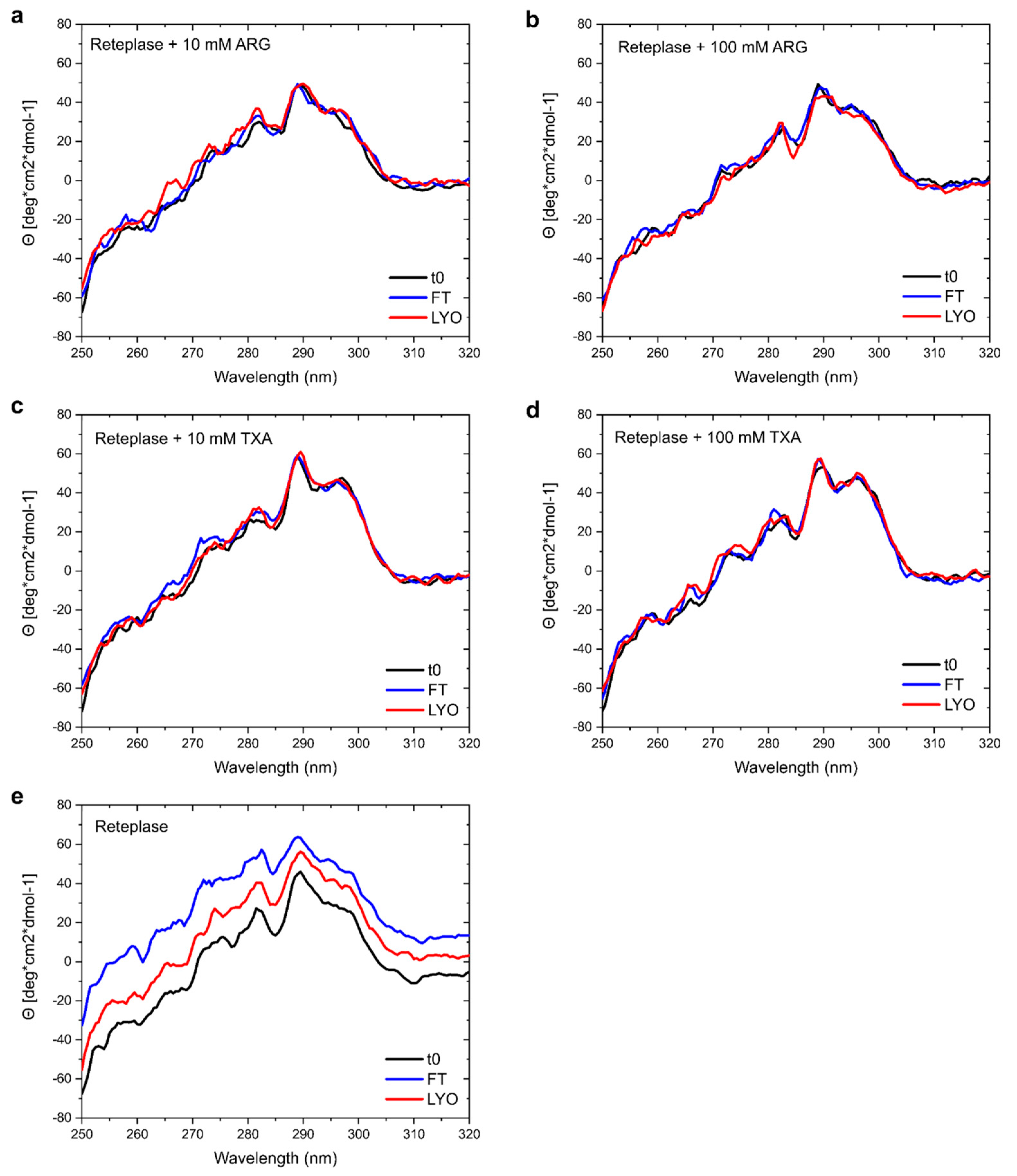
3.4. Experimental Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, W. Lyophilization and Development of Solid Protein Pharmaceuticals. Int. J. Pharm. 2000, 203, 1–60. [Google Scholar] [CrossRef] [PubMed]
- Seifert, I.; Friess, W. 3 Established and Novel Excipients for Freeze-Drying of Proteins. In Freeze Drying of Pharmaceutical Products; CRC Press: Boca Raton, FL, USA, 2019; p. 33. [Google Scholar]
- Arsiccio, A.; Pisano, R. Stability of Proteins in Carbohydrates and Other Additives during Freezing: The Human Growth Hormone as a Case Study. J. Phys. Chem. B 2017, 121, 8652–8660. [Google Scholar] [CrossRef]
- Arsiccio, A.; Pisano, R. Clarifying the Role of Cryo- and Lyo-Protectants in the Biopreservation of Proteins. Phys. Chem. Chem. Phys. 2018, 20, 8267–8277. [Google Scholar] [CrossRef] [PubMed]
- Arsiccio, A.; Pisano, R. Water Entrapment and Structure Ordering as Protection Mechanisms for Protein Structural Preservation. J. Chem. Phys. 2018, 148, 055102. [Google Scholar] [CrossRef] [PubMed]
- Arsiccio, A.; Pisano, R. Surfactants as Stabilizers for Biopharmaceuticals: An Insight into the Molecular Mechanisms for Inhibition of Protein Aggregation. Eur. J. Pharm. Biopharm. 2018, 128, 98–106. [Google Scholar] [CrossRef]
- Arsiccio, A.; Pisano, R. The Preservation of Lyophilized Human Growth Hormone Activity: How Do Buffers and Sugars Interact? Pharm. Res. 2018, 35, 131. [Google Scholar] [CrossRef]
- Arsiccio, A.; McCarty, J.; Pisano, R.; Shea, J.E. Effect of Surfactants on Surface-Induced Denaturation of Proteins: Evidence of an Orientation-Dependent Mechanism. J. Phys. Chem. B 2018, 122, 11390–11399. [Google Scholar] [CrossRef]
- Arsiccio, A.; Paladini, A.; Pattarino, F.; Pisano, R. Designing the Optimal Formulation for Biopharmaceuticals: A New Approach Combining Molecular Dynamics and Experiments. J. Pharm. Sci. 2019, 108, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Arsiccio, A.; McCarty, J.; Pisano, R.; Shea, J.-E. Heightened Cold-Denaturation of Proteins at the Ice–Water Interface. J. Am. Chem. Soc. 2020, 142, 5722–5730. [Google Scholar] [CrossRef]
- Rospiccio, M.; Arsiccio, A.; Winter, G.; Pisano, R. The Role of Cyclodextrins against Interface-Induced Denaturation in Pharmaceutical Formulations: A Molecular Dynamics Approach. Mol. Pharm. 2021, 18, 2322–2333. [Google Scholar] [CrossRef]
