
Pharmacological Treatments and Therapeutic Drug Monitoring in Patients with Chronic Pain
 , , , , and
, , , , and
Abstract
:1. Pain Definition, Classification and Action Mechanism
- Nociceptive: pain generated by tissue damage following an injurious event [32].
- Neuropathic: pain caused by damage or dysfunction of the peripheral or central nervous system [33].
- Nociplastic: pain induced by activation of nociception in the absence of damage tissue, real or potential [34].
- Mixed: pain presenting a complex overlap of components previously described in any combination [35].
2. The Pharmacological Approach
2.1. Adjuvant Drugs
2.2. Non-Narcotic Analgesics
2.2.1. Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
2.2.2. Paracetamol

{kind=link}
{kind=link}
{kind=link}
| Basic Drug | Use | Mechanism of Action |
|---|---|---|
| Acetylsalicylic acid (ASA) | It relieves mild to moderate acute pain [82]. | Non selective non-steroidal anti-inflammatory drugs (nsNSAIDs) [71]. |
| Ibuprofen | It is preferable as a drug of first choice to provide relief from musculoskeletal pain in children. It is used in several clinical conditions, such as dysmenorrhea, dental pain, headache and migraine, soft tissue pain, and fever [83]. | Non selective non-steroidal anti-inflammatory drugs (nsNSAIDs) [71]. |
| Naproxen | It is used for post operative pain/acute pain [84]. It is the first-line treatment for acute gouty arthritis, osteoarthritis, musculoskeletal pain, inflammation, and dysmenorrhea [85]. | Non selective non-steroidal anti-inflammatory drugs (nsNSAIDs) [71]. |
| Celecoxib | It is administered before surgery because it decreases the post operative pain intensity of arthroscopy [86]. It seems to have a superior efficacy compared with paracetamol in chronic nonspecific lower back pain [87]. | Selective cyclo-oxygenase 2 NSAIDs (COXIBs) [71]. |
| Paracetamol | It provides pain relief in chronic osteoarthritic pain and lower back pain [77]. Furthermore, it may be used in combination with opioids for cancer pain [77]. It is the first-line treatment for the majority of mild to moderate acute pains [88]. Paracetamol is also effective for acute renal colic pain [81,89,90,91]. | Partial Selective cyclo-oxygenase 2 NSAIDs (COXIBs) [71]. |
2.3. Opioids
2.3.1. Opioids for Mild to Moderate Pain
2.3.2. Opioids for Moderate to Strong/Severe Pain
| Basic Drug | Use | Mechanism of Action |
|---|---|---|
| Codeine | It is used for mild to moderate pain in the treatment of acute and chronic noncancer pain [103]. The combination paracetamol/codeine may be used to treat postoperative pain, osteoarthritis related pain, cancer pain and polytrauma pain [105]. | Weak affinity to µ receptors [93,103]. |
| Tramadol | It is used for mild to moderate pain alone or in combination with nonopioid analgesic drugs. Several studies show the efficacy for the treatment of lower back pain, neuropathic pain, pain related to osteoarthritis and rheumatoid arthritis, acute and postoperative pain [107,108,109,110,111,112,113,114,115]. | Weak affinity to µ receptors, but it binds to monoaminergic, serotonergic receptors and ion channel receptors (muscarinic, nicotinic and K+ channels) [106]. It inhibits NE and 5-HT reuptake and it reduces the levels of PGE2 and TNF-α [106]. |
