
Metal-Based Nanoparticles for Cancer Metalloimmunotherapy
 , ,
, ,
Abstract
:1. Introduction
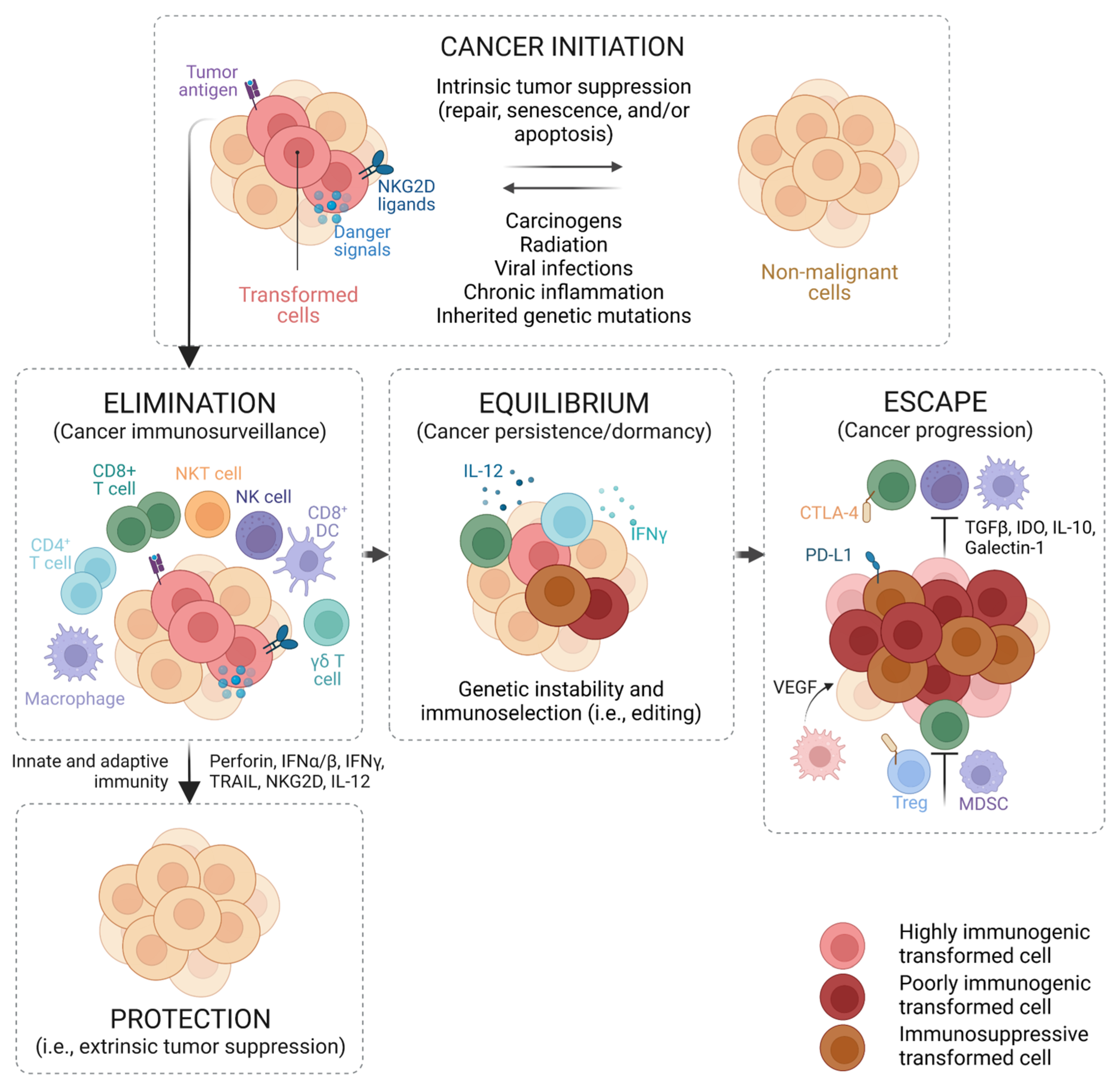
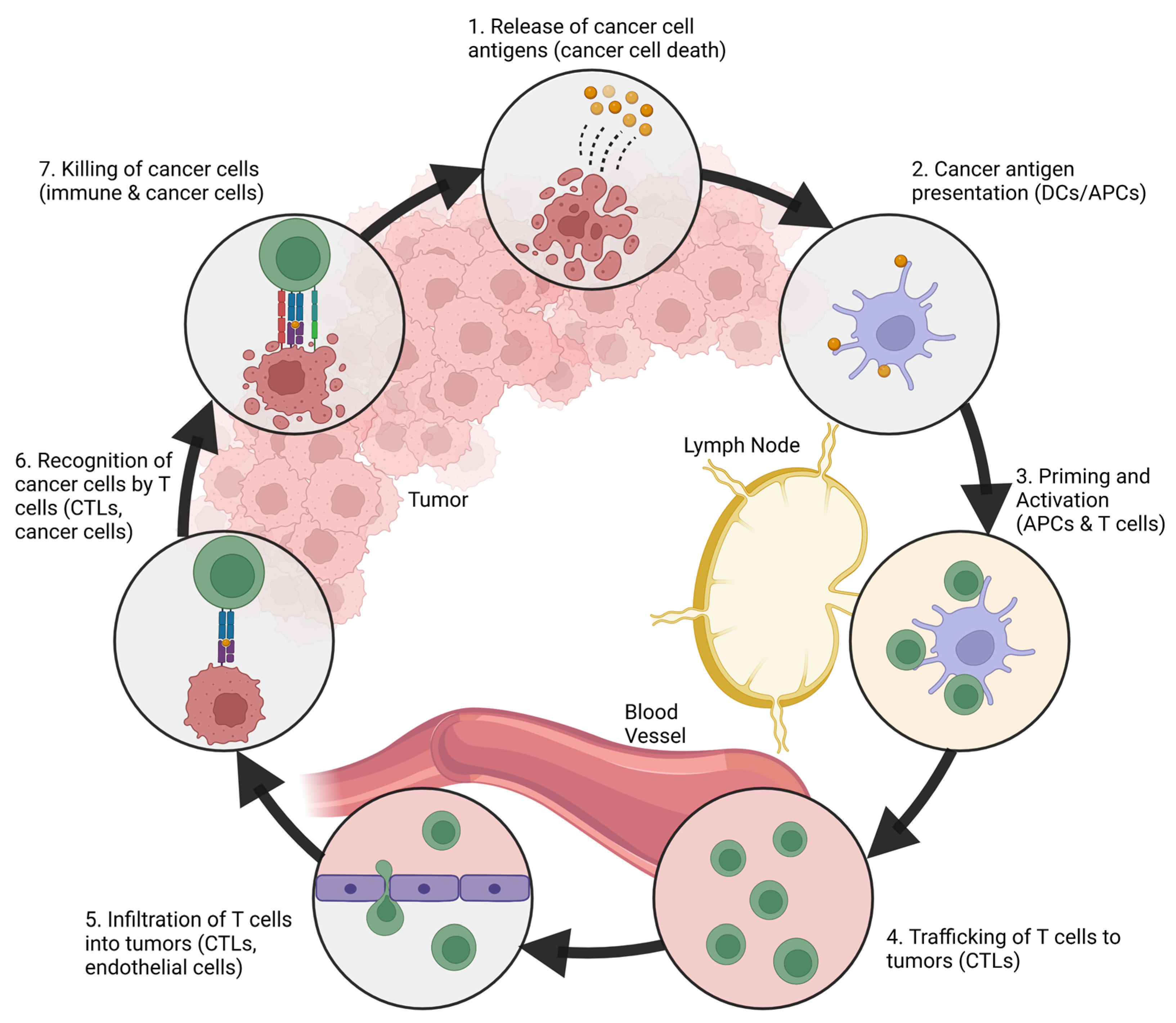
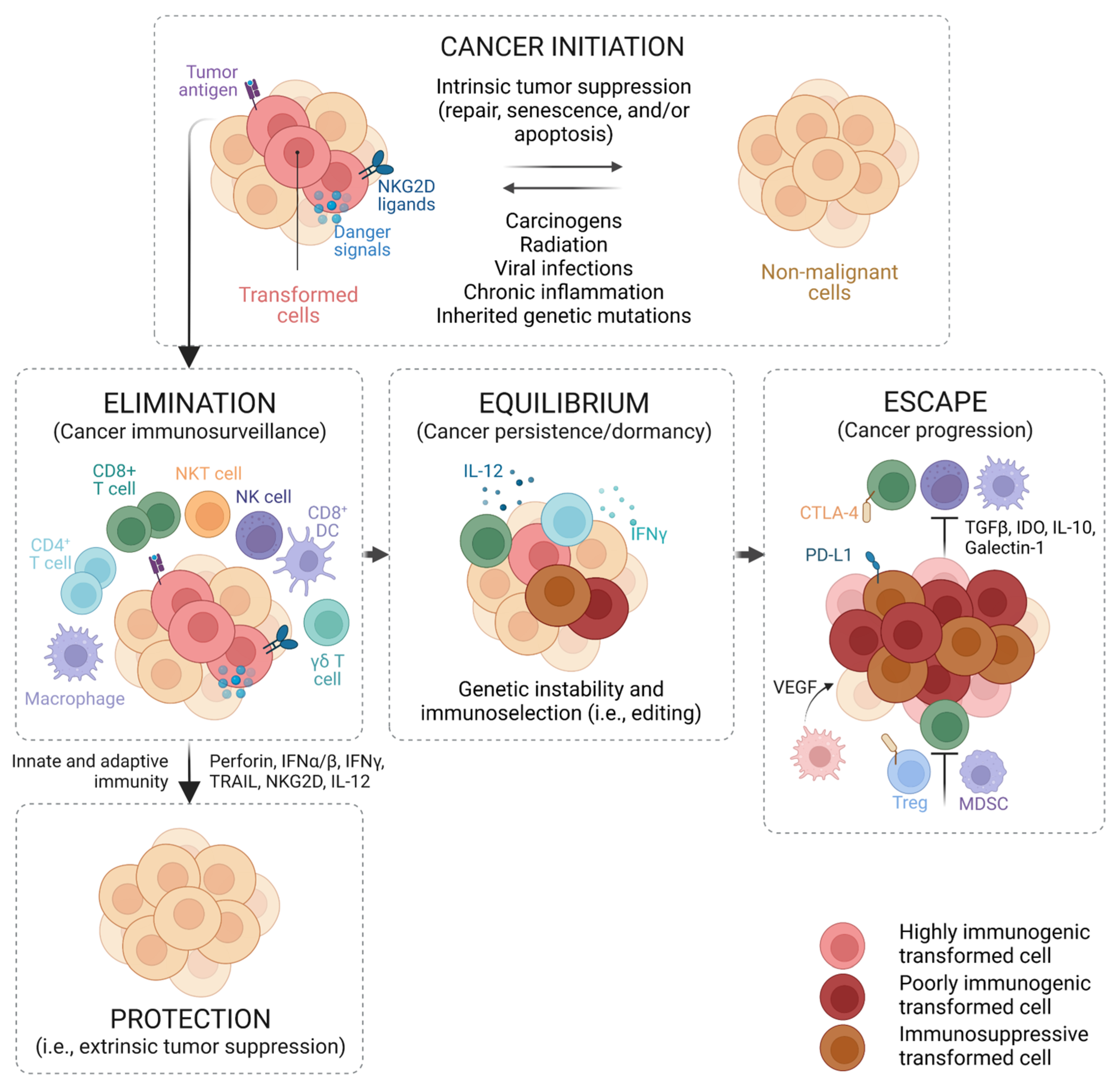
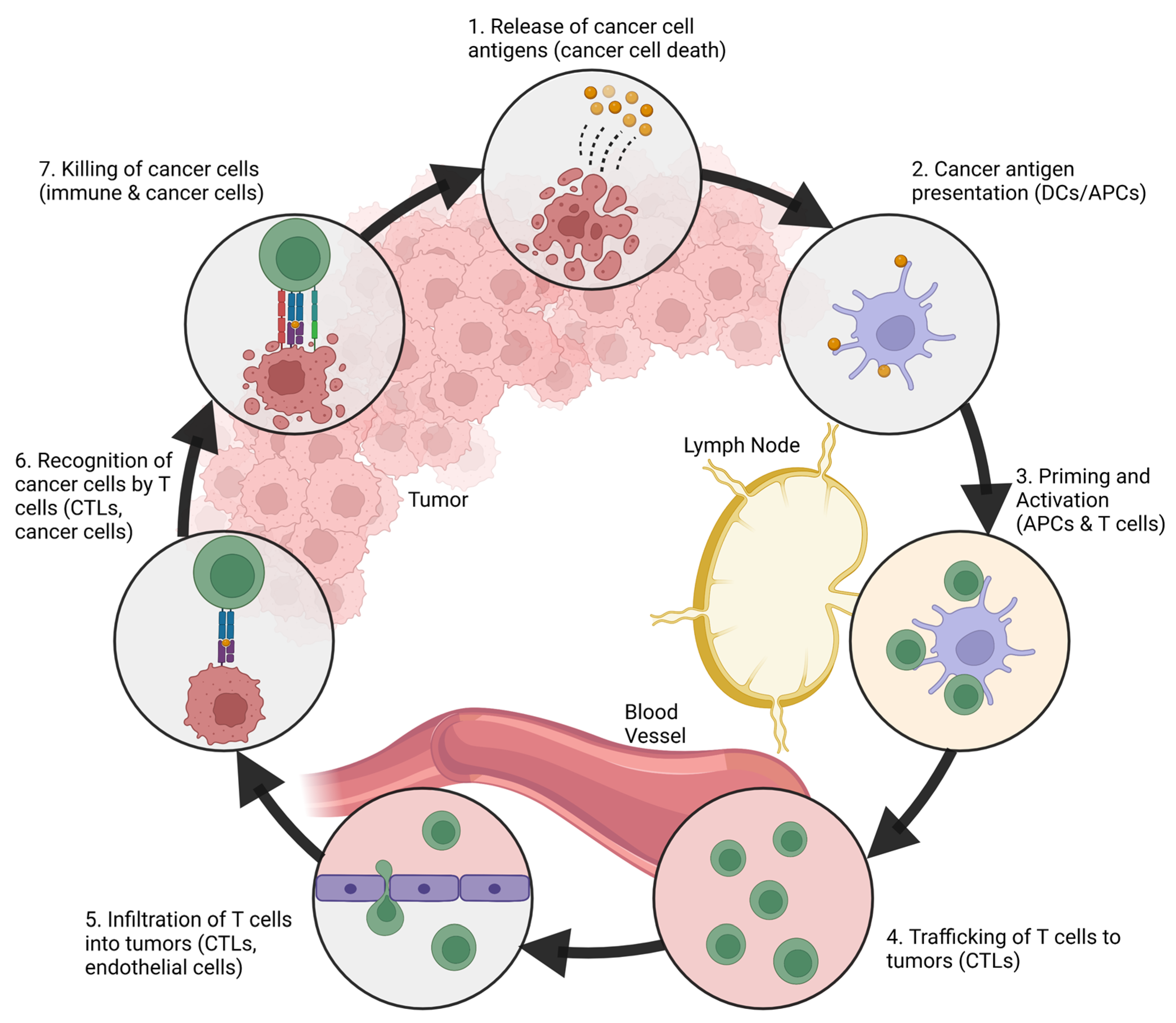
2. The Immune System and Cancer
2.1. Antigen-Presenting Cells
2.2. Effector Immune Cells
2.3. Immunosuppressive Cells
3. Metal Ions in the Host Response and Immune Modulation
4. Metal-Based Nanoparticles for Cancer Metalloimmunotherapy
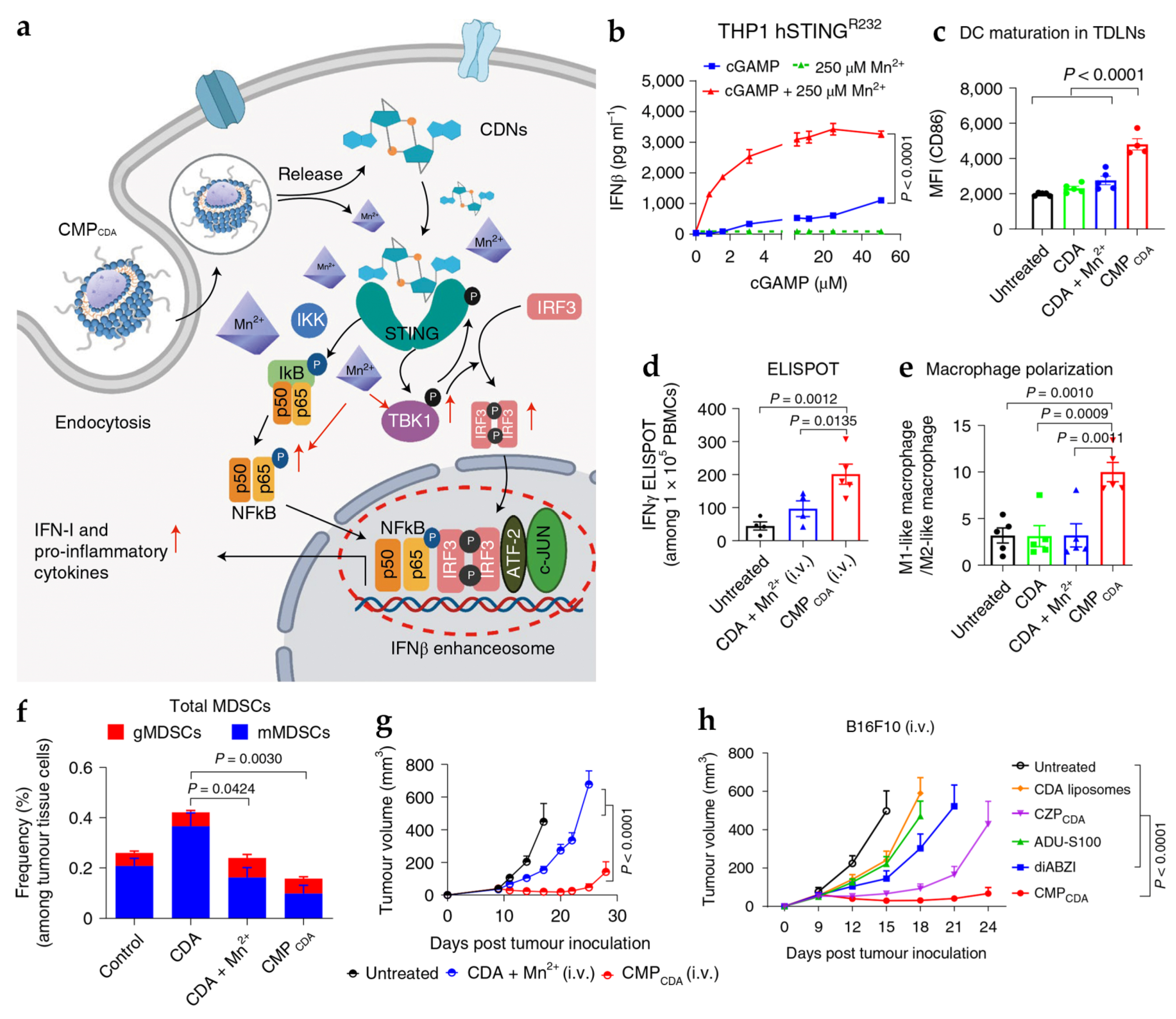
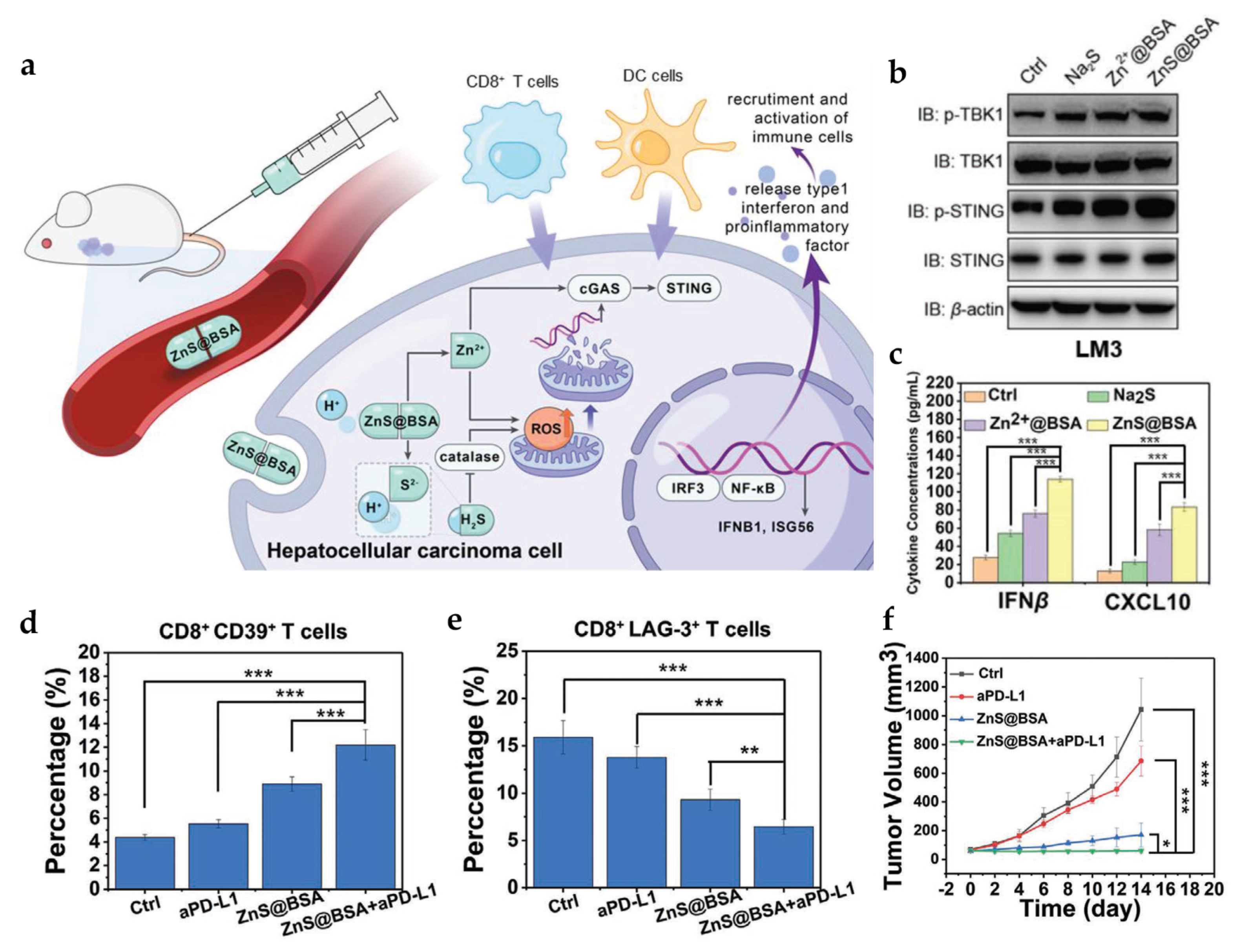
4.1. Manganese- and Zinc-Based Nanoparticles for cGAS-STING Activation
4.2. Iron- and Copper-Based Nanoparticles for M1 Macrophage Polarization
4.3. Calcium- and Sodium-Based Nanoparticles for Ion Overloading Effect

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal | Nanoparticle Formulation | Surface Modification and Drug Loading | Nanoparticle Characteristics | Target Cells | Route and Dose (In Vivo) | Properties and Outcomes | References |
|---|---|---|---|---|---|---|---|
| Mn | CMPCDA | SM: PEG-lipid bilayer | PS: 118 ± 41 nm PDI: 0.107 ZP: −2.49 ± 6.42 mV | In vitro DC: BMDCs In vitro tumor cells: THP1 In vivo tumor model: CT26, B16F10, NOOC1 mice | i.t.: CDA 5 µg, Mn2+ 2.5 µg i.v.: CDA 20 µg, Mn2+ 10 µg | Deliver STING agonist to immune cells and enhances its activity through the activation of the cGAS-STING pathway | [9] |
| Bio-MnO2 NPs + RT | - | PS: 101 nm PDI: 0.122 | In vitro tumor cells: A549, PC9, H520 In vivo tumor model: LLC mice | i.t.: 2 mg/kg + IR: 8 Gy | Relieve tumor hypoxia, activate the cGAS-STING pathway, and sensitize RT | [67] | |
| PL/APMP-DOX NPs | SM: PL layer DL: DOX | PS: ~180 nm PDI: 0.232 ZP: −23.8 ± 0.6 mV Pore Size: 3.88 nm | In vivo tumor model: 4T1 mice | i.v.: APMP 2.50 mg/kg, DOX 2.50 mg/kg | Activate the cGAS-STING pathway and carry DOX to induce DNA damage | [68] | |
| Zn | ZnO/DOX | DL: DOX | In DMEM/10%FBS medium: PS: 62.02 ± 0.98 nm PDI: 0.31 ± 0.03 ZP: −16.06 ± 0.09 mV | In vitro tumor cells: DOX-sensitive (MDA-MB-231 and HeLa), DOX-resistant (NCI/ADR-RES and MES-SA/Dx5) In vitro macrophage: RAW264.7 | - | Generate ROS and activate caspase 3/7, polarize macrophages into M1 phenotype, downregulate the CD44 expression of stem-like cancer cells, and carry DOX to kill cancer cells synergistically | [70] |
| ZnS@BSA | - | PS ≈ 100 nm | In vitro tumor cells: HCC (LM3 and Hepa1-6) In vivo tumor model: Hepa1-6 mice | i.v. | Cooperate with H2S to accumulate ROS and activate the cGAS-STING pathway | [69] | |
| Zn-LDH | - | - | In vitro tumor cells: B16F10 and 4T1 In vitro DC: DC2.4 In vitro macrophage: RAW264.7 In vivo tumor model: B16F10 and 4T1 mice | PT: 1 mg | Activate the cGAS-STING pathway, block the autophagy pathway, and neutralize TME acidity | [71] | |
| Fe | Ferumoxytol (Feraheme) | - | PS 30 nm | In vitro tumor cells: MMTV-PyMT In vitro macrophage: RAW264.7/BMDMs In vivo tumor model: MMTV-PyMT mice In vivo metastatic model: KP1 mice | MFP injection: 100 µL at a conc. of 2.73 mg Fe/mL i.v.: 10 mg Fe/kg | Polarize macrophages into M1 phenotype | [76] |