- Kohnert, U.; Rudolph, R.; Verheijen, J.H.; Jacoline, E.; Weening-Verhoeff, D.; Stern, A.; Opitz, U.; Martin, U.; Lill, H.; Prinz, H. Biochemical Properties of the Kringle 2 and Protease Domains Are Maintained in the Refolded T-PA Deletion Variant BM 06.022. Protein Eng. Des. Sel. 1992, 5, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Gurman, P.; Miranda, O.R.; Nathan, A.; Washington, C.; Rosen, Y.; Elman, N.M. Recombinant Tissue Plasminogen Activators (RtPA): A Review. Clin. Pharmacol. Ther. 2015, 97, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Tischer, A.; Lilie, H.; Rudolph, R.; Lange, C. L-Arginine Hydrochloride Increases the Solubility of Folded and Unfolded Recombinant Plasminogen Activator RPA. Protein Sci. 2010, 19, 1783–1795. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, S.B.; Fazeli, A.; Shojaosadati, S.A.; Fazeli, M.R.; Hashemi-Najafabadi, S. Purification and Efficient Refolding Process for Recombinant Tissue-Type Plasminogen Activator Derivative (Reteplase) Using Glycerol and Tranexamic Acid. Process Biochem. 2017, 53, 135–144. [Google Scholar] [CrossRef]
- De Serrano, V.S.; Sehl, L.C.; Castellino, F.J. Direct Identification of Lysine-33 as the Principal Cationic Center of the ω-Amino Acid Binding Site of the Recombinant Kringle 2 Domain of Tissue-Type Plasminogen Activator. Arch. Biochem. Biophys. 1992, 292, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Byeon, I.-J.L.; Kelley, R.F.; Mulkerrin, M.G.; An, S.S.A.; Llinas, M. Ligand Binding to the Tissue-Type Plasminogen Activator Kringle 2 Domain: Structural Characterization by 1H-NMR. Biochemistry 1995, 34, 2739–2750. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.K. Molecular Understanding of the Effects of Stabilizing Excipient during Lyophilization of Biopharmaceuticals. Ph.D. Thesis, DTU Chemistry, Lyngby, Denmark, 2021. [Google Scholar]
- Waller, M.; Kohnert, U. Reteplase, a Recombinant Plasminogen Activator. In Biopharmaceuticals, An Industrial Perspective; Springer: Berlin/Heidelberg, Germany, 1999; pp. 185–216. [Google Scholar]
- Byeon, I.-J.L.; Llinás, M. Solution Structure of the Tissue-Type Plasminogen Activator Kringle 2 Domain Complexed to 6-Aminohexanoic Acid an Antifibrinolytic Drug. J. Mol. Biol. 1991, 222, 1035–1051. [Google Scholar] [CrossRef]
- De Vos, A.M.; Ultsch, M.H.; Kelley, R.F.; Padmanabhan, K.; Tulinsky, A.; Westbrook, M.L.; Kossiakoff, A.A. Crystal Structure of the Kringle 2 Domain of Tissue Plasminogen Activator at 2.4—A Resolution. Biochemistry 1992, 31, 270–279. [Google Scholar] [CrossRef]
- Renatus, M.; Engh, R.A.; Stubbs, M.T.; Huber, R.; Fischer, S.; Kohnert, U.; Bode, W. Lysine 156 Promotes the Anomalous Proenzyme Activity of TPA: X-ray Crystal Structure of Single-chain Human TPA. EMBO J. 1997, 16, 4797–4805. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Darré, L.; Machado, M.R.; Brandner, A.F.; González, H.C.; Ferreira, S.; Pantano, S. SIRAH: A Structurally Unbiased Coarse-Grained Force Field for Proteins with Aqueous Solvation and Long-Range Electrostatics. J. Chem. Theory Comput. 2015, 11, 723–739. [Google Scholar] [CrossRef]