| Morphine | It can be administered through different routes of administration: intravenous, intramuscular, subcutaneous, oral, rectal, epidural and intrathecal [121]. It is used for moderate to severe pain in the treatment of acute and chronic noncancer pain and cancer pain [39,40,41,42,122,123]. | High affinity to the µ receptor, while the binding is weaker than the δ and κ receptors [117]. |
| Buprenorphine | It is used in opioid addiction [135,136,137,138]. The oral forms are used to treat BTP [139]. The subdermal or subcutaneous implant, intravenous or intramuscular injections, and transdermal patches are used for the treatment of chronic noncancer pain and cancer pain [135]. Transdermal buprenorphine is not approved for children, while the parenteral form is frequently used in the perioperative setting [141]. | It is a weak κ receptor antagonist and δ receptor agonist [135] and it is a partial (or low efficacy) agonist of the µ receptor [136]. |
| Fentanyl | The patch is available for the management of chronic noncancer pain and chronic cancer pain [145]. Rapid onset transmucosal fentanyl preparations have been developed for BTP [145], while intravenous formulation is widely used for anesthesia and analgesia, often in operating rooms and intensive care units [143,145]. | It has lower affinity for δ and κ opioid receptors, but it has high affinity for the µ receptor of which it is an agonist [137,143]. |
| Oxycodone | It is widely used in clinical practice to control postoperative pain, neuropathic pain and cancer pain [148,149]. Oxycodone is mainly used in the form of controlled-release tablets for chronic pain, whereas the immediate-release solution and tablets are used for acute pain or for BTP [146]. It is also available for intravenous, intramuscular, intranasal, subcutaneous and rectal routes, which are good alternatives when opioids cannot be administered orally [146,148]. The oral combination oxycodone–paracetamol has shown an adequate analgesia management for moderate–severe cancer pain [150,151]. | It is an agonist of μ opioid receptor and it also binds the δ and κ-opioid receptors [146,147]. |
| Tapentadol | Unlike conventional opioids, it has shown a favorable long-term safety profile in studies evaluating specific adverse events such as seizures, gastrointestinal events, hypertension, pulmonary dysfunction, serotonin syndrome, and endocrine toxicity [152]. It is a drug that has recently been used in chronic therapies for both cancer and non-cancer pain, for different age groups, such as the elderly and children [152,155,156]. It is administered orally in immediate-release and extended-release formulations and is used for the treatment of chronic neuropathic and mixed pain [152,155,156]. It is often used in combination with anticonvulsant drugs (e.g., gabapentin or pregabalin) to treat severe and mixed neuropathic pain [155,156]. | It is an agonist of µ-opioid receptor, but it is also a strong NE reuptake inhibitor and a weak 5-HT reuptake inhibitor [154,155]. |
| Drug | Volume of Distribution | Protein Binding | Clearance | Log P | Binding Affinity for Opioid Receptor (Ki) (Median) |
|---|---|---|---|---|---|