| Positively and Negatively Charged SPIONs | - | For S+: PS: 19.4 ± 0.8 nm ZP: +44.72 mV For S-: PS: 21.3 ± 1.6 nm ZP: −27.31 mV | In vitro tumor cells: HT1080 In vitro macrophage: RAW264.7 In vivo tumor model: HT1080 mice | i.t. | Polarize macrophages into M1 phenotype | [87] | |
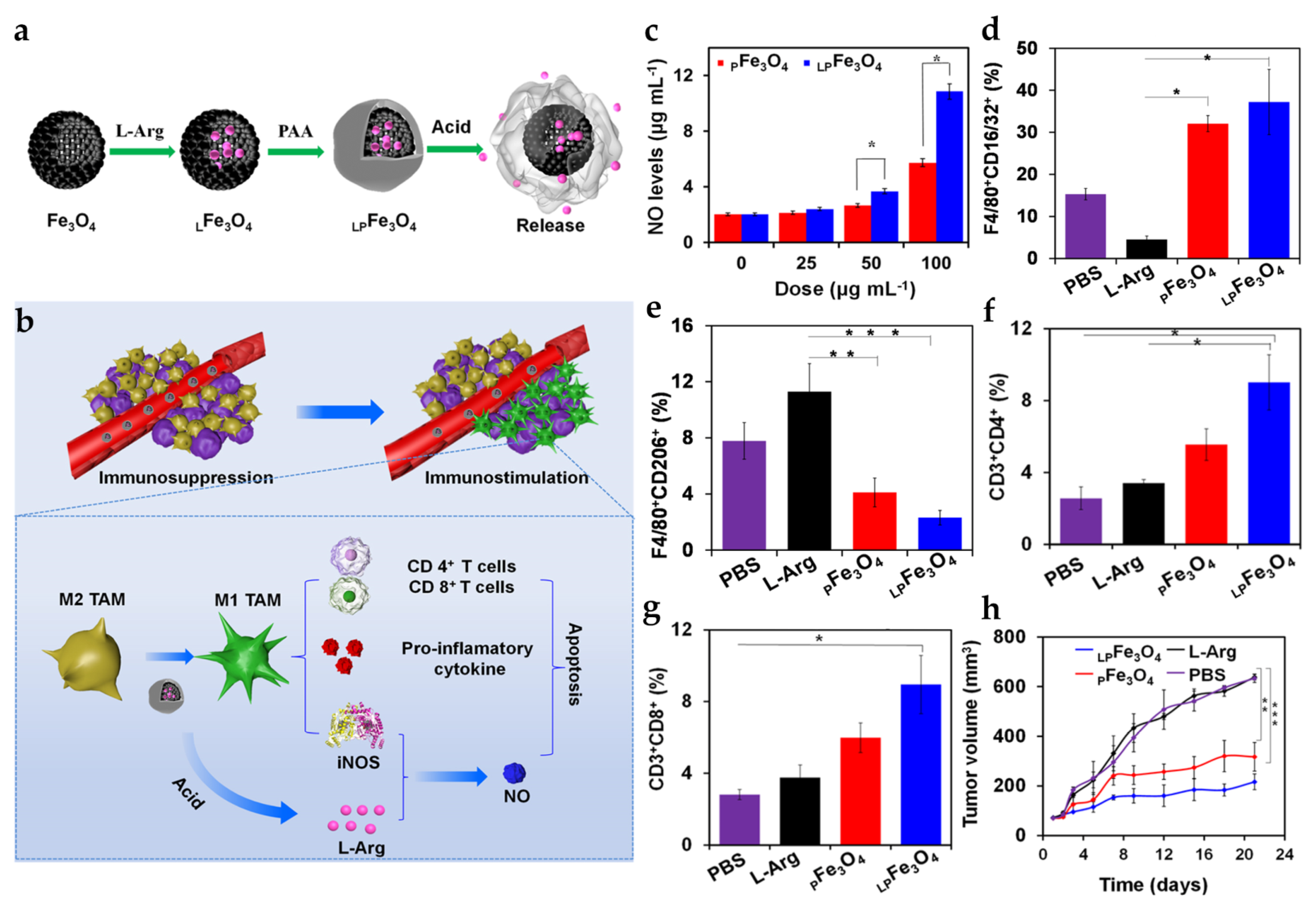
| LPFe3O4 NPs | SM: PAA layer DL: L-Arg | PS: 229.4 ± 5.0 nm ZP: −31.1 ± 0.6 mV Pore Size: 3.5 nm | In vitro tumor cells: 4T1 In vitro macrophage: RAW264.7/BMDMs In vivo tumor model: 4T1 mice | i.v.: 20 mg Fe3O4/kg | Polarize macrophages into M1 phenotype and carry L-Arg to induce NO production | [78] | |
| IO-LPMONs-OVA vaccine + IO-LPMONs | - | PS: 360 nm Pore Size: 6.3 nm | In vitro tumor cells: SCC7 In vitro macrophage: RAW264.7 In vivo tumor model: EG7-OVA mice | s.c.: IO-LPMONs-OVA vaccine (100 µL, conc. 1 mg/mL) + i.t.: IO-LPMONs (100 µL, conc. 0.2 mg/mL) | Polarize macrophages into M1 phenotype and carry OVA for vaccination | [75] | |
| Cu | CuS NPs | SM: PEG | PS ≈ 17 nm | In vivo tumor model: B16F10 mice | i.t. | Polarize BMDMs ex vivo into M1 phenotype for adoptive macrophage therapy | [81] |
| Cu | CuS@OVA | DL: OVA | PS: 16.3 ± 0.2 nm | In vitro tumor cells: B16-OVA In vitro DC: BMDCs In vitro macrophage: BMDMs In vivo tumor model: B16-OVA and orthotopic uveal B16-Luc mice | i.t.: CuS-OVA (10 or 2 µL, conc. 1 mg/mL) + i.p.: aPD-1 10 mg/kg | Polarize macrophages into M1 phenotype and activate DCs, serve as a photosensitizer for PTT, and augment the antitumor efficacy of aPD-1 | [82] |
| Ca | MTX + HOCN | - | PS: 250 nm ZP: −7.6 mV | In vitro tumor cells: CT26 In vitro DC: DC2.4 In vivo tumor model: CT26 mice | i.v.: 40 mg/kg | Overcome the multiple barriers of DCs by disrupting autophagy inhibition, attenuating TME acidity, and releasing DAMPs | [83] |
| CaNMs | DL: CUR | - | In vitro tumor cells: 4T1 In vivo tumor model: 4T1 mice | i.t. | Induce Ca2+ overloading-mediated pyroptosis to stimulate antitumor immune responses | [84] | |
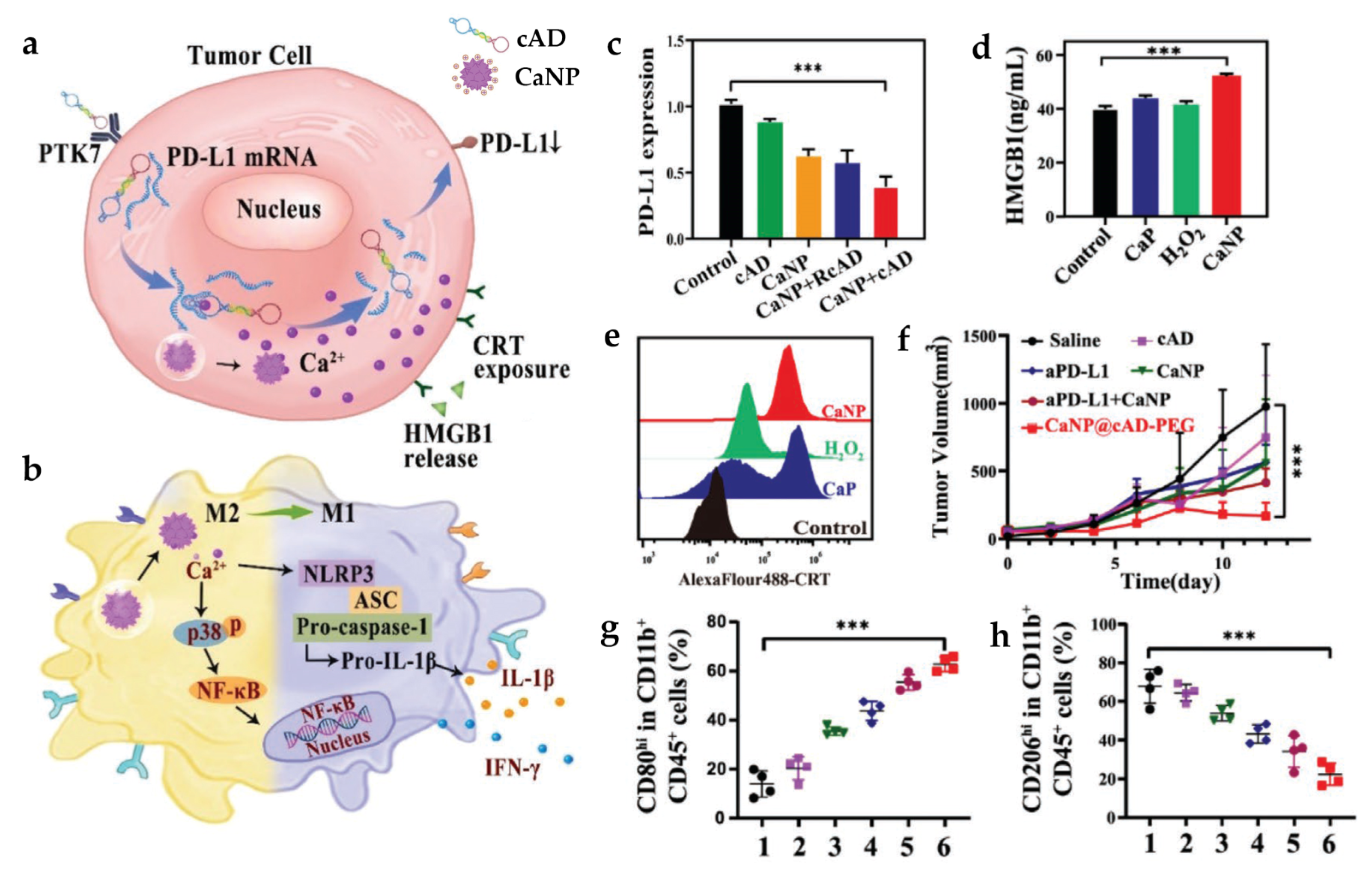
| CaNP@cAD-PEG | SM: DSPE-PEG2000 DL: cAD | PS: 180 nm | In vitro tumor cells: B16 In vitro macrophage: BMDMs In vivo tumor model: B16 and CT26 mice | i.v. | Induce TAM re-education through multiple inflammation-related signaling pathways as well as NLRP3-inflammasome, promote cancer antigen release, and suppress PD-L1 expression | [85] | |
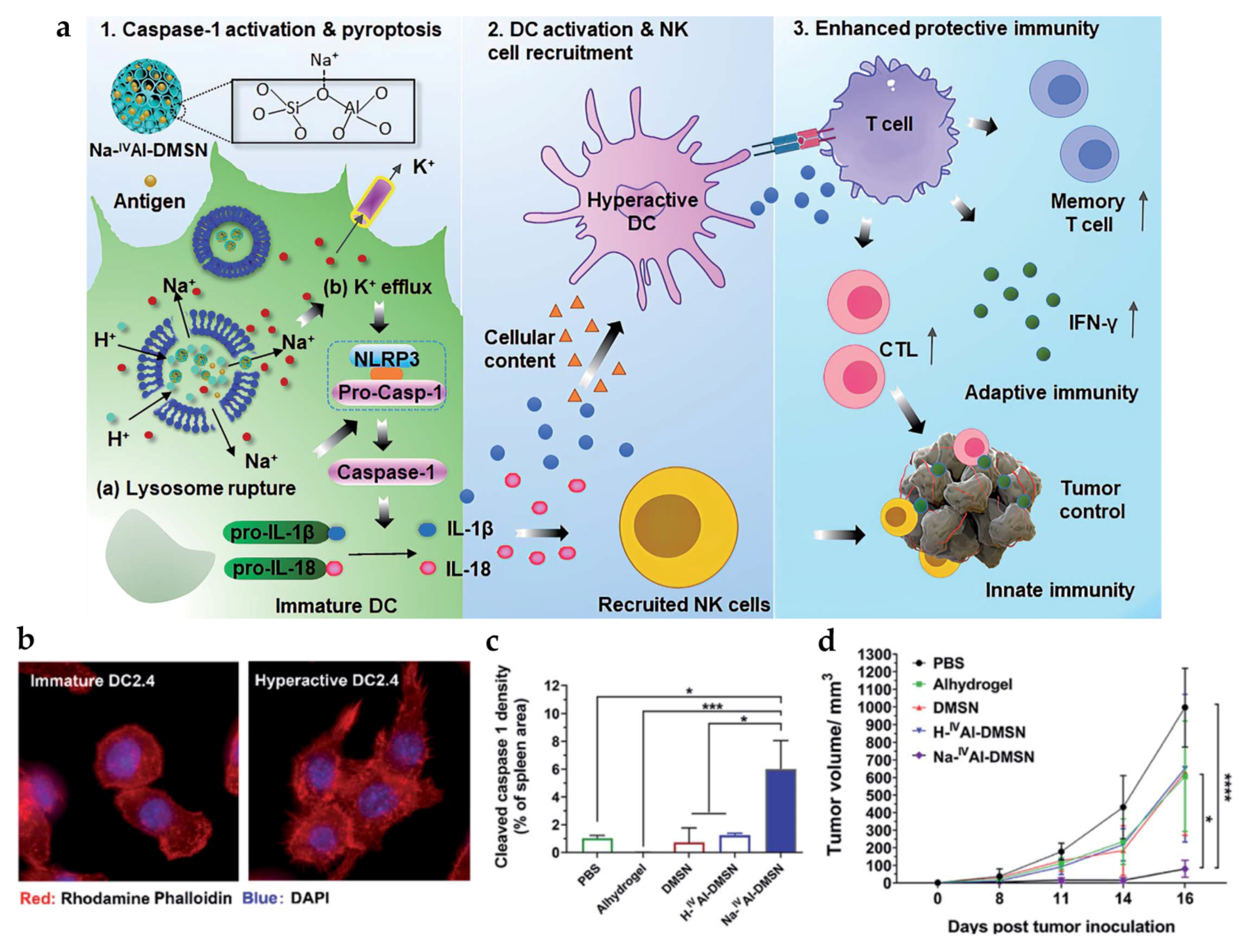
| Na | Na-IVAl-DMSN | - | PS: ~240 nm | In vitro DC: DC2.4 In vivo tumor model: CT26 mice | s.c. | Induce intracellular ion perturbation for DC pyroptosis and hyperactivation, provoking enhanced NK cell-mediated innate immunity and both cellular and humoral adaptive immune responses | [86] |
5. Conclusions