- Machado, M.R.; Pantano, S. SIRAH Tools: Mapping, Backmapping and Visualization of Coarse-Grained Models. Bioinformatics 2016, 32, 1568–1570. [Google Scholar] [CrossRef] [Green Version]
- Machado, M.R.; Barrera, E.E.; Klein, F.; Sónora, M.; Silva, S.; Pantano, S. The SIRAH 2.0 Force Field: Altius, Fortius, Citius. J. Chem. Theory Comput. 2019, 15, 2719–2733. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Ko, S.K.; Berner, C.; Kulakova, A.; Schneider, M.; Antes, I.; Winter, G.; Harris, P.; Peters, G.H.J. Investigation of the PH-Dependent Aggregation Mechanisms of GCSF Using Low Resolution Protein Characterization Techniques and Advanced Molecular Dynamics Simulations. Comput. Struct. Biotechnol. J. 2022, 20, 1439–1455. [Google Scholar] [CrossRef] [PubMed]
- Darré, L.; Machado, M.R.; Dans, P.D.; Herrera, F.E.; Pantano, S. Another Coarse Grain Model for Aqueous Solvation: WAT FOUR? J. Chem. Theory Comput. 2010, 6, 3793–3807. [Google Scholar] [CrossRef]
- Arakawa, T.; Tsumoto, K.; Kita, Y.; Chang, B.; Ejima, D. Biotechnology Applications of Amino Acids in Protein Purification and Formulations. Amino Acids 2007, 33, 587–605. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Urbassek, H.M.; Thirumuruganandham, S.P. Water Evaporation and Conformational Changes from Partially Solvated Ubiquitin. Biochem. Res. Int. 2010, 2010, 213936. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Giambasu, G. Amber 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Schneider, T.; Stoll, E. Molecular-Dynamics Study of a Three-Dimensional One-Component Model for Distortive Phase Transitions. Phys. Rev. B 1978, 17, 1302. [Google Scholar] [CrossRef]
- Åqvist, J.; Wennerström, P.; Nervall, M.; Bjelic, S.; Brandsdal, B.O. Molecular Dynamics Simulations of Water and Biomolecules with a Monte Carlo Constant Pressure Algorithm. Chem. Phys. Lett. 2004, 384, 288–294. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Chéron, N.; Naepels, M.; Pluharova, E.; Laage, D. Protein Preferential Solvation in Water: Glycerol Mixtures. J. Phys. Chem. B 2020, 124, 1424–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloutier, T.; Sudrik, C.; Sathish, H.A.; Trout, B.L. Kirkwood–Buff-Derived Alcohol Parameters for Aqueous Carbohydrates and Their Application to Preferential Interaction Coefficient Calculations of Proteins. J. Phys. Chem. B 2018, 122, 9350–9360. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lee, J.; Jo, S.; Brooks III, C.L.; Lee, H.S.; Im, W. CHARMM-GUI Ligand Reader and Modeler for CHARMM Force Field Generation of Small Molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an Improved Four-Site Water Model for Biomolecular Simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abascal, J.L.F.; Sanz, E.; Fernández, R.G.; Vega, C. A Potential Model for the Study of Ices and Amorphous Water: TIP4P/Ice. J. Chem. Phys. 2005, 122, 234511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Yagasaki, T.; Tanaka, H. GenIce: Hydrogen-Disordered Ice Generator. J. Comput. Chem. 2017, 39, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.J.; Matejtschuk, P.; Longstaff, C.; Dalby, P.A. Selective Stabilization and Destabilization of Protein Domains in Tissue-Type Plasminogen Activator Using Formulation Excipients. Mol. Pharm. 2018, 16, 744–755. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriata, A.; Iglesias, V.; Pujols, J.; Kurcinski, M.; Kmiecik, S.; Ventura, S. Aggrescan3D (A3D) 2.0: Prediction and Engineering of Protein Solubility. Nucleic Acids Res. 2019, 47, W300–W307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, T.; Minatovicz, B.; Bai, J.; Shin, D.; Mohammadiarani, H.; Chaudhuri, B. Molecular Dynamics Simulation to Uncover the Mechanisms of Protein Instability during Freezing. J. Pharm. Sci. 2021, 110, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Bottaro, S.; Lindorff-Larsen, K. Biophysical Experiments and Biomolecular Simulations: A Perfect Match? Science 2018, 361, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Pikal, M.J. Design of Freeze-Drying Processes for Pharmaceuticals: Practical Advice. Pharm. Res. 2004, 21, 191–200. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Excipient | Concentration | Number of Excipient Molecules | Number of Ions |
|---|---|---|---|
| No Excipient | No Excipient | No Excipient | 1 Na+ |
| ARG | 10% w/w | 284 | 283 Cl− |
| ARG | 100 mM | 51 | 50 Cl− |
| ARG | 10 mM | 5 | 4 Cl− |
| TXA | 10% w/w | 314 | 1 Na+ |
| TXA | 100 mM | 51 | 1 Na+ |
| TXA | 10 mM | 5 | 1 Na+ |
| SUC | 10% w/w | 144 | 1 Na+ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, S.K.; Björkengren, G.; Berner, C.; Winter, G.; Harris, P.; Peters, G.H.J. Combining Molecular Dynamics Simulations and Biophysical Characterization to Investigate Protein-Specific Excipient Effects on Reteplase during Freeze Drying. Pharmaceutics 2023, 15, 1854. https://doi.org/10.3390/pharmaceutics15071854
Ko SK, Björkengren G, Berner C, Winter G, Harris P, Peters GHJ. Combining Molecular Dynamics Simulations and Biophysical Characterization to Investigate Protein-Specific Excipient Effects on Reteplase during Freeze Drying. Pharmaceutics. 2023; 15(7):1854. https://doi.org/10.3390/pharmaceutics15071854
Chicago/Turabian StyleKo, Suk Kyu, Gabriella Björkengren, Carolin Berner, Gerhard Winter, Pernille Harris, and Günther H. J. Peters. 2023. "Combining Molecular Dynamics Simulations and Biophysical Characterization to Investigate Protein-Specific Excipient Effects on Reteplase during Freeze Drying" Pharmaceutics 15, no. 7: 1854. https://doi.org/10.3390/pharmaceutics15071854