| Morphine | 2.1–4.0 L/kg [157]. | 35%; 10% for M3G and 15% for M6G [120]. | 1600 mL/min (intravenous or subcutaneous) [158]. | 0.9 [159]. | µ = 14 nM, ĸ = 47 nM, δ = 140 nM [117]. |
| Oxycodone | 2.6 L/kg [160]. | 45%, primarily serum albumin and, to a lesser extent, α1 acid glycoprotein. [161]. | 1400 mL/min [162]. | 0.7 [163]. | μ = 18 ± 4 nM, δ = 958 ± 499 nM, κ = 677 ± 326 nM [146]. |
| Buprenorphine | 188–335 L [164]. | 96%, primarily to α- and β-globulin. [164]. | 1042–1280 mL/min [164]. | 4.5 [163]. | μ = 0.2157 nM [165]. |
| Fentanyl | 4 L/kg [166]. | 80–85%. It is unclear whether fentanyl binds primarily to albumin (ALB) or α1 acid glycoprotein (AAG) [167]. | 500–1200 mL/min [168]. | 3.8 [163]. | Μ = 1.35 nM [165]. |
| Tapentadol | 540 ± 98 L [169]. | 20% [169]. | 1530 ± 177 mL/min [170]. | 2.87 [171]. | μ = 160 nM [172]. |
2.3.3. Pharmacogenomics of Opioids
| Phenotypes | Activity Score Range a | Exemples of CYP2D6 Diplotypes | Implications | Recommendations |
|---|---|---|---|---|
| CYP2D6 ultrarapid metabolizer | >2.25 | *1/*1 × N, *1/*2 × N, *2/*2 × N | Increased formation of morphine leading to higher risk of toxicity | Avoid codeine use because of potential for serious toxicity. If opioid use is warranted, consider a non-tramadol opioid. |
| CYP2D6 normal metabolizer | 1.25 ≤ × ≤ 2.25 | *1/*10 *1/*41, *1/*9 *10/*41 × 3 *1/*1, *1/*2 *2×2/*10 | Expected morphine Formation. | Use codeine label recommended age-specific or weight-specific dosing. |
| CYP2D6 intermediate metabolizer | 0 < × < 1.25 | *4/*10 *4/*41, *10/*10 *10/*41 *41/*41, *1/*5 | Reduced morphine Formation. | Use codeine label recommended age-specific or weight-specific dosing. If no response and opioid use is warranted, consider a non-tramadol opioid. |
| CYP2D6 poor metabolizer | 0 | *3/*4, *4/*4, *5/*5, *5/*6 | Greatly reduced morphine formation leading to diminished analgesia | Avoid codeine use due to the possibility of diminished analgesia. If opioid use is warranted, consider a non-tramadol opioid. |
| Phenotypes | Activity Score Range a | Examples of CYP2D6 Diplotypes | Implications | Recommendations |
|---|---|---|---|---|
| CYP2D6 ultrarapid metabolizer | >2.25 | *1/*1 × N, *1/*2 × N, *2/*2 × N | Increased formation of O-desmethyltramadol (active metabolite) leading to higher risk of toxicity | Avoid tramadol use because of potential for toxicity. If opioid use is warranted, consider a non-codeine opioid. |
| CYP2D6 normal metabolizer | 1.25 ≤ × ≤ 2.25 | *1/*10 *1/*41, *1/*9 *10/*41 × 3 *1/*1, *1/*2 *2 × 2/*10 | Expected O-desmethyltramadol (active metabolite) formation | Use tramadol label recommended age-specific or weight-specific dosing. |
| CYP2D6 intermediate metabolizer | 0 < × < 1.25 | *4/*10 *4/*41, *10/*10 *10/*41 *41/*41, *1/*5 | Reduced O-desmethyltramadol (active metabolite) formation | Use tramadol label recommended age-specific or weight-specific dosing. If no response and opioid use is warranted, consider non-codeine opioid. |
| CYP2D6 poor metabolizer | 0 | *3/*4, *4/*4, *5/*5, *5/*6 | Greatly reduced O-desmethyltramadol (active metabolite) formation leading to diminished analgesia | Avoid tramadol use because of possibility of diminished analgesia. If opioid use is warranted, consider a non-codeine opioid. |
3. Complications Related to Prolonged Treatment
4. Therapeutic Drug Monitoring of Opioids, Challenges and Potentials
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ilana, E. Pain terms: A list with definitions and notes on usage. Pain 1979, 6, 249. [Google Scholar]
- Editorial The need of a taxonomy. Pain 1979, 6, 247–252. [CrossRef] [PubMed]
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Alcock, M.M. Defining pain: Past, present, and future. Pain 2017, 158, 761–762. [Google Scholar] [CrossRef]
- Aydede, M. Defending the IASP Definition of Pain. Monist 2017, 100, 439–464. [Google Scholar] [CrossRef] [Green Version]
- Aydede, M. Does the IASP definition of pain need updating? Pain Rep. 2019, 4, e777. [Google Scholar] [CrossRef]
- Craig, K.D.; de C Williams, A.C. Reply. Pain 2017, 158, 363–365. [Google Scholar] [CrossRef]
- Cunningham, N. Comments on Derbyshire. Pain 1998, 74, 102–106. [Google Scholar] [CrossRef]
- Osborn, M. Situating pain in a more helpful place. Pain Rep. 2018, 3, e642. [Google Scholar] [CrossRef]
- Tesarz, J.; Eich, W. A conceptual framework for “updating the definition of pain”. Pain 2017, 158, 1177–1178. [Google Scholar] [CrossRef]
- Treede, R.D. The International Association for the Study of Pain definition of pain: As valid in 2018 as in 1979, but in need of regularly updated footnotes. Pain Rep. 2018, 3, e643. [Google Scholar] [CrossRef] [PubMed]
- Wright, A. A Criticism of the IASPs Definition of Pain. J. Conscious. Stud. 2011, 18, 9–10. [Google Scholar]
- Wright, A.; Aydede, M. Critical comments on Williams and Craig’s recent proposal for revising the definition of pain. Pain 2017, 158, 362–363. [Google Scholar] [CrossRef]
- Anand, K.J.S.; Rovnaghi, C.; Walden, M.; Churchill, J. Consciousness, behavior, and clinical impact of the definition of pain. Pain Forum 1999, 8, 64–73. [Google Scholar] [CrossRef]
- Cunningham, N. Primary requirements for an ethical definition of pain. Pain Forum 1999, 8, 93–99. [Google Scholar] [CrossRef]
- Williams, A.C.C.; Craig, K.D. Updating the definition of pain. Pain 2016, 157, 2420–2423. [Google Scholar] [CrossRef]
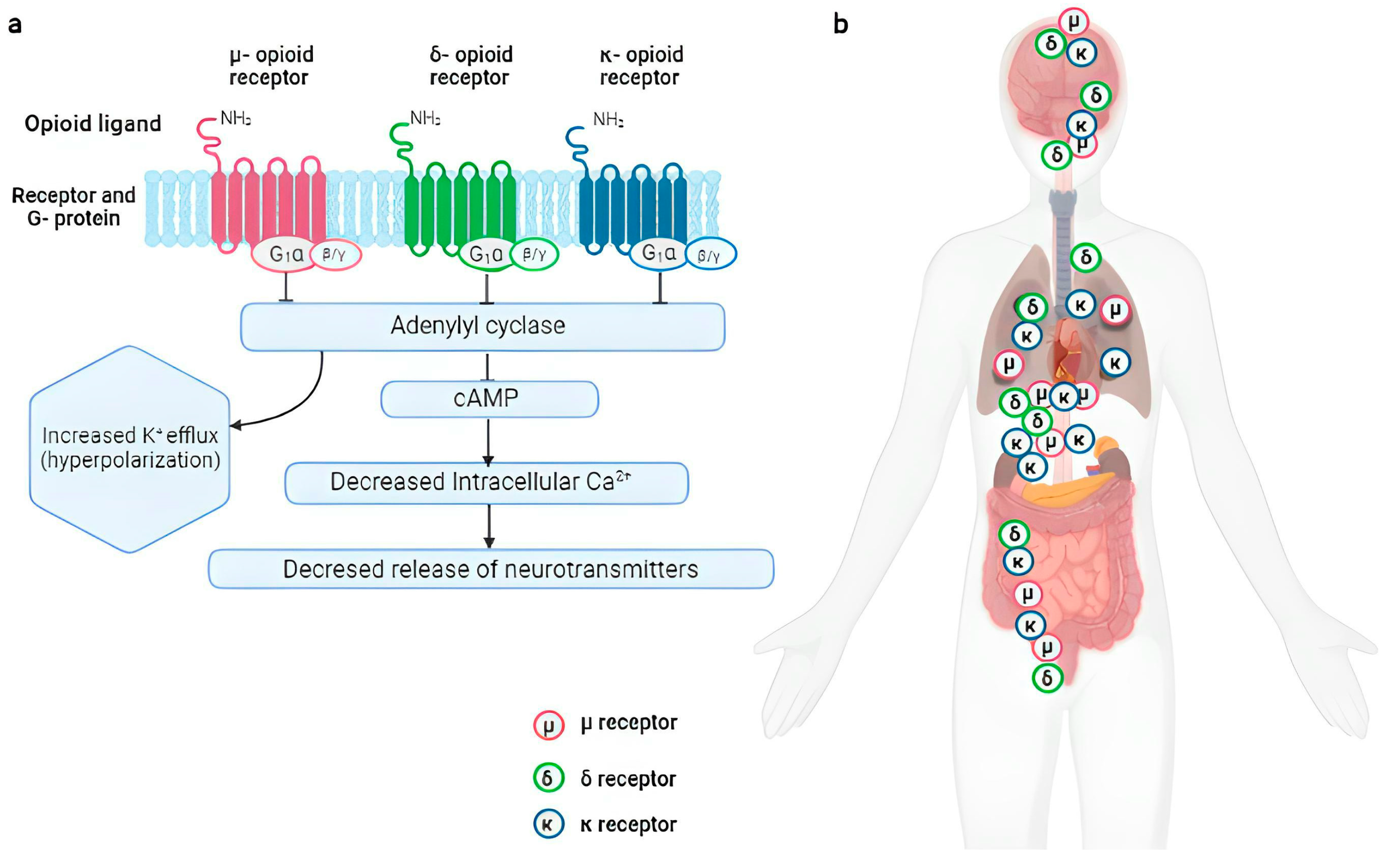
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toubia, T.; Khalife, T. The Endogenous Opioid System: Role and Dysfunction Caused by Opioid Therapy. Clin. Obstet. Gynecol. 2019, 62, 3–10. [Google Scholar] [CrossRef]
- Dhaliwal, A.; Gupta, M. Physiology, Opioid Receptor. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Connor, M.; Christie, M.D. Opioid receptor signalling mechanisms. Clin. Exp. Pharmacol. Physiol. 1999, 26, 493–499. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedema, H.P.; Grace, A.A. Corticotropin-releasing hormone directly activates noradrenergic neurons of the locus ceruleus recorded in vitro. J. Neurosci. 2004, 24, 9703–9713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, A.D.; Holt, C.B.; Downes, T.J.; Ruggeri, E.; Del Vecchio, S.; De Giorgio, R. Pathophysiology, diagnosis, and management of opioid-induced constipation. Lancet Gastroenterol. Hepatol. 2018, 3, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Hang, A.; Wang, Y.J.; He, L.; Liu, J.G. The role of the dynorphin/κ opioid receptor system in anxiety. Acta Pharmacol. Sin. 2015, 36, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Kiyatkin, E.A. Respiratory depression and brain hypoxia induced by opioid drugs: Morphine, oxycodone, heroin, and fentanyl. Neuropharmacology 2019, 151, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarski, P.; Karuga, F.F.; Szmyd, B.; Sochal, M.; Białasiewicz, P.; Strzelecki, D.; Gabryelska, A. The Role of Inflammation, Hypoxia, and Opioid Receptor Expression in Pain Modulation in Patients Suffering from Obstructive Sleep Apnea. Int. J. Mol. Sci. 2022, 23, 9080. [Google Scholar] [CrossRef]
- Leppert, W. The impact of opioid analgesics on the gastrointestinal tract function and the current management possibilities. Contemp. Oncol. 2012, 16, 125–131. [Google Scholar] [CrossRef]
- Chen, A.; Ashburn, M.A. Cardiac Effects of Opioid Therapy. Pain Med. 2015, 16, S27–S31. [Google Scholar] [CrossRef] [Green Version]
- Tyan, P.; Carey, E.T. Physiological Response to Opioids. Clin. Obstet. Gynecol. 2019, 62, 11–21. [Google Scholar] [CrossRef]
- Roy, S.; Loh, H.H. Effects of opioids on the immune system. Neurochem. Res. 1996, 21, 1375–1386. [Google Scholar] [CrossRef]
- Salute.gov.it. Available online: https://www.salute.gov.it/imgs/C_17_pubblicazioni_1257_allegato.pdf (accessed on 1 March 2023).
- Dunne, F.J.; Getachew, H.; Cullenbrooke, F.; Dunne, C. Pain and pain syndromes. Br. J. Hosp. Med. 2018, 79, 449–453. [Google Scholar] [CrossRef]
- Bouhassira, D. Neuropathic pain: Definition, assessment and epidemiology. Rev. Neurol. 2019, 175, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Fitzcharles, M.A.; Cohen, S.P.; Clauw, D.J.; Littlejohn, G.; Usui, C.; Häuser, W. Nociplastic pain: Towards an understanding of prevalent pain conditions. Lancet 2021, 397, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Freynhagen, R.; Parada, H.A.; Calderon-Ospina, C.A.; Chen, J.; Rakhmawati Emril, D.; Fernández-Villacorta, F.J.; Franco, H.; Ho, K.Y.; Lara-Solares, A.; Li, C.C.; et al. Current understanding of the mixed pain concept: A brief narrative review. Curr. Med. Res. Opin. 2019, 35, 1011–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjermstad, M.J.; Gibbins, J.; Haugen, D.F.; Caraceni, A.; Loge, J.H.; Kaasa, S.; EPCRC, European Palliative Care Research Collaborative. Pain assessment tools in palliative care: An urgent need for consensus. Palliat. Med. 2008, 22, 895–903. [Google Scholar] [CrossRef]
- Turk, D.C.; Dworkin, R.H.; Allen, R.R.; Bellamy, N.; Brandenburg, N.; Carr, D.B.; Cleeland, C.; Dionne, R.; Farrar, J.T.; Galer, B.S.; et al. Core outcome domains for chronic pain clinical trials: IMMPACT recommendations. Pain 2003, 106, 337–345. [Google Scholar] [CrossRef]
- Thong, I.S.K.; Jensen, M.P.; Miró, J.; Tan, G. The validity of pain intensity measures: What do the NRS, VAS, VRS, and FPS-R measure? Scand. J. Pain 2018, 18, 99–107. [Google Scholar] [CrossRef]
- Ventafridda, V.; Saita, L.; Ripamonti, C.; De Conno, F. WHO guidelines for the use of analgesics in cancer pain. Int. J. Tissue React. 1985, 7, 93–96. [Google Scholar]
- World Health Organization. Cancer Pain Relief, Second Edition, with a Guide to Opioid Availability, 2nd ed.; World Health Organization: Geneva, Switzerland, 1996. [Google Scholar]
- WHO Guidelines for the Pharmacological and Radiotherapeutic Management of Cancer Pain in Adults and Adolescents; World Health Organization: Geneva, Switzerland, 2018.
- National Collaborating Centre for Cancer. Opioids in Palliative Care: Safe and Effective Prescribing of Strong Opioids for Pain in Palliative Care of Adults; National Collaborating Centre for Cancer: Cardiff, UK, 2012. [Google Scholar]
- PDQ Supportive and Palliative Care Editorial Board. Cancer Pain (PDQ®): Health Professional Version. In PDQ Cancer Information Summaries [Internet]; National Cancer Institute: Bethesda, MD, USA, 2002. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Neufeld, N.J.; Elnahal, S.M.; Alvarez, R.H. Cancer pain: A review of epidemiology, clinical quality and value impact. Future Oncol. 2017, 13, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Hylands-White, N.; Duarte, R.V.; Raphael, J.H. An overview of treatment approaches for chronic pain management. Rheumatol. Int. 2017, 37, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwan, J.; Sclafani, J.; Tawfik, V.L. Chronic Pain Management in the Elderly. Anesthesiol. Clin. 2019, 37, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Gai, N.; Naser, B.; Hanley, J.; Peliowski, A.; Hayes, J.; Aoyama, K. A practical guide to acute pain management in children. J. Anesth. 2020, 34, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.N.; Elsner, F.; Filbet, M.J.; Porta-Sales, J.; Ripamonti, C.; Santini, D.; Webber, K. Breakthrough cancer pain (BTcP) management: A review of international and national guidelines. BMJ Support. Palliat. Care 2018, 8, 241–249. [Google Scholar] [CrossRef]
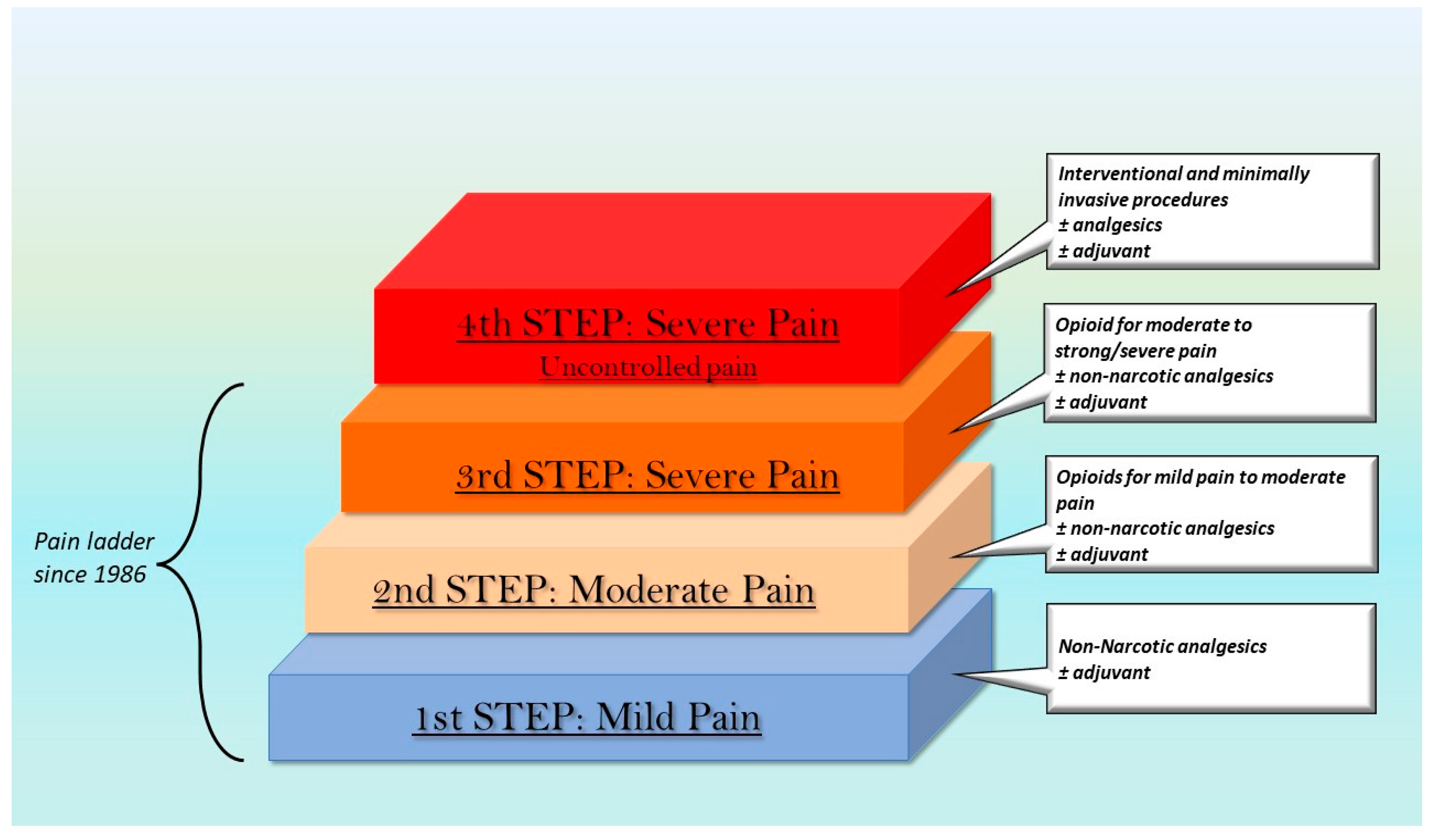
- Anekar, A.A.; Cascella, M. WHO Analgesic Ladder. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Araujo, A.M.; Gómez, M.; Pascual, J.; Castañeda, M.; Pezonaga, L.; Borque, J.L. Tratamiento del dolor en el paciente oncológico [Treatment of pain in the oncology patient]. An. Sist. Sanit. Navar. 2004, 3, 63–75. [Google Scholar]
- Di Napoli, R.; Esposito, G.; Cascella, M. Intrathecal Catheter. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK549790/ (accessed on 6 February 2023).
- Cascella, M.; Muzio, M.R.; Viscardi, D.; Cuomo, A. Features and Role of Minimally Invasive Palliative Procedures for Pain Management in Malignant Pelvic Diseases: A Review. Am. J. Hosp. Palliat. Care 2017, 34, 524–531. [Google Scholar] [CrossRef]
- Kanpolat, Y. Percutaneous destructive pain procedures on the upper spinal cord and brain stem in cancer pain: CT-guided techniques, indications and results. Adv. Tech. Stand. Neurosurg. 2007, 32, 147–173. [Google Scholar] [CrossRef]
- Cahana, A.; Mavrocordatos, P.; Geurts, J.W.; Groen, G.J. Do minimally invasive procedures have a place in the treatment of chronic lower back pain? Expert Rev. Neurother. 2004, 4, 479–490. [Google Scholar] [CrossRef]
- Raffa, R.B.; Pergolizzi, J.V., Jr. A modern analgesics pain ‘pyramid’. J. Clin. Pharm. Ther. 2014, 39, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Fillingim, R.B. Individual differences in pain: Understanding the mosaic that makes pain personal. Pain 2017, 158, S11–S18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, E.; Oka, K.; Koshikawa, F. Dorsolateral prefrontal cortex sensing analgesia. Biophys. Physicobiol. 2022, 19, e190014. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J. What is this thing called pain? J. Clin. Investig. 2010, 120, 3742–3744. [Google Scholar] [CrossRef] [PubMed]
- Dale, R.; Stacey, B. Multimodal Treatment of Chronic Pain. Med. Clin. N. Am. 2016, 100, 55–64. [Google Scholar] [CrossRef]
- Dailey, D.L.; Vance, C.G.T.; Rakel, B.A.; Zimmerman, M.B.; Embree, J.; Merriwether, E.N.; Geasland, K.M.; Chimenti, R.; Williams, J.M.; Golchha, M.; et al. Transcutaneous Electrical Nerve Stimulation Reduces Movement-Evoked Pain and Fatigue: A Randomized, Controlled Trial. Arthritis Rheumatol. 2020, 72, 824–836. [Google Scholar] [CrossRef] [Green Version]
- Mitra, R.; Jones, S. Adjuvant analgesics in cancer pain: A review. Am. J. Hosp. Palliat. Care 2012, 29, 70–79. [Google Scholar] [CrossRef]
- Meserve, J.R.; Kaye, A.D.; Prabhakar, A.; Urman, R.D. The role of analgesics in cancer propagation. Best Pract. Res. Clin. Anaesthesiol. 2014, 28, 139–151. [Google Scholar] [CrossRef]
- Lucchinetti, E.; Awad, A.E.; Rahman, M.; Feng, J.; Lou, P.H.; Ionescu, L.; Lemieux, H.; Thèbaud, B.; Zaugg, M. Antiproliferative effects of local anesthetics on mesenchymal stem cells: Potential implications for tumor spreading and wound healing. Anesthesiology 2012, 116, 841–856. [Google Scholar] [CrossRef] [Green Version]
- Romero-Sandoval, E.A.; Kolano, A.L.; Alvarado-Vázquez, P.A. Cannabis and Cannabinoids for Chronic Pain. Curr. Rheumatol. Rep. 2017, 19, 67. [Google Scholar] [CrossRef]
- Meng, H.; Dai, T.; Hanlon, J.G.; Downar, J.; Alibhai, S.M.H.; Clarke, H. Cannabis and cannabinoids in cancer pain management. Curr. Opin. Support. Palliat. Care 2020, 14, 87–93. [Google Scholar] [CrossRef] [PubMed]
- International Association for the Study of Pain. IASP Position Statement on the Use of Cannabinoids to Treat Pain. Available online: https://www.iasp-pain.org/publications/iasp-news/iasp-position-statement-on-the-use-of-cannabinoids-to-treat-pain/ (accessed on 12 January 2023).
- Bacchi, S.; Palumbo, P.; Sponta, A.; Coppolino, M.F. Clinical pharmacology of non-steroidal anti-inflammatory drugs: A review. Antiinflamm. Anti-Allergy Agents Med. Chem. 2012, 11, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.C.; Zhang, J.L.; Ge, C.T.; Yu, Y.Y.; Wang, P.; Yuan, T.F.; Fu, C.Y. Advances in cancer pain from bone metastasis. Drug Des. Devel. Ther. 2015, 9, 4239–4245. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Bah, M. NSAIDs in the Treatment of Postoperative Pain. Curr. Pain Headache Rep. 2016, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- da Costa, B.R.; Reichenbach, S.; Keller, N.; Nartey, L.; Wandel, S.; Juni, P.; Trelle, S. Effectiveness of non-steroidal anti-inflammatory drugs for the treatment of pain in knee and hip osteoarthritis:a network meta-analysis. Lancet 2017, 390, e21–33. [Google Scholar] [CrossRef] [PubMed]
- Eccleston, C.; Cooper, T.E.; Fisher, E.; Anderson, B.; Wilkinson, N.M. Non-steroidal anti-inflammatory drugs (NSAIDs) for chronic non-cancer pain in children and adolescents. Cochrane Database Syst. Rev. 2017, 8, CD012537. [Google Scholar] [CrossRef]
- Vardy, J.; Agar, M. Nonopioid drugs in the treatment of cancer pain. J. Clin. Oncol. 2014, 32, 1677–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, G.G.; Davies, M.J.; Day, R.O.; Mohamudally, A.; Scott, K.F. The modern pharmacology of paracetamol: Therapeutic actions, mechanism of action, metabolism, toxicity and recent pharmacological findings. Inflammopharmacology 2013, 21, 201–232. [Google Scholar] [CrossRef]
- Toda, K. Is acetaminophen safe in pregnancy? Scand. J. Pain 2017, 17, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Bloukh, S.; Wazaify, M.; Matheson, C. Paracetamol: Unconventional uses of a well-known drug. Int. J. Pharm. Pract. