
Targeting Potential of Innate Lymphoid Cells in Melanoma and Other Cancers
 ,
,
Abstract
:1. Introduction
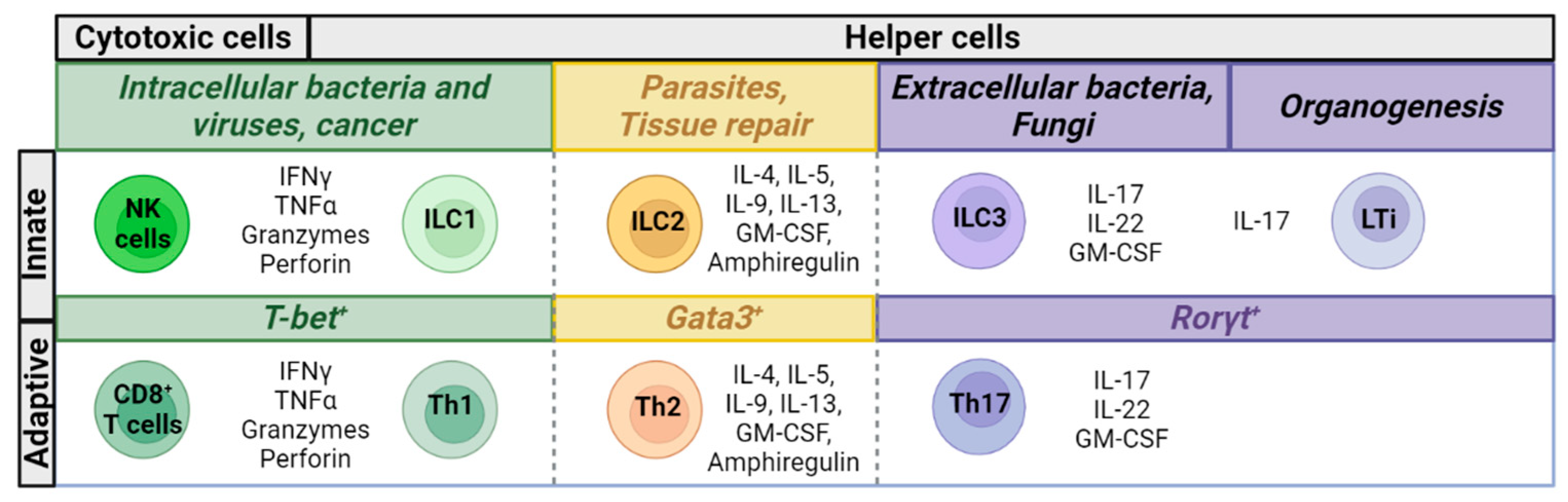
2. Characteristics of ILCs
3. ILCs and the Immune Response to Tumors
4. ILCs and Immune Checkpoint Molecules
4.1. PD-1 Expression Patterns

{kind=link}
{kind=link}
| Population | Mouse Expression | Human Expression | Function | References |
|---|---|---|---|---|
| NK cells | Expressed particularly on tumor infiltrating populations | Negative regulator | [28,100,101,102,103,104,105,106,107,108] | |
| ILC1s | Low level at steady state ILC1-subsets in tumors express intermediate levels | High levels in tumor infiltrating populations Expressed in PBMCs of cancer patients | Potential inhibitory role | [28,51,97] |
| ILC2s | Expressed in tumor infiltrating populations 20–40% of lung ILC2s, increases upon inflammation Substantially expressed in the colon | Significantly expressed in tumor-infiltrating populations Expressed in PBMCs of cancer patients | Regulates airway hypersensitivity Negative regulator of ILC2 function | [28,60,62,99,111,113,114,115,116,117,118,119] |
| ILC3s | Expressed in mouse lung, colon, decidual tissues Expressed substantially on LTi cells residing in the colon and in other gut tissues, upregulated upon activation | Expressed in the decidua Expressed in breast and GI tumors Low expression in PBMCs of cancer patients | Mediating immune tolerance during pregnancy Promoting metabolism and maintaining barrier function in the intestine Potential inhibitory role in cancer | [28,97,120,121] |
4.2. Immune Checkpoint Targeting Potential of ILCs
5. Novel ILC Targeting Approaches in Cancer
5.1. Targeting Cytokines and Their Receptors
5.2. Influencing ILC Plasticity and Transdifferentiation
5.3. ILC Differentiation, Expansion, and Engineering: Chimeric Antigen Receptors and TCRs
5.4. Bi- and Tri-Specific Vectors
5.5. Potential of ILC-Based Immunotherapies in Melanoma
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Den Boogaard, W.M.C.; Komninos, D.S.J.; Vermeij, W.P. Chemotherapy Side-Effects: Not All DNA Damage Is Equal. Cancers 2022, 14, 627. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D.; Tsang, D.S.; Tinkle, C.L.; Olch, A.J.; Kremer, L.C.; Ronckers, C.M.; Gibbs, I.C.; Constine, L.S. Late effects of radiation therapy in pediatric patients and survivorship. Pediatr. Blood Cancer 2021, 68, e28349. [Google Scholar] [CrossRef]
- Muster, T.; Subbarao, E.K.; Enami, M.; Murphy, B.R.; Palese, P. An influenza A virus containing influenza B virus 5′ and 3′ noncoding regions on the neuraminidase gene is attenuated in mice. Proc. Natl. Acad. Sci. USA 1991, 88, 5177–5181. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The Next Decade of Immune Checkpoint Therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef]
- Chocarro, L.; Bocanegra, A.; Blanco, E.; Fernández-Rubio, L.; Arasanz, H.; Echaide, M.; Garnica, M.; Ramos, P.; Piñeiro-Hermida, S.; Vera, R.; et al. Cutting-Edge: Preclinical and Clinical Development of the First Approved Lag-3 Inhibitor. Cells 2022, 11, 2351. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef]
- Huang, A.C.; Zappasodi, R. A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, R.J.; Weber, J.S. Immune-related toxicities of checkpoint inhibitors: Mechanisms and mitigation strategies. Nat. Rev. Drug Discov. 2021, 21, 495–508. [Google Scholar] [CrossRef]
- Sibaud, V. Dermatologic Reactions to Immune Checkpoint Inhibitors: Skin Toxicities and Immunotherapy. Am. J. Clin. Dermatol. 2018, 19, 345–361. [Google Scholar] [CrossRef]
- Wang, D.Y.; Mooradian, M.J.; Kim, D.; Shah, N.J.; Fenton, E.S.; Conry, R.M.; Mehta, R.; Silk, A.W.; Zhou, A.; Compton, M.L.; et al. Clinical characterization of colitis arising from anti-PD-1 based therapy. Oncoimmunology 2019, 8, e1524695. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, S.S.; Fradley, M.G.; Cohen, J.V.; Nohria, A.; Reynolds, K.L.; Heinzerling, L.M.; Sullivan, R.J.; Damrongwatanasuk, R.; Chen, C.L.; Gupta, D.; et al. Myocarditis in Patients Treated With Immune Checkpoint Inhibitors. J. Am. Coll. Cardiol. 2018, 71, 1755–1764. [Google Scholar] [CrossRef]
- Eberl, G.; Colonna, M.; Di Santo, J.P.; McKenzie, A.N.J. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, aaa6566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, C.S.N.; Artis, D. Innate lymphoid cells control signaling circuits to regulate tissue-specific immunity. Cell Res. 2020, 30, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M. Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity 2018, 48, 1104–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Bal, S.M.; Golebski, K.; Spits, H. Plasticity of innate lymphoid cell subsets. Nat. Rev. Immunol. 2020, 20, 552–565. [Google Scholar] [CrossRef]
- Kansler, E.R.; Li, M.O. Innate lymphocytes—Lineage, localization and timing of differentiation. Cell. Mol. Immunol. 2019, 16, 627–633. [Google Scholar] [CrossRef]
- Xiong, L.; Nutt, S.L.; Seillet, C. Innate lymphoid cells: More than just immune cells. Front. Immunol. 2022, 13, 1033904. [Google Scholar] [CrossRef] [PubMed]
- Vély, F.; Barlogis, V.; Vallentin, B.; Neven, B.; Piperoglou, C.; Ebbo, M.; Perchet, T.; Petit, M.; Yessaad, N.; Touzot, F.; et al. Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 2016, 17, 1291–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Zotto, G.; Vacca, P.; Moretta, L.; Quatrini, L. CPHEN-15: Comprehensive phenotyping of human peripheral blood helper-ILCs by flow cytometry. Cytom. Part A 2023, 103, 378–382. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, C.S.N.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef]
- Shannon, J.P.; Vrba, S.M.; Reynoso, G.V.; Wynne-Jones, E.; Kamenyeva, O.; Malo, C.S.; Cherry, C.R.; McManus, D.T.; Hickman, H.D. Group 1 innate lymphoid-cell-derived interferon-γ maintains anti-viral vigilance in the mucosal epithelium. Immunity 2021, 54, 276–290.e5. [Google Scholar] [CrossRef]
- Jacquelot, N.; Seillet, C.; Vivier, E.; Belz, G.T. Innate lymphoid cells and cancer. Nat. Immunol. 2022, 23, 371–379. [Google Scholar] [CrossRef]
- Salimi, M.; Wang, R.; Yao, X.; Li, X.; Wang, X.; Hu, Y.; Chang, X.; Fan, P.; Dong, T.; Ogg, G. Activated innate lymphoid cell populations accumulate in human tumour tissues. BMC Cancer 2018, 18, 341. [Google Scholar] [CrossRef]
- Bruchard, M.; Spits, H. The role of ILC subsets in cancer. Semin. Immunol. 2022, 61–64, 101654. [Google Scholar] [CrossRef]
- Chung, D.C.; Jacquelot, N.; Ghaedi, M.; Warner, K.; Ohashi, P.S. Innate Lymphoid Cells: Role in Immune Regulation and Cancer. Cancers 2022, 14, 2071. [Google Scholar] [CrossRef]
- Sivori, S.; Pende, D.; Quatrini, L.; Pietra, G.; Della Chiesa, M.; Vacca, P.; Tumino, N.; Moretta, F.; Mingari, M.C.; Locatelli, F.; et al. NK cells and ILCs in tumor immunotherapy. Mol. Asp. Med. 2020, 80, 100870. [Google Scholar] [CrossRef] [PubMed]
- Lopes, N.; Vivier, E.; Narni-Mancinelli, E. Natural killer cells and type 1 innate lymphoid cells in cancer. Semin. Immunol. 2023, 66, 101709. [Google Scholar] [CrossRef] [PubMed]
- Cursons, J.; Souza-Fonseca-Guimaraes, F.; Foroutan, M.; Anderson, A.; Hollande, F.; Hediyeh-Zadeh, S.; Behren, A.; Huntington, N.D.; Davis, M.J. A Gene Signature Predicting Natural Killer Cell Infiltration and Improved Survival in Melanoma Patients. Cancer Immunol. Res. 2019, 7, 1162–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messaoudene, M.; Fregni, G.; Fourmentraux-Neves, E.; Chanal, J.; Maubec, E.; Mazouz-Dorval, S.; Couturaud, B.; Girod, A.; Sastre-Garau, X.; Albert, S.; et al. Mature Cytotoxic CD56bright/CD16+ Natural Killer Cells Can Infiltrate Lymph Nodes Adjacent to Metastatic Melanoma. Cancer Res. 2014, 74, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquelot, N.; Roberti, M.P.; Enot, D.P.; Rusakiewicz, S.; Semeraro, M.; Jégou, S.; Flores, C.; Chen, L.; Kwon, B.S.; Borg, C.; et al. Immunophenotyping of Stage III Melanoma Reveals Parameters Associated with Patient Prognosis. J. Investig. Dermatol. 2016, 136, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Messaoudene, M.; Périer, A.; Fregni, G.; Neves, E.; Zitvogel, L.; Cremer, I.; Chanal, J.; Sastre-Garau, X.; Deschamps, L.; Marinho, E.; et al. Characterization of the Microenvironment in Positive and Negative Sentinel Lymph Nodes from Melanoma Patients. PLoS ONE 2015, 10, e0133363. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Liu, L.; Huang, Q.; Liu, H.; Huang, M.; Wang, J.; Wen, H.; Lin, R.; Qu, K.; Li, K.; et al. Accumulation of Tumor-Infiltrating CD49a+ NK Cells Correlates with Poor Prognosis for Human Hepatocellular Carcinoma. Cancer Immunol. Res. 2019, 7, 1535–1546. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, Y.; Tang, B.; Zhao, Q.; Wang, D.; Liu, Y.; Guo, M.; Zhao, S.; Qi, Y.; Zhang, Y.; et al. IL-6-induced CD39 expression on tumor-infiltrating NK cells predicts poor prognosis in esophageal squamous cell carcinoma. Cancer Immunol. Immunother. 2020, 69, 2371–2380. [Google Scholar] [CrossRef]
- Duault, C.; Kumar, A.; Khani, A.T.; Lee, S.J.; Yang, L.; Huang, M.; Hurtz, C.; Manning, B.; Ghoda, L.Y.; McDonald, T.; et al. Activated natural killer cells predict poor clinical prognosis in high-risk B- and T-cell acute lymphoblastic leukemia. Blood 2021, 138, 1465–1480. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Q.; Yang, J.; Li, X.; Xian, L.; Li, W.; Lin, T.; Cheng, J.; Lin, Q.; Xu, X.; et al. Increased TIGIT expressing NK cells with dysfunctional phenotype in AML patients correlated with poor prognosis. Cancer Immunol. Immunother. 2022, 71, 277–287. [Google Scholar] [CrossRef]
- Crome, S.; Nguyen, L.T.; Lopez-Verges, S.; Yang, S.Y.C.; Martin, B.; Yam, J.Y.; Johnson, D.J.; Nie, J.; Pniak, M.; Yen, P.H.; et al. A distinct innate lymphoid cell population regulates tumor-associated T cells. Nat. Med. 2017, 23, 368–375. [Google Scholar] [CrossRef]
- Neo, S.Y.; Yang, Y.; Record, J.; Ma, R.; Chen, X.; Chen, Z.; Tobin, N.P.; Blake, E.; Seitz, C.; Thomas, R.; et al. CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J. Clin. Investig. 2020, 130, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- Kansler, E.R.; Dadi, S.; Krishna, C.; Nixon, B.G.; Stamatiades, E.G.; Liu, M.; Kuo, F.; Zhang, J.; Zhang, X.; Capistrano, K.; et al. Cytotoxic innate lymphoid cells sense cancer cell-expressed interleukin-15 to suppress human and murine malignancies. Nat. Immunol. 2022, 23, 904–915. [Google Scholar] [CrossRef]
- Dadi, S.; Chhangawala, S.; Whitlock, B.M.; Franklin, R.A.; Luo, C.T.; Oh, S.A.; Toure, A.; Pritykin, Y.; Huse, M.; Leslie, C.S.; et al. Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell 2016, 164, 365–377. [Google Scholar] [CrossRef] [Green Version]
- Nixon, B.G.; Chou, C.; Krishna, C.; Dadi, S.; Michel, A.O.; Cornish, A.E.; Kansler, E.R.; Do, M.H.; Wang, X.; Capistrano, K.J.; et al. Cytotoxic granzyme C–expressing ILC1s contribute to antitumor immunity and neonatal autoimmunity. Sci. Immunol. 2022, 7, eabi8642. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ma, R.; Ma, S.; Tian, L.; Lu, T.; Zhang, J.; Mundy-Bosse, B.L.; Zhang, B.; Marcucci, G.; Caligiuri, M.A.; et al. ILC1s control leukemia stem cell fate and limit development of AML. Nat. Immunol. 2022, 23, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- He, Y.-F.; Wang, X.-H.; Zhang, G.-M.; Chen, H.-T.; Zhang, H.; Feng, Z.-H. Sustained low-level expression of interferon-γ promotes tumor development: Potential insights in tumor prevention and tumor immunotherapy. Cancer Immunol. Immunother. 2005, 54, 891–897. [Google Scholar] [CrossRef]
- Song, M.; Ping, Y.; Zhang, K.; Yang, L.; Li, F.; Zhang, C.; Cheng, S.; Yue, D.; Maimela, N.R.; Qu, J.; et al. Low-Dose IFNγ Induces Tumor Cell Stemness in Tumor Microenvironment of Non–Small Cell Lung Cancer. Cancer Res. 2019, 79, 3737–3748. [Google Scholar] [CrossRef]
- Ercolano, G.; Garcia-Garijo, A.; Salomé, B.; Gomez-Cadena, A.; Vanoni, G.; Mastelic-Gavillet, B.; Ianaro, A.; Speiser, D.E.; Romero, P.; Trabanelli, S.; et al. Immunosuppressive Mediators Impair Proinflammatory Innate Lymphoid Cell Function in Human Malignant Melanoma. Cancer Immunol. Res. 2020, 8, 556–564. [Google Scholar] [CrossRef]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.S.; Ulland, T.K.; Cervantes-Barragan, L.; Bando, J.K.; Robinette, M.L.; Wang, Q.; White, A.J.; Gilfillan, S.; Cella, M.; Colonna, M. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat. Immunol. 2017, 18, 995–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabanelli, S.; Chevalier, M.F.; Martinez-Usatorre, A.; Gomez-Cadena, A.; Salomé, B.; Lecciso, M.; Salvestrini, V.; Verdeil, G.; Racle, J.; Papayannidis, C.; et al. Tumour-derived PGD2 and NKp30-B7H6 engagement drives an immunosuppressive ILC2-MDSC axis. Nat. Commun. 2017, 8, 593. [Google Scholar] [CrossRef] [Green Version]
- Jou, E.; Rodriguez-Rodriguez, N.; Ferreira, A.-C.F.; Jolin, H.E.; Clark, P.A.; Sawmynaden, K.; Ko, M.; Murphy, J.E.; Mannion, J.; Ward, C.; et al. An innate IL-25–ILC2–MDSC axis creates a cancer-permissive microenvironment for Apc mutation–driven intestinal tumorigenesis. Sci. Immunol. 2022, 7, eabn0175. [Google Scholar] [CrossRef]
- Chevalier, M.F.; Trabanelli, S.; Racle, J.; Salomé, B.; Cesson, V.; Gharbi, D.; Bohner, P.; Domingos-Pereira, S.; Dartiguenave, F.; Fritschi, A.-S.; et al. ILC2-modulated T cell–to-MDSC balance is associated with bladder cancer recurrence. J. Clin. Investig. 2017, 127, 2916–2929. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Jin, K.; Liao, Z.; Xiao, Z.; Xu, H.; Lin, X.; Li, H.; Li, T.; Zhang, W.; Han, X.; et al. Hypoxia-reprogrammed regulatory group 2 innate lymphoid cells promote immunosuppression in pancreatic cancer. Ebiomedicine 2022, 79, 104016. [Google Scholar] [CrossRef] [PubMed]
- Ricardo-Gonzalez, R.R.; Van Dyken, S.J.; Schneider, C.; Lee, J.; Nussbaum, J.C.; Liang, H.-E.; Vaka, D.; Eckalbar, W.L.; Molofsky, A.B.; Erle, D.J.; et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol. 2018, 19, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Long, A.; Dominguez, D.; Qin, L.; Chen, S.; Fan, J.; Zhang, M.; Fang, D.; Zhang, Y.; Kuzel, T.M.; Zhang, B. Type 2 Innate Lymphoid Cells Impede IL-33–Mediated Tumor Suppression. J. Immunol. 2018, 201, 3456–3464. [Google Scholar] [CrossRef] [Green Version]
- Schuijs, M.J.; Png, S.; Richard, A.C.; Tsyben, A.; Hamm, G.; Stockis, J.; Garcia, C.; Pinaud, S.; Nicholls, A.; Ros, X.R.; et al. ILC2-driven innate immune checkpoint mechanism antagonizes NK cell antimetastatic function in the lung. Nat. Immunol. 2020, 21, 998–1009. [Google Scholar] [CrossRef]
- Jacquelot, N.; Seillet, C.; Wang, M.; Pizzolla, A.; Liao, Y.; Hediyeh-Zadeh, S.; Grisaru-Tal, S.; Louis, C.; Huang, Q.; Schreuder, J.; et al. Blockade of the co-inhibitory molecule PD-1 unleashes ILC2-dependent antitumor immunity in melanoma. Nat. Immunol. 2021, 22, 851–864. [Google Scholar] [CrossRef]
- Wagner, M.; Ealey, K.N.; Tetsu, H.; Kiniwa, T.; Motomura, Y.; Moro, K.; Koyasu, S. Tumor-Derived Lactic Acid Contributes to the Paucity of Intratumoral ILC2s. Cell Rep. 2020, 30, 2743–2757.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moral, J.A.; Leung, J.; Rojas, L.A.; Ruan, J.; Zhao, J.; Sethna, Z.; Ramnarain, A.; Gasmi, B.; Gururajan, M.; Redmond, D.; et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 2020, 579, 130–135. [Google Scholar] [CrossRef]
- Huang, Q.; Jacquelot, N.; Preaudet, A.; Hediyeh-Zadeh, S.; Souza-Fonseca-Guimaraes, F.; McKenzie, A.N.J.; Hansbro, P.M.; Davis, M.J.; Mielke, L.A.; Putoczki, T.L.; et al. Type 2 Innate Lymphoid Cells Protect against Colorectal Cancer Progression and Predict Improved Patient Survival. Cancers 2021, 13, 559. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Wu, Y.; Huang, L.; Tian, Y.; Ji, X.; Abdelaziz, M.H.; Cai, W.; Dineshkumar, K.; Lei, Y.; Yao, S.; et al. ILC2-derived IL-9 inhibits colorectal cancer progression by activating CD8+ T cells. Cancer Lett. 2021, 502, 34–43. [Google Scholar] [CrossRef]
- JSiegler, J.-J.; Correia, M.P.; Hofman, T.; Prager, I.; Birgin, E.; Rahbari, N.N.; Watzl, C.; Stojanovic, A.; Cerwenka, A. Human ILC3 Exert TRAIL-Mediated Cytotoxicity Towards Cancer Cells. Front. Immunol. 2022, 13, 742571. [Google Scholar] [CrossRef]
- Nussbaum, K.; Burkhard, S.H.; Ohs, I.; Mair, F.; Klose, C.S.; Arnold, S.J.; Diefenbach, A.; Tugues, S.; Becher, B. Tissue microenvironment dictates the fate and tumor-suppressive function of type 3 ILCs. J. Exp. Med. 2017, 214, 2331–2347. [Google Scholar] [CrossRef] [PubMed]
- Bruchard, M.; Geindreau, M.; Perrichet, A.; Truntzer, C.; Ballot, E.; Boidot, R.; Racoeur, C.; Barsac, E.; Chalmin, F.; Hibos, C.; et al. Recruitment and activation of type 3 innate lymphoid cells promote antitumor immune responses. Nat. Immunol. 2022, 23, 262–274. [Google Scholar] [CrossRef]
- Goc, J.; Lv, M.; Bessman, N.J.; Flamar, A.-L.; Sahota, S.; Suzuki, H.; Teng, F.; Putzel, G.G.; Eberl, G.; Withers, D.R.; et al. Dysregulation of ILC3s unleashes progression and immunotherapy resistance in colon cancer. Cell 2021, 184, 5015–5030.e16. [Google Scholar] [CrossRef]
- Huang, Q.; Cao, W.; Mielke, L.; Seillet, C.; Belz, G.T.; Jacquelot, N. Innate Lymphoid Cells in Colorectal Cancers: A Double-Edged Sword. Front. Immunol. 2020, 10, 3080. [Google Scholar] [CrossRef] [Green Version]
- Huber, S.; Gagliani, N.; Zenewicz, L.A.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor, W.; Murphy, A.J.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Roediger, B.; Kyle, R.; Yip, K.H.; Sumaria, N.; Guy, T.V.; Kim, B.S.; Mitchell, A.J.; Tay, S.S.; Jain, R.; Forbes-Blom, E.; et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 564–573. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Koyasu, S. Innate Lymphoid Cells in Skin Homeostasis and Malignancy. Front. Immunol. 2021, 12, 758522. [Google Scholar] [CrossRef] [PubMed]
- Jacquelot, N.; Ghaedi, M.; Warner, K.; Chung, D.C.; Crome, S.Q.; Ohashi, P.S. Immune Checkpoints and Innate Lymphoid Cells—New Avenues for Cancer Immunotherapy. Cancers 2021, 13, 5967. [Google Scholar] [CrossRef]
- Pesce, S.; Trabanelli, S.; Di Vito, C.; Greppi, M.; Obino, V.; Guolo, F.; Minetto, P.; Bozzo, M.; Calvi, M.; Zaghi, E.; et al. Cancer Immunotherapy by Blocking Immune Checkpoints on Innate Lymphocytes. Cancers 2020, 12, 3504. [Google Scholar] [CrossRef]
- Garofalo, C.; Cerantonio, A.; Muscoli, C.; Mollace, V.; Viglietto, G.; De Marco, C.; Cristiani, C.M. Helper Innate Lymphoid Cells—Unappreciated Players in Melanoma Therapy. Cancers 2023, 15, 933. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, F.R.; Quatrini, L.; Munari, E.; Vacca, P.; Moretta, L. Innate Lymphoid Cells: Expression of PD-1 and Other Checkpoints in Normal and Pathological Conditions. Front. Immunol. 2019, 10, 910. [Google Scholar] [CrossRef] [PubMed]
- Cameron, F.; Whiteside, G.; Perry, C. Ipilimumab: First Global Approval. Drugs 2011, 71, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; Liu, J.; Subudhi, S.K.; Poon, C.; Gant, K.L.; et al. Immune checkpoint therapy—Current perspectives and future directions. Cell 2023, 186, 1652–1669. [Google Scholar] [CrossRef]
- Keam, S.J. Tremelimumab: First Approval. Drugs 2022, 83, 93–102. [Google Scholar] [CrossRef]
- Raedler, L.A. Keytruda (Pembrolizumab): First PD-1 Inhibitor Approved for Previously Treated Unresectable or Metastatic Melanoma. Am. Health Drug Benefits 2015, 8, 96–100. [Google Scholar]
- Pai-Scherf, L.; Blumenthal, G.M.; Li, H.; Subramaniam, S.; Mishra-Kalyani, P.S.; He, K.; Zhao, H.; Yu, J.; Paciga, M.; Goldberg, K.B.; et al. FDA Approval Summary: Pembrolizumab for Treatment of Metastatic Non-Small Cell Lung Cancer: First-Line Therapy and Beyond. Oncologist 2017, 22, 1392–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Raedler, L. Opdivo (Nivolumab): Second PD-1 Inhibitor Receives FDA Approval for Unresectable or Metastatic Melanoma. Am. Health Drug Benefits 2015, 8, 180–183. [Google Scholar] [PubMed]
- Xu, J.X.; Maher, V.E.; Zhang, L.; Tang, S.; Sridhara, R.; Ibrahim, A.; Kim, G.; Pazdur, R. FDA Approval Summary: Nivolumab in Advanced Renal Cell Carcinoma After Anti-Angiogenic Therapy and Exploratory Predictive Biomarker Analysis. Oncologist 2017, 22, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Kazandjian, D.; Suzman, D.L.; Blumenthal, G.; Mushti, S.; He, K.; Libeg, M.; Keegan, P.; Pazdur, R. FDA Approval Summary: Nivolumab for the Treatment of Metastatic Non-Small Cell Lung Cancer With Progression On or After Platinum-Based Chemotherapy. Oncologist 2016, 21, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasamon, Y.L.; de Claro, R.A.; Wang, Y.; Shen, Y.L.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Nivolumab for the Treatment of Relapsed or Progressive Classical Hodgkin Lymphoma. Oncologist 2017, 22, 585–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, A.; Duggan, S. Cemiplimab: First Global Approval. Drugs 2018, 78, 1841–1846. [Google Scholar] [CrossRef]
- Davis, C.M.; Lewis, K.D. Brief overview: Cemiplimab for the treatment of advanced basal cell carcinoma: PD-1 strikes again. Ther. Adv. Med Oncol. 2022, 14, 175883592110661. [Google Scholar] [CrossRef]
- Gogishvili, M.; Melkadze, T.; Makharadze, T.; Giorgadze, D.; Dvorkin, M.; Penkov, K.; Laktionov, K.; Nemsadze, G.; Nechaeva, M.; Rozhkova, I.; et al. Cemiplimab plus chemotherapy versus chemotherapy alone in non-small cell lung cancer: A randomized, controlled, double-blind phase 3 trial. Nat. Med. 2022, 28, 2374–2380. [Google Scholar] [CrossRef]
- Costa, B.; Vale, N. Dostarlimab: A Review. Biomolecules 2022, 12, 1031. [Google Scholar] [CrossRef]
- Ning, Y.-M.; Suzman, D.; Maher, V.E.; Zhang, L.; Tang, S.; Ricks, T.; Palmby, T.; Fu, W.; Liu, Q.; Goldberg, K.B.; et al. FDA Approval Summary: Atezolizumab for the Treatment of Patients with Progressive Advanced Urothelial Carcinoma after Platinum-Containing Chemotherapy. Oncologist 2017, 22, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Weinstock, C.; Khozin, S.; Suzman, D.; Zhang, L.; Tang, S.; Wahby, S.; Goldberg, K.B.; Kim, G.; Pazdur, R.U.S. Food and Drug Administration Approval Summary: Atezolizumab for Metastatic Non–Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 4534–4539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyck, J.E.T.; Kahlon, N.; Masih, S.; Hamouda, D.M.; Petros, F.G. Clinical Evaluation of Avelumab in the Treatment of Advanced Urothelial Carcinoma: Focus on Patient Selection and Outcomes. Cancer Manag. Res. 2022, 14, 729–738. [Google Scholar] [CrossRef]
- Kim, E.S. Avelumab: First Global Approval. Drugs 2017, 77, 929–937. [Google Scholar] [CrossRef]
- Syed, Y.Y. Durvalumab: First Global Approval. Drugs 2017, 77, 1369–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA approves anti-LAG3 checkpoint. Nat. Biotechnol. 2022, 40, 625. [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Yu, Y.; Tsang, J.C.H.; Wang, C.; Clare, S.; Wang, J.; Chen, X.; Brandt, C.; Kane, L.; Campos, L.S.; Lu, L.; et al. Single-cell RNA-seq identifies a PD-1hi ILC progenitor and defines its development pathway. Nature 2016, 539, 102–106. [Google Scholar] [CrossRef]
- Seillet, C.; Mielke, L.A.; Amann-Zalcenstein, D.B.; Su, S.; Gao, J.; Almeida, F.F.; Shi, W.; Ritchie, M.E.; Naik, S.H.; Huntington, N.D.; et al. Deciphering the Innate Lymphoid Cell Transcriptional Program. Cell Rep. 2016, 17, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.; Huang, Y.; Mallett, G.; Stathopoulou, C.; Felizardo, T.C.; Sun, M.-A.; Martin, E.L.; Zhu, N.; Woodward, E.L.; Elias, M.S.; et al. PD-1 regulates KLRG1+ group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1663–1678. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.-C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346.e3. [Google Scholar] [CrossRef] [Green Version]
- Davis, Z.; Felices, M.; Lenvik, T.; Badal, S.; Walker, J.T.; Hinderlie, P.; Riley, J.L.; Vallera, D.A.; Blazar, B.R.; Miller, J.S. Low-density PD-1 expression on resting human natural killer cells is functional and upregulated after transplantation. Blood Adv. 2021, 5, 1069–1080. [Google Scholar] [CrossRef]
- Vari, F.; Arpon, D.; Keane, C.; Hertzberg, M.S.; Talaulikar, D.; Jain, S.; Cui, Q.; Han, E.; Tobin, J.; Bird, R.; et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood 2018, 131, 1809–1819. [Google Scholar] [CrossRef] [Green Version]
- Beldi-Ferchiou, A.; Lambert, M.; Dogniaux, S.; Vély, F.; Vivier, E.; Olive, D.; Dupuy, S.; Levasseur, F.; Zucman, D.; Lebbé, C.; et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 2016, 7, 72961–72977. [Google Scholar] [CrossRef] [Green Version]
- Niu, C.; Li, M.; Zhu, S.; Chen, Y.; Zhou, L.; Xu, D.; Xu, J.; Li, Z.; Li, W.; Cui, J. PD-1-positive Natural Killer Cells have a weaker antitumor function than that of PD-1-negative Natural Killer Cells in Lung Cancer. Int. J. Med. Sci. 2020, 17, 1964–1973. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017, 36, 6143–6153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumino, N.; Martini, S.; Munari, E.; Scordamaglia, F.; Besi, F.; Mariotti, F.R.; Bogina, G.; Mingari, M.C.; Vacca, P.; Moretta, L. Presence of innate lymphoid cells in pleural effusions of primary and metastatic tumors: Functional analysis and expression of PD-1 receptor. Int. J. Cancer 2019, 145, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Di, G.; Bian, M. Dysfunction of natural killer cells mediated by PD-1 and Tim-3 pathway in anaplastic thyroid cancer. Int. Immunopharmacol. 2018, 64, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Judge, S.J.; Dunai, C.; Aguilar, E.G.; Vick, S.C.; Sturgill, I.R.; Khuat, L.T.; Stoffel, K.M.; Van Dyke, J.; Longo, D.L.; Darrow, M.A.; et al. Minimal PD-1 expression in mouse and human NK cells under diverse conditions. J. Clin. Investig. 2020, 130, 3051–3068. [Google Scholar] [CrossRef] [PubMed]
- Hasim, M.S.; Marotel, M.; Hodgins, J.J.; Vulpis, E.; Makinson, O.J.; Asif, S.; Shih, H.-Y.; Scheer, A.K.; MacMillan, O.; Alonso, F.G.; et al. When killers become thieves: Trogocytosed PD-1 inhibits NK cells in cancer. Sci. Adv. 2022, 8, eabj3286. [Google Scholar] [CrossRef]
- Helou, D.G.; Shafiei-Jahani, P.; Lo, R.; Howard, E.; Hurrell, B.P.; Galle-Treger, L.; Painter, J.D.; Lewis, G.; Soroosh, P.; Sharpe, A.H.; et al. PD-1 pathway regulates ILC2 metabolism and PD-1 agonist treatment ameliorates airway hyperreactivity. Nat. Commun. 2020, 11, 3998. [Google Scholar] [CrossRef] [PubMed]
- Falquet, M.; Ercolano, G.; Jandus, P.; Jandus, C.; Trabanelli, S. Healthy and Patient Type 2 Innate Lymphoid Cells are Differently Affected by in vitro Culture Conditions. J. Asthma Allergy 2021, 14, 773–783. [Google Scholar] [CrossRef]
- Oldenhove, G.; Boucquey, E.; Taquin, A.; Acolty, V.; Bonetti, L.; Ryffel, B.; Le Bert, M.; Englebert, K.; Boon, L.; Moser, M. PD-1 Is Involved in the Dysregulation of Type 2 Innate Lymphoid Cells in a Murine Model of Obesity. Cell Rep. 2018, 25, 2053–2060.e4. [Google Scholar] [CrossRef] [Green Version]
- Akama, Y.; Park, E.J.; Satoh-Takayama, N.; Gaowa, A.; Ito, A.; Kawamoto, E.; Darkwah, S.; Appiah, M.G.; Myint, P.K.; Ohno, H.; et al. Sepsis Induces Deregulation of IL-13 Production and PD-1 Expression in Lung Group 2 Innate Lymphoid Cells. Shock 2021, 55, 357–370. [Google Scholar] [CrossRef]
- Maggi, E.; Veneziani, I.; Moretta, L.; Cosmi, L.; Annunziato, F. Group 2 Innate Lymphoid Cells: A Double-Edged Sword in Cancer? Cancers 2020, 12, 3452. [Google Scholar] [CrossRef] [PubMed]
- Kiniwa, T.; Moro, K. Localization and site-specific cell–cell interactions of group 2 innate lymphoid cells. Int. Immunol. 2021, 33, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qu, Y.; Xia, P.; Chen, Y.; Zhu, X.; Zhang, J.; Wang, G.; Tian, Y.; Ying, J.; Fan, Z. Transdifferentiation of tumor infiltrating innate lymphoid cells during progression of colorectal cancer. Cell Res. 2020, 30, 610–622. [Google Scholar] [CrossRef]
- Heinrich, B.; Gertz, E.M.; Schäffer, A.A.; Craig, A.; Ruf, B.; Subramanyam, V.; McVey, J.C.; Diggs, L.P.; Heinrich, S.; Rosato, U.; et al. The tumour microenvironment shapes innate lymphoid cells in patients with hepatocellular carcinoma. Gut 2021, 71, 1161–1175. [Google Scholar] [CrossRef]
- Shen, C.; Liu, C.; Zhang, Z.; Ping, Y.; Shao, J.; Tian, Y.; Yu, W.; Qin, G.; Liu, S.; Wang, L.; et al. PD-1 Affects the Immunosuppressive Function of Group 2 Innate Lymphoid Cells in Human Non-Small Cell Lung Cancer. Front. Immunol. 2021, 12, 680055. [Google Scholar] [CrossRef]
- Di Wu, D.; Hu, L.; Han, M.; Deng, Y.; Zhang, Y.; Ren, G.; Zhao, X.; Li, Z.; Li, P.; Zhang, Y.; et al. PD-1 signaling facilitates activation of lymphoid tissue inducer cells by restraining fatty acid oxidation. Nat. Metab. 2022, 4, 867–882. [Google Scholar] [CrossRef]
- Vacca, P.; Pesce, S.; Greppi, M.; Fulcheri, E.; Munari, E.; Olive, D.; Mingari, M.C.; Moretta, A.; Moretta, L.; Marcenaro, E. PD-1 is expressed by and regulates human group 3 innate lymphoid cells in human decidua. Mucosal Immunol. 2019, 12, 624–631. [Google Scholar] [CrossRef]
- Dębska-Zielkowska, J.; Moszkowska, G.; Zieliński, M.; Zielińska, H.; Dukat-Mazurek, A.; Trzonkowski, P.; Stefańska, K. KIR Receptors as Key Regulators of NK Cells Activity in Health and Disease. Cells 2021, 10, 1777. [Google Scholar] [CrossRef] [PubMed]
- Kärre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H–2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678. [Google Scholar] [CrossRef] [PubMed]
- SSivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Sternberg-Simon, M.; Brodin, P.; Pickman, Y.; Önfelt, B.; Kärre, K.; Malmberg, K.-J.; Höglund, P.; Mehr, R. Natural Killer Cell Inhibitory Receptor Expression in Humans and Mice: A Closer Look. Front. Immunol. 2013, 4, 65. [Google Scholar] [CrossRef] [Green Version]
- Anfossi, N.; André, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK Cell Education by Inhibitory Receptors for MHC Class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Yawata, M.; Yawata, N.; Draghi, M.; Partheniou, F.; Little, A.-M.; Parham, P. MHC class I–specific inhibitory receptors and their ligands structure diverse human NK-cell repertoires toward a balance of missing self-response. Blood 2008, 112, 2369–2380. [Google Scholar] [CrossRef] [Green Version]
- Fauriat, C.; Ivarsson, M.A.; Ljunggren, H.-G.; Malmberg, K.-J.; Michaëlsson, J. Education of human natural killer cells by activating killer cell immunoglobulin-like receptors. Blood 2010, 115, 1166–1174. [Google Scholar] [CrossRef] [Green Version]
- Höglund, P.; Brodin, P. Current perspectives of natural killer cell education by MHC class I molecules. Nat. Rev. Immunol. 2010, 10, 724–734. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, J.; Chen, S.; Yang, H.; Dong, Z. Synergized regulation of NK cell education by NKG2A and specific Ly49 family members. Nat. Commun. 2019, 10, 5010. [Google Scholar] [CrossRef] [Green Version]
- Vahlne, G.; Lindholm, K.; Meier, A.; Wickström, S.; Lakshmikanth, T.; Brennan, F.; Wilken, M.; Nielsen, R.; Romagné, F.; Wagtmann, N.R.; et al. In vivo tumor cell rejection induced by NK cell inhibitory receptor blockade: Maintained tolerance to normal cells even in the presence of IL-2. Eur. J. Immunol. 2010, 40, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Sola, C.; André, P.; Lemmers, C.; Fuseri, N.; Bonnafous, C.; Bléry, M.; Wagtmann, N.R.; Romagné, F.; Vivier, E.; Ugolini, S. Genetic and antibody-mediated reprogramming of natural killer cell missing-self recognition in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 12879–12884. [Google Scholar] [CrossRef]
- Romagné, F.; André, P.; Spee, P.; Zahn, S.; Anfossi, N.; Gauthier, L.; Capanni, M.; Ruggeri, L.; Benson, D.M., Jr.; Blaser, B.W.; et al. Preclinical characterization of 1-7F9, a novel human anti–KIR receptor therapeutic antibody that augments natural killer–mediated killing of tumor cells. Blood 2009, 114, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M.; Bakan, C.E.; Zhang, S.; Collins, S.M.; Liang, J.; Srivastava, S.; Hofmeister, C.C.; Efebera, Y.; Andre, P.; Romagne, F.; et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood 2011, 118, 6387–6391. [Google Scholar] [CrossRef] [Green Version]
- Carlsten, M.; Korde, N.; Kotecha, R.; Reger, R.; Bor, S.; Kazandjian, D.; Landgren, O.; Childs, R.W. Checkpoint Inhibition of KIR2D with the Monoclonal Antibody IPH2101 Induces Contraction and Hyporesponsiveness of NK Cells in Patients with Myeloma. Clin. Cancer Res. 2016, 22, 5211–5222. [Google Scholar] [CrossRef] [PubMed]
- NVey, N.; Karlin, L.; Sadot-Lebouvier, S.; Broussais, F.; Berton-Rigaud, D.; Rey, J.; Charbonnier, A.; Marie, D.; André, P.; Paturel, C.; et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D.; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 2018, 9, 17675–17688. [Google Scholar] [CrossRef] [Green Version]
- Yalniz, F.F.; Daver, N.; Rezvani, K.; Kornblau, S.; Ohanian, M.; Borthakur, G.; DiNardo, C.D.; Konopleva, M.; Burger, J.; Gasior, Y.; et al. A Pilot Trial of Lirilumab With or Without Azacitidine for Patients With Myelodysplastic Syndrome. Clin. Lymphoma Myeloma Leuk. 2018, 18, 658–663.e2. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Lesokhin, A.; Borrello, I.; Timmerman, J.; Gutierrez, M.; Zhu, L.; McKiver, M.P.; Ansell, S.M. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leukemia 2021, 35, 777–786. [Google Scholar] [CrossRef]
- Liu, H.; Zhou, S.; Liu, J.; Chen, F.; Zhang, Y.; Liu, M.; Min, S.; Wang, H.; Wang, X.; Wu, N. Lirilumab and Avelumab Enhance Anti-HPV+ Cervical Cancer Activity of Natural Killer Cells via Vav1-Dependent NF-κB Disinhibition. Front. Oncol. 2022, 12, 747482. [Google Scholar] [CrossRef]
- Hanna, G.J.; O’Neill, A.; Shin, K.-Y.; Wong, K.; Jo, V.Y.; Quinn, C.T.; Cutler, J.M.; Flynn, M.; Lizotte, P.H.; Annino, J.D.J.; et al. Neoadjuvant and Adjuvant Nivolumab and Lirilumab in Patients with Recurrent, Resectable Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2022, 28, 468–478. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [Green Version]
- Colevas, D.; Misiukiewicz, K.; Pearson, A.; Fayette, J.; Bauman, J.; Cupissol, D.; Saada-Bouzid, E.; Adkins, D.; Marie, D.; Cornen, S.; et al. 123MO Monalizumab, cetuximab and durvalumab in first-line treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (R/M SCCHN): A phase II trial. Ann. Oncol. 2021, 32, S1432. [Google Scholar] [CrossRef]
- Segal, N.H.; Naidoo, J.; Curigliano, G.; Patel, S.; Sahebjam, S.; Papadopoulos, K.P.; Gordon, M.S.; Wang, D.; Rueda, A.G.; Song, X.; et al. First-in-human dose escalation of monalizumab plus durvalumab, with expansion in patients with metastatic microsatellite-stable colorectal cancer. J. Clin. Oncol. 2018, 36, 3540. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Majem, M.; Barlesi, F.; Carcereny, E.; Chu, Q.; Monnet, I.; Sanchez-Hernandez, A.; Dakhil, S.; Camidge, D.R.; Winzer, L.; et al. COAST: An Open-Label, Phase II, Multidrug Platform Study of Durvalumab Alone or in Combination With Oleclumab or Monalizumab in Patients With Unresectable, Stage III Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Barlesi, F.; Goldberg, S.; Mann, H.; Gopinathan, A.; Newton, M.; Aggarwal, C. P1.10-01 Phase 3 Study of Durvalumab Combined with Oleclumab or Monalizumab in Patients with Unresectable Stage III NSCLC (PACIFIC-9). J. Thorac. Oncol. 2022, 17, S107. [Google Scholar] [CrossRef]
- Patel, P.; Alrifai, D.; McDonald, F.; Forster, M.; AstraZeneca UK Limited. Beyond chemoradiotherapy: Improving treatment outcomes for patients with stage III unresectable non-small-cell lung cancer through immuno-oncology and durvalumab (Imfinzi®▼, AstraZeneca UK Limited). Br. J. Cancer 2020, 123, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Juliá, E.P.; Amante, A.; Pampena, M.B.; Mordoh, J.; Levy, E.M. Avelumab, an IgG1 anti-PD-L1 Immune Checkpoint Inhibitor, Triggers NK Cell-Mediated Cytotoxicity and Cytokine Production Against Triple Negative Breast Cancer Cells. Front. Immunol. 2018, 9, 2140. [Google Scholar] [CrossRef]
- Dong, W.; Wu, X.; Ma, S.; Wang, Y.; Nalin, A.P.; Zhu, Z.; Zhang, J.; Benson, D.M.; He, K.; Caligiuri, M.A.; et al. The Mechanism of Anti–PD-L1 Antibody Efficacy against PD-L1–Negative Tumors Identifies NK Cells Expressing PD-L1 as a Cytolytic Effector. Cancer Discov. 2019, 9, 1422–1437. [Google Scholar] [CrossRef]
- Cristiani, C.M.; Capone, M.; Garofalo, C.; Madonna, G.; Mallardo, D.; Tuffanelli, M.; Vanella, V.; Greco, M.; Foti, D.P.; Viglietto, G.; et al. Altered Frequencies and Functions of Innate Lymphoid Cells in Melanoma Patients Are Modulated by Immune Checkpoints Inhibitors. Front. Immunol. 2022, 13, 811131. [Google Scholar] [CrossRef]
- Howard, E.; Hurrell, B.P.; Helou, D.G.; Quach, C.; Painter, J.D.; Shafiei-Jahani, P.; Fung, M.; Gill, P.S.; Soroosh, P.; Sharpe, A.H.; et al. PD-1 Blockade on Tumor Microenvironment-Resident ILC2s Promotes TNF-α Production and Restricts Progression of Metastatic Melanoma. Front. Immunol. 2021, 12, 733136. [Google Scholar] [CrossRef]
- Okuyama, Y.; Okajima, A.; Sakamoto, N.; Hashimoto, A.; Tanabe, R.; Kawajiri, A.; Kawabe, T.; Ishii, N. IL-33-ILC2 axis promotes anti-tumor CD8+ T cell responses via OX40 signaling. Biochem. Biophys. Res. Commun. 2022, 637, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Andreone, S.; Gambardella, A.R.; Mancini, J.; Loffredo, S.; Marcella, S.; La Sorsa, V.; Varricchi, G.; Schiavoni, G.; Mattei, F. Anti-Tumorigenic Activities of IL-33: A Mechanistic Insight. Front. Immunol. 2020, 11, 571593. [Google Scholar] [CrossRef] [PubMed]
- Ercolano, G.; Gomez-Cadena, A.; Dumauthioz, N.; Vanoni, G.; Kreutzfeldt, M.; Wyss, T.; Michalik, L.; Loyon, R.; Ianaro, A.; Ho, P.-C.; et al. PPARɣ drives IL-33-dependent ILC2 pro-tumoral functions. Nat. Commun. 2021, 12, 2538. [Google Scholar] [CrossRef] [PubMed]
- Chatila, T.A. Interleukin-4 receptor signaling pathways in asthma pathogenesis. Trends Mol. Med. 2004, 10, 493–499. [Google Scholar] [CrossRef]
- Matsunaga, K.; Katoh, N.; Fujieda, S.; Izuhara, K.; Oishi, K. Dupilumab: Basic aspects and applications to allergic diseases. Allergol. Int. 2020, 69, 187–196. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Chiles, R. First Therapeutic Approval for Eosinophilic Esophagitis. Gastroenterol. Insights 2022, 13, 238–244. [Google Scholar] [CrossRef]
- Gil Yosipovitch, G.; Mollanazar, N.; Ständer, S.; Kwatra, S.G.; Kim, B.S.; Laws, E.; Mannent, L.P.; Amin, N.; Akinlade, B.; Staudinger, H.W.; et al. Dupilumab in patients with prurigo nodularis: Two randomized, double-blind, placebo-controlled phase 3 trials. Nat. Med. 2023, 29, 1180–1190. [Google Scholar] [CrossRef]
- Patel, G.; Pasha, M.A.; D’Souza, S.; Yang, Q. Group 2 Innate Lymphoid Cells in Patients with Severe Atopic Dermatitis on Dupilumab. J. Allergy Clin. Immunol. 2019, 143, AB19. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Kusakabe, M.; Nagai, M.; Yasuda, K.; Yamanishi, K. Dupilumab Effects on Innate Lymphoid Cell and Helper T Cell Populations in Patients with Atopic Dermatitis. JID Innov. 2021, 1, 100003. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Cella, M.; Gamini, R.; Sécca, C.; Collins, P.L.; Zhao, S.; Peng, V.; Robinette, M.L.; Schettini, J.; Zaitsev, K.; Gordon, W.; et al. Subsets of ILC3−ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat. Immunol. 2019, 20, 980–991. [Google Scholar] [CrossRef]
- Bal, S.; Bernink, J.H.; Nagasawa, M.; Groot, J.; Shikhagaie, M.M.; Golebski, K.; Van Drunen, C.M.; Lutter, R.; Jonkers, E.R.; Hombrink, P.; et al. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat. Immunol. 2016, 17, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.I.; Menegatti, S.; Bustamante, J.; Le Bourhis, L.; Allez, M.; Rogge, L.; Casanova, J.-L.; Yssel, H.; Di Santo, J.P. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J. Exp. Med. 2016, 213, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Guo, L.; Qiu, J.; Chen, X.; Hu-Li, J.; Siebenlist, U.; Williamson, P.R.; Urban, J.F., Jr.; Paul, E.W. IL-25-responsive, lineage-negative KLRG1hi cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat. Immunol. 2015, 16, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernink, J.H.; Ohne, Y.; Teunissen, M.B.M.; Wang, J.; Wu, J.; Krabbendam, L.; Guntermann, C.; Volckmann, R.; Koster, J.; van Tol, S.; et al. c-Kit-positive ILC2s exhibit an ILC3-like signature that may contribute to IL-17-mediated pathologies. Nat. Immunol. 2019, 20, 992–1003. [Google Scholar] [CrossRef]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.-J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Miller, B.C.; Maus, M.V. CD19-Targeted CAR T Cells: A New Tool in the Fight against B Cell Malignancies. Oncol. Res. Treat. 2015, 38, 683–690. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Berrien-Elliott, M.M.; Jacobs, M.T.; Fehniger, T.A. Allogeneic natural killer cell therapy. Blood 2023, 141, 856–868. [Google Scholar] [CrossRef]
- Shah, N.; Li, L.; McCarty, J.; Kaur, I.; Yvon, E.; Shaim, H.; Muftuoglu, M.; Liu, E.; Orlowski, R.Z.; Cooper, L.; et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br. J. Haematol. 2017, 177, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Meng, Y.; Feng, X.; Han, Z. CAR-NK cells for cancer immunotherapy: From bench to bedside. Biomark. Res. 2022, 10, 12. [Google Scholar] [CrossRef]
- Li, H.; Song, W.; Li, Z.; Zhang, M. Preclinical and clinical studies of CAR-NK-cell therapies for malignancies. Front. Immunol. 2022, 13, 992232. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, T.J.; Biederstädt, A.; Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 2022, 22, 557–575. [Google Scholar] [CrossRef] [PubMed]
- Kilgour, M.K.; Bastin, D.J.; Lee, S.-H.; Ardolino, M.; McComb, S.; Visram, A. Advancements in CAR-NK therapy: Lessons to be learned from CAR-T therapy. Front. Immunol. 2023, 14, 1166038. [Google Scholar] [CrossRef] [PubMed]
- Karahan, Z.S.; Aras, M.; Sütlü, T. TCR-NK Cells: A Novel Source for Adoptive Immunotherapy of Cancer. Turk. J. Haematol. 2023, 40, 1–10. [Google Scholar] [CrossRef]
- Mensali, N.; Dillard, P.; Hebeisen, M.; Lorenz, S.; Theodossiou, T.; Myhre, M.R.; Fåne, A.; Gaudernack, G.; Kvalheim, G.; Myklebust, J.H.; et al. NK cells specifically TCR-dressed to kill cancer cells. Ebiomedicine 2019, 40, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Ueda, N.; Uemura, Y.; Zhang, R.; Kitayama, S.; Iriguchi, S.; Kawai, Y.; Yasui, Y.; Tatsumi, M.; Ueda, T.; Liu, T.-Y.; et al. Generation of TCR-Expressing Innate Lymphoid-like Helper Cells that Induce Cytotoxic T Cell-Mediated Anti-leukemic Cell Response. Stem Cell Rep. 2018, 10, 1935–1946. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, C.S.; Montel-Hagen, A.; Chen, H.-C.; Lopez, S.; Zhou, O.; Dai, K.; Tsai, S.; Satyadi, W.; Botero, C.; et al. Strength of CAR signaling determines T cell versus ILC differentiation from pluripotent stem cells. Cell Rep. 2023, 42, 112241. [Google Scholar] [CrossRef]
- Ueda, T.; Kumagai, A.; Iriguchi, S.; Yasui, Y.; Miyasaka, T.; Nakagoshi, K.; Nakane, K.; Saito, K.; Takahashi, M.; Sasaki, A.; et al. Non–clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti–glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020, 111, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Lotze, M.T.; Zhu, Z.; Storkus, W.J.; Song, X.-T. Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy. Biomedicines 2020, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Bailis, J.M.; Britten, C.D.; Klinger, M.; Nagorsen, D.; Coxon, A.; Egen, J.G.; Martin, F. Targeting Solid Tumors with Bispecific T Cell Engager Immune Therapy. Annu. Rev. Cancer Biol. 2022, 6, 17–34. [Google Scholar] [CrossRef]
- Sidaway, P. Blinatumomab improves outcomes in infant ALL. Nat. Rev. Clin. Oncol. 2023, 20, 426. [Google Scholar] [CrossRef]
- Phung, S.K.; Miller, J.S.; Felices, M. Bi-specific and Tri-specific NK Cell Engagers: The New Avenue of Targeted NK Cell Immunotherapy. Mol. Diagn. Ther. 2021, 25, 577–592. [Google Scholar] [CrossRef]
- Ellwanger, K.; Reusch, U.; Fucek, I.; Wingert, S.; Ross, T.; Müller, T.; Schniegler-Mattox, U.; Haneke, T.; Rajkovic, E.; Koch, J.; et al. Redirected optimized cell killing (ROCK®): A highly versatile multispecific fit-for-purpose antibody platform for engaging innate immunity. mAbs 2019, 11, 899–918. [Google Scholar] [CrossRef] [Green Version]
- Arulanandam, A.; Lin, L.; Chang, H.-M.; Cerutti, M.; Choblet, S.; Gao, P.; Rath, A.; Bensussan, A.; Kadouche, J.; Teper, D.; et al. Derivation and Preclinical Characterization of CYT-303, a Novel NKp46-NK Cell Engager Targeting GPC3. Cells 2023, 12, 996. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2020, 18, 85–100. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, T.R.; Kodal, B.; Lenvik, A.J.; Hinderlie, P.; Bendzick, L.E.; Schirm, D.K.; Kaminski, M.F.; McElmurry, R.T.; Geller, M.A.; et al. Potent Cytolytic Activity and Specific IL15 Delivery in a Second-Generation Trispecific Killer Engager. Cancer Immunol. Res. 2020, 8, 1139–1149. [Google Scholar] [CrossRef]
- Vallera, D.A.; Ferrone, S.; Kodal, B.; Hinderlie, P.; Bendzick, L.; Ettestad, B.; Hallstrom, C.; Zorko, N.A.; Rao, A.; Fujioka, N.; et al. NK-Cell-Mediated Targeting of Various Solid Tumors Using a B7-H3 Tri-Specific Killer Engager In Vitro and In Vivo. Cancers 2020, 12, 2659. [Google Scholar] [CrossRef]
- Cristiani, C.M.; Garofalo, C.; Passacatini, L.C.; Carbone, E. New avenues for melanoma immunotherapy: Natural Killer cells? Scand. J. Immunol. 2019, 91, e12861. [Google Scholar] [CrossRef]
- Kubick, B.J.; Fan, X.; Crouch, A.; McCarthy, R.; Roop, D.R. Tracing the Equilibrium Phase of Cancer Immunoediting in Epidermal Neoplasms via Longitudinal Intravital Imaging. J. Investig. Dermatol. 2020, 140, 891–900.e10. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Quek, C.; Silva, I.; Tasker, A.; Batten, M.; Rizos, H.; Lim, S.Y.; Gide, T.N.; Shang, P.; Attrill, G.H.; et al. Integrated molecular and immunophenotypic analysis of NK cells in anti-PD-1 treated metastatic melanoma patients. Oncoimmunology 2018, 8, e1537581. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Da Silva, I.P.; Palendira, U.; Scolyer, R.A.; Long, G.V.; Wilmott, J.S. Targeting NK Cells to Enhance Melanoma Response to Immunotherapies. Cancers 2021, 13, 1363. [Google Scholar] [CrossRef]
- van Vliet, A.A.; Georgoudaki, A.-M.; Raimo, M.; de Gruijl, T.D.; Spanholtz, J. Adoptive NK Cell Therapy: A Promising Treatment Prospect for Metastatic Melanoma. Cancers 2021, 13, 4722. [Google Scholar] [CrossRef]
- Garofalo, C.; De Marco, C.; Cristiani, C.M. NK Cells in the Tumor Microenvironment as New Potential Players Mediating Chemotherapy Effects in Metastatic Melanoma. Front. Oncol. 2021, 11, 754541. [Google Scholar] [CrossRef]

| Antibody | Target | FDA Approval with Initial Indication | Subsequent Indications | References |
|---|---|---|---|---|
| ipilimumab | CTLA-4 | 2011 for unresectable or metastatic melanoma | melanoma (alone or w/nivolumab) other solid tumors (w/nivolumab) | [77,78] |
| tremelimumab | CTLA-4 | 2022 for unresectable HCC (w/durvalumab), and metastatic NSCLC (w/durvalumab and platinum-based chemotherapy) | [79] | |
| pembrolizumab | PD-1 | 2014 (accelerated) for unresectable or metastatic melanoma 2015 (accelerated) for metastatic NSCLC | many solid tumors cHL, PMBCL | [78,80,81] |
| nivolumab | PD-1 | 2014 (accelerated) for unresectable or metastatic melanoma 2015 for advanced RCC and metastatic squamous and non-squamous NSCLC 2016 (accelerated) for relapsed or progressive cHL | many solid tumors | [78,82,83,84,85] |
| cemiplimab | PD-1 | 2018 for locally advanced or metastatic CSCC | BCC, NSCLC | [86,87,88] |
| dostarlimab | PD-1 | 2021 (accelerated) for dMMR recurrent or advanced endometrial cancer | [89] | |
| atezolizumab | PD-L1 | 2016 (accelerated) for locally advanced or metastatic urothelial carcinoma 2016 for metastatic NSCLC | other solid tumors | [78,90,91] |
| avelumab | PD-L1 | 2017 (accelerated) for metastatic MCC and locally advanced or metastatic urothelial carcinoma | RCC | [78,92,93] |
| durvalumab | PD-L1 | 2017 (accelerated) for locally advanced or metastatic urothelial carcinoma | NSCLC, SCLC, BTC | [78,94] |
| relatlimab | LAG-3 | 2022 for unresectable or metastatic melanoma (w/nivolumab, market name Opdualag) | [95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, H.; Verma, A.; Kinzel, M.; Huang, Q.; Mahoney, D.J.; Jacquelot, N. Targeting Potential of Innate Lymphoid Cells in Melanoma and Other Cancers. Pharmaceutics 2023, 15, 2001. https://doi.org/10.3390/pharmaceutics15072001
Seo H, Verma A, Kinzel M, Huang Q, Mahoney DJ, Jacquelot N. Targeting Potential of Innate Lymphoid Cells in Melanoma and Other Cancers. Pharmaceutics. 2023; 15(7):2001. https://doi.org/10.3390/pharmaceutics15072001
Chicago/Turabian StyleSeo, Hobin, Amisha Verma, Megan Kinzel, Qiutong Huang, Douglas J. Mahoney, and Nicolas Jacquelot. 2023. "Targeting Potential of Innate Lymphoid Cells in Melanoma and Other Cancers" Pharmaceutics 15, no. 7: 2001. https://doi.org/10.3390/pharmaceutics15072001