6. Perspectives and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Shariatzadeh, S.; Moghimi, N.; Khalafi, F.; Shafiee, S.; Mehrabi, M.; Ilkhani, S.; Tosan, F.; Nakhaei, P.; Alizadeh, A.; Varma, R.S.; et al. Metallic Nanoparticles for the Modulation of Tumor Microenvironment; A New Horizon. Front. Bioeng. Biotechnol. 2022, 10, 847433. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, A.; Sathiyamoorthy, P.; Park, I.-K. Metallic Nanoparticle-Mediated Immune Cell Regulation and Advanced Cancer Immunotherapy. Pharmaceutics 2021, 13, 1867. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-R.; Wu, X.-L.; Sun, Y.-L. Therapeutic Targets and Biomarkers of Tumor Immunotherapy: Response versus Non-Response. Signal Transduct. Target. Ther. 2022, 7, 331. [Google Scholar] [CrossRef]
- Mishra, A.K.; Ali, A.; Dutta, S.; Banday, S.; Malonia, S.K. Emerging Trends in Immunotherapy for Cancer. Diseases 2022, 10, 60. [Google Scholar] [CrossRef]
- Peterson, C.; Denlinger, N.; Yang, Y. Recent Advances and Challenges in Cancer Immunotherapy. Cancers 2022, 14, 3972. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, R.; Wei, X.; Lv, M.; Jiang, Z. Metalloimmunology: The Metal Ion-Controlled Immunity. Adv. Immunol. 2020, 145, 187–241. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, Y.; Li, J.; Park, K.S.; Han, K.; Zhou, X.; Xu, Y.; Nam, J.; Xu, J.; Shi, X.; et al. Amplifying STING Activation by Cyclic Dinucleotide–Manganese Particles for Local and Systemic Cancer Metalloimmunotherapy. Nat. Nanotechnol. 2021, 16, 1260–1270. [Google Scholar] [CrossRef]
- Nam, J.; Son, S.; Park, K.S.; Zou, W.; Shea, L.D.; Moon, J.J. Cancer Nanomedicine for Combination Cancer Immunotherapy. Nat. Rev. Mater. 2019, 4, 398–414. [Google Scholar] [CrossRef]
- Surendran, S.P.; Moon, M.J.; Park, R.; Jeong, Y.Y. Bioactive Nanoparticles for Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3877. [Google Scholar] [CrossRef] [Green Version]
- Pandya, P.H.; Murray, M.E.; Pollok, K.E.; Renbarger, J.L. The Immune System in Cancer Pathogenesis: Potential Therapeutic Approaches. J. Immunol. Res. 2016, 2016, e4273943. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S.; Baker, D.L. Cellular and Molecular Immunology, 10th ed.; Elsevier: Philadelphia, PA, USA, 2022; ISBN 978-0-323-75748-5. [Google Scholar]
- Chaplin, D.D. Overview of the Immune Response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An Introduction to Immunology and Immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zeng, G. Cancer and Innate Immune System Interactions: Translational Potentials for Cancer Immunotherapy. J. Immunother. 2012, 35, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Janssen, L.M.E.; Ramsay, E.E.; Logsdon, C.D.; Overwijk, W.W. The Immune System in Cancer Metastasis: Friend or Foe? J. Immunother. Cancer 2017, 5, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swann, J.B.; Smyth, M.J. Immune Surveillance of Tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; de Mingo Pulido, Á.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katopodi, T.; Petanidis, S.; Charalampidis, C.; Chatziprodromidou, I.; Eskitzis, P.; Tsavlis, D.; Zarogoulidis, P.; Kosmidis, C.; Matthaios, D.; Porpodis, K. Tumor-Infiltrating Dendritic Cells: Decisive Roles in Cancer Immunosurveillance, Immunoediting, and Tumor T Cell Tolerance. Cells 2022, 11, 3183. [Google Scholar] [CrossRef]
- Del Prete, A.; Sozio, F.; Barbazza, I.; Salvi, V.; Tiberio, L.; Laffranchi, M.; Gismondi, A.; Bosisio, D.; Schioppa, T.; Sozzani, S. Functional Role of Dendritic Cell Subsets in Cancer Progression and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 3930. [Google Scholar] [CrossRef]
- Noubade, R.; Majri-Morrison, S.; Tarbell, K.V. Beyond CDC1: Emerging Roles of DC Crosstalk in Cancer Immunity. Front. Immunol. 2019, 10, 1014. [Google Scholar] [CrossRef] [Green Version]
- Kumari, N.; Choi, S.H. Tumor-Associated Macrophages in Cancer: Recent Advancements in Cancer Nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41, 68. [Google Scholar] [CrossRef]
- Galli, F.; Aguilera, J.V.; Palermo, B.; Markovic, S.N.; Nisticò, P.; Signore, A. Relevance of Immune Cell and Tumor Microenvironment Imaging in the New Era of Immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 89. [Google Scholar] [CrossRef]
- Lorenzo-Sanz, L.; Muñoz, P. Tumor-Infiltrating Immunosuppressive Cells in Cancer-Cell Plasticity, Tumor Progression and Therapy Response. Cancer Microenviron. 2019, 12, 119–132. [Google Scholar] [CrossRef]
- Duan, Z.; Luo, Y. Targeting Macrophages in Cancer Immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 127. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T Cells in Cancer and Cancer Immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munhoz, R.R.; Postow, M.A. Recent Advances in Understanding Antitumor Immunity. F1000Research 2016, 5, 2545. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.; Gao, F.; Yan, M.; Zhao, S.; Yan, Z.; Shi, B.; Liu, Y. Natural Killer Cells: A Promising Immunotherapy for Cancer. J. Transl. Med. 2022, 20, 240. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat4, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK Cell-Based Cancer Immunotherapy: From Basic Biology to Clinical Development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK Cells for Cancer Immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, T.; Gao, F.; Zhou, Z.; Shu, G.; Zou, Y.; Yin, G. Natural Killer Cells: A Promising Kit in the Adoptive Cell Therapy Toolbox. Cancers 2022, 14, 5657. [Google Scholar] [CrossRef] [PubMed]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Payer, Á.R.; Gonzalez, S.; López-Soto, A. Mechanisms of Apoptosis Resistance to NK Cell-Mediated Cytotoxicity in Cancer. Int. J. Mol. Sci. 2020, 21, 3726. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cao, X. Immunosuppressive Cells in Tumor Immune Escape and Metastasis. J. Mol. Med. 2016, 94, 509–522. [Google Scholar] [CrossRef]
- Shi, L.; Gu, H. Emerging Nanoparticle Strategies for Modulating Tumor-Associated Macrophage Polarization. Biomolecules 2021, 11, 1912. [Google Scholar] [CrossRef] [PubMed]
- Tie, Y.; Tang, F.; Wei, Y.; Wei, X. Immunosuppressive Cells in Cancer: Mechanisms and Potential Therapeutic Targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Dongye, S.; Liu, X.; Xu, X.; Wang, L.; Jin, C.Q.; Yao, M.; Gong, Z.; Jiang, D.; Zhang, K.; et al. Immunotherapy of Targeting MDSCs in Tumor Microenvironment. Front. Immunol. 2022, 13, 990463. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, T.; Zhu, W.; Xie, S.; Zhao, Z.; Feng, B.; Guo, H.; Yang, R. Targeting MDSC for Immune-Checkpoint Blockade in Cancer Immunotherapy: Current Progress and New Prospects. Clin. Med. Insights Oncol. 2021, 15, 11795549211035540. [Google Scholar] [CrossRef]
- Gao, Y.; Ouyang, Z.; Yang, C.; Song, C.; Jiang, C.; Song, S.; Shen, M.; Shi, X. Overcoming T Cell Exhaustion via Immune Checkpoint Modulation with a Dendrimer-Based Hybrid Nanocomplex. Adv. Healthc. Mater. 2021, 10, 2100833. [Google Scholar] [CrossRef]
- Verma, N.K.; Wong, B.H.S.; Poh, Z.S.; Udayakumar, A.; Verma, R.; Goh, R.K.J.; Duggan, S.P.; Shelat, V.G.; Chandy, K.G.; Grigoropoulos, N.F. Obstacles for T-Lymphocytes in the Tumour Microenvironment: Therapeutic Challenges, Advances and Opportunities beyond Immune Checkpoint. eBioMedicine 2022, 83, 104216. [Google Scholar] [CrossRef]
- Bayati, F.; Mohammadi, M.; Valadi, M.; Jamshidi, S.; Foma, A.M.; Sharif-Paghaleh, E. The Therapeutic Potential of Regulatory T Cells: Challenges and Opportunities. Front. Immunol. 2021, 11, 585819. [Google Scholar] [CrossRef]
- Chen, B.-J.; Zhao, J.-W.; Zhang, D.-H.; Zheng, A.-H.; Wu, G.-Q. Immunotherapy of Cancer by Targeting Regulatory T Cells. Int. Immunopharmacol. 2022, 104, 108469. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic Cell-Based Immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [Green Version]
- Shurin, G.V.; Ma, Y.; Shurin, M.R. Immunosuppressive Mechanisms of Regulatory Dendritic Cells in Cancer. Cancer Microenviron. 2013, 6, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Serra, M.; Columbano, A.; Ammarah, U.; Mazzone, M.; Menga, A. Understanding Metal Dynamics Between Cancer Cells and Macrophages: Competition or Synergism? Front. Oncol. 2020, 10, 646. [Google Scholar] [CrossRef]
- Monteith, A.J.; Skaar, E.P. The Impact of Metal Availability on Immune Function during Infection. Trends Endocrinol. Metab. 2021, 32, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Hood, M.I.; Skaar, E.P. Nutritional Immunity: Transition Metals at the Pathogen–Host Interface. Nat. Rev. Microbiol. 2012, 10, 525–537. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, P.; Benedetti, G.; Albarède, F.; Miossec, P. Zinc and Its Role in Immunity and Inflammation. Autoimmun. Rev. 2015, 14, 277–285. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schöneberg, T.; Schaefer, M.; Krügel, U.; et al. Extracellular Ca2+ Is a Danger Signal Activating the NLRP3 Inflammasome through G Protein-Coupled Calcium Sensing Receptors. Nat. Commun. 2012, 3, 1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scambler, T.; Jarosz-Griffiths, H.H.; Lara-Reyna, S.; Pathak, S.; Wong, C.; Holbrook, J.; Martinon, F.; Savic, S.; Peckham, D.; McDermott, M.F. ENaC-Mediated Sodium Influx Exacerbates NLRP3-Dependent Inflammation in Cystic Fibrosis. eLife 2019, 8, e49248. [Google Scholar] [CrossRef]
- Du, M.; Chen, Z.J. DNA-Induced Liquid Phase Condensation of CGAS Activates Innate Immune Signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef]
- Haase, H. Innate Immune Cells Speak Manganese. Immunity 2018, 48, 616–618. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Bi, Y.; Yang, W.; Guo, X.; Jiang, Y.; Wan, C.; Li, L.; Bai, Y.; Guo, J.; Wang, Y.; et al. Ca2+ Regulates T-Cell Receptor Activation by Modulating the Charge Property of Lipids. Nature 2013, 493, 111–115. [Google Scholar] [CrossRef]
- Vodnala, S.K.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.-H.; Lee, P.-H.; Shin, M.; Patel, S.J.; Yu, Z.; et al. T Cell Stemness and Dysfunction in Tumors Are Triggered by a Common Mechanism. Science 2019, 363, eaau0135. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.W.; Martinez, E.; Louka, P.; Wingett, D.G. Zinc Oxide Nanoparticles for Selective Destruction of Tumor Cells and Potential for Drug Delivery Applications. Expert Opin. Drug Deliv. 2010, 7, 1063–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irvine, D.J.; Dane, E.L. Enhancing Cancer Immunotherapy with Nanomedicine. Nat. Rev. Immunol. 2020, 20, 321–334. [Google Scholar] [CrossRef]
- Wang, C.; Guan, Y.; Lv, M.; Zhang, R.; Guo, Z.; Wei, X.; Du, X.; Yang, J.; Li, T.; Wan, Y.; et al. Manganese Increases the Sensitivity of the CGAS-STING Pathway for Double-Stranded DNA and Is Required for the Host Defense against DNA Viruses. Immunity 2018, 48, 675–687.e7. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Chen, M.; Zhang, R.; Zhang, W.; Wang, C.; Zhang, Y.; Wei, X.; Guan, Y.; Liu, J.; Feng, K.; et al. Manganese Is Critical for Antitumor Immune Responses via CGAS-STING and Improves the Efficacy of Clinical Immunotherapy. Cell Res. 2020, 30, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kifle, M.T.; Xie, H.; Xu, L.; Luo, M.; Li, Y.; Huang, Z.; Gong, Y.; Wu, Y.; Xie, C. Biomineralized Manganese Oxide Nanoparticles Synergistically Relieve Tumor Hypoxia and Activate Immune Response with Radiotherapy in Non-Small Cell Lung Cancer. Nanomaterials 2022, 12, 3138. [Google Scholar] [CrossRef]
- Hou, L.; Tian, C.; Yan, Y.; Zhang, L.; Zhang, H.; Zhang, Z. Manganese-Based Nanoactivator Optimizes Cancer Immunotherapy via Enhancing Innate Immunity. ACS Nano 2020, 14, 3927–3940. [Google Scholar] [CrossRef]
- Cen, D.; Ge, Q.; Xie, C.; Zheng, Q.; Guo, J.; Zhang, Y.; Wang, Y.; Li, X.; Gu, Z.; Cai, X. ZnS@BSA Nanoclusters Potentiate Efficacy of Cancer Immunotherapy. Adv. Mater. 2021, 33, 2104037. [Google Scholar] [CrossRef]
- Wang, J.; Lee, J.S.; Kim, D.; Zhu, L. Exploration of Zinc Oxide Nanoparticles as a Multitarget and Multifunctional Anticancer Nanomedicine. ACS Appl. Mater. Interfaces 2017, 9, 39971–39984. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, J.; Hu, X.; Wang, C.; Jia, Y.; Zhu, C.; Xie, S.; Lee, J.; Li, F.; Ling, D. A Peritumorally Injected Immunomodulating Adjuvant Elicits Robust and Safe Metalloimmunotherapy against Solid Tumors. Adv. Mater. 2022, 34, 2206915. [Google Scholar] [CrossRef]
- Dadfar, S.M.; Roemhild, K.; Drude, N.I.; von Stillfried, S.; Knüchel, R.; Kiessling, F.; Lammers, T. Iron Oxide Nanoparticles: Diagnostic, Therapeutic and Theranostic Applications. Adv. Drug Deliv. Rev. 2019, 138, 302–325. [Google Scholar] [CrossRef]
- Montiel Schneider, M.G.; Martín, M.J.; Otarola, J.; Vakarelska, E.; Simeonov, V.; Lassalle, V.; Nedyalkova, M. Biomedical Applications of Iron Oxide Nanoparticles: Current Insights Progress and Perspectives. Pharmaceutics 2022, 14, 204. [Google Scholar] [CrossRef]
- Arias, L.; Pessan, J.; Vieira, A.; Lima, T.; Delbem, A.; Monteiro, D. Iron Oxide Nanoparticles for Biomedical Applications: A Perspective on Synthesis, Drugs, Antimicrobial Activity, and Toxicity. Antibiotics 2018, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Ma, X.; Dang, M.; Dong, H.; Hu, H.; Su, X.; Liu, W.; Wang, Q.; Mou, Y.; Teng, Z. Simultaneous T Cell Activation and Macrophage Polarization to Promote Potent Tumor Suppression by Iron Oxide-Embedded Large-Pore Mesoporous Organosilica Core–Shell Nanospheres. Adv. Healthc. Mater. 2019, 8, 1900039. [Google Scholar] [CrossRef]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M.; et al. Iron Oxide Nanoparticles Inhibit Tumour Growth by Inducing Pro-Inflammatory Macrophage Polarization in Tumour Tissues. Nat. Nanotechnol. 2016, 11, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Cui, W.; Wang, W.; Zhang, H.; Liu, L.; Zhang, Z.; Li, Z.; Ying, G.; Zhang, N.; et al. Visualization of Caspase-3-like Activity in Cells Using a Genetically Encoded Fluorescent Biosensor Activated by Protein Cleavage. Nat. Commun. 2013, 4, 2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Cheng, Y.; Zheng, R.; Xu, K.; Yan, J.; Song, P.; Wang, Y.; Rauf, A.; Pan, Y.; Zhang, H. Immunomodulation of Tumor Microenvironment by Arginine-Loaded Iron Oxide Nanoparticles for Gaseous Immunotherapy. ACS Appl. Mater. Interfaces 2021, 13, 19825–19835. [Google Scholar] [CrossRef] [PubMed]
- Díez-Tercero, L.; Delgado, L.M.; Bosch-Rué, E.; Perez, R.A. Evaluation of the Immunomodulatory Effects of Cobalt, Copper and Magnesium Ions in a pro Inflammatory Environment. Sci. Rep. 2021, 11, 11707. [Google Scholar] [CrossRef] [PubMed]
- Díez-Tercero, L.; Delgado, L.M.; Perez, R.A. Modulation of Macrophage Response by Copper and Magnesium Ions in Combination with Low Concentrations of Dexamethasone. Biomedicines 2022, 10, 764. [Google Scholar] [CrossRef]
- Xu, J.; Zheng, B.; Zhang, S.; Liao, X.; Tong, Q.; Wei, G.; Yu, S.; Chen, G.; Wu, A.; Gao, S.; et al. Copper Sulfide Nanoparticle-Redirected Macrophages for Adoptive Transfer Therapy of Melanoma. Adv. Funct. Mater. 2021, 31, 2008022. [Google Scholar] [CrossRef]
- Ge, Y.; Zhang, J.; Jin, K.; Ye, Z.; Wang, W.; Zhou, Z.; Ye, J. Multifunctional Nanoparticles Precisely Reprogram the Tumor Microenvironment and Potentiate Antitumor Immunotherapy after Near-Infrared-II Light-Mediated Photothermal Therapy. Acta Biomater. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Zhang, K.; Wang, B.; Wu, S.; Wang, Y.; Zhang, H.; Zhang, Z.; Liu, J.; Shi, J. Nanoenabled Disruption of Multiple Barriers in Antigen Cross-Presentation of Dendritic Cells via Calcium Interference for Enhanced Chemo-Immunotherapy. ACS Nano 2020, 14, 7639–7650. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Ding, B.; Zhu, G.; Li, C.; Lin, J. Biodegradable Ca2+ Nanomodulators Activate Pyroptosis through Mitochondrial Ca2+ Overload for Cancer Immunotherapy. Angew. Chem. Int. Ed. 2022, 61, e202204904. [Google Scholar] [CrossRef]
- An, J.; Liu, M.; Zhao, L.; Lu, W.; Wu, S.; Zhang, K.; Liu, J.; Zhang, Z.; Shi, J. Boosting Tumor Immunotherapy by Bioactive Nanoparticles via Ca 2+ Interference Mediated TME Reprogramming and Specific PD-L1 Depletion. Adv. Funct. Mater. 2022, 32, 2201275. [Google Scholar] [CrossRef]
- Tang, J.; Yang, Y.; Qu, J.; Ban, W.; Song, H.; Gu, Z.; Yang, Y.; Cai, L.; Theivendran, S.; Wang, Y.; et al. Mesoporous Sodium Four-Coordinate Aluminosilicate Nanoparticles Modulate Dendritic Cell Pyroptosis and Activate Innate and Adaptive Immunity. Chem. Sci. 2022, 13, 8507–8517. [Google Scholar] [CrossRef]
- Zhang, W.; Cao, S.; Liang, S.; Tan, C.H.; Luo, B.; Xu, X.; Saw, P.E. Differently Charged Super-Paramagnetic Iron Oxide Nanoparticles Preferentially Induced M1-Like Phenotype of Macrophages. Front. Bioeng. Biotechnol. 2020, 8, 537. [Google Scholar] [CrossRef]
- Tan, S.; Li, D.; Zhu, X. Cancer Immunotherapy: Pros, Cons and Beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef]
- Sanità, G.; Carrese, B.; Lamberti, A. Nanoparticle Surface Functionalization: How to Improve Biocompatibility and Cellular Internalization. Front. Mol. Biosci. 2020, 7, 587012. [Google Scholar] [CrossRef]
- Cao, Z.; Li, W.; Liu, R.; Li, X.; Li, H.; Liu, L.; Chen, Y.; Lv, C.; Liu, Y. PH- and Enzyme-Triggered Drug Release as an Important Process in the Design of Anti-Tumor Drug Delivery Systems. Biomed. Pharmacother. 2019, 118, 109340. [Google Scholar] [CrossRef]
- Zhang, N.; Xiong, G.; Liu, Z. Toxicity of Metal-Based Nanoparticles: Challenges in the Nano Era. Front. Bioeng. Biotechnol. 2022, 10, 1001572. [Google Scholar] [CrossRef]
- Thapa, R.K.; Kim, J.O. Nanomedicine-Based Commercial Formulations: Current Developments and Future Prospects. J. Pharm. Investig. 2023, 53, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suliman, I.H.; Kim, K.; Chen, W.; Kim, Y.; Moon, J.-H.; Son, S.; Nam, J. Metal-Based Nanoparticles for Cancer Metalloimmunotherapy. Pharmaceutics 2023, 15, 2003. https://doi.org/10.3390/pharmaceutics15072003
Suliman IH, Kim K, Chen W, Kim Y, Moon J-H, Son S, Nam J. Metal-Based Nanoparticles for Cancer Metalloimmunotherapy. Pharmaceutics. 2023; 15(7):2003. https://doi.org/10.3390/pharmaceutics15072003
Chicago/Turabian StyleSuliman, Ivan Hardianto, Kidong Kim, Weihsuan Chen, Yubin Kim, Jeong-Hyun Moon, Sejin Son, and Jutaek Nam. 2023. "Metal-Based Nanoparticles for Cancer Metalloimmunotherapy" Pharmaceutics 15, no. 7: 2003. https://doi.org/10.3390/pharmaceutics15072003
APA StyleSuliman, I. H., Kim, K., Chen, W., Kim, Y., Moon, J.-H., Son, S., & Nam, J. (2023). Metal-Based Nanoparticles for Cancer Metalloimmunotherapy. Pharmaceutics, 15(7), 2003. https://doi.org/10.3390/pharmaceutics15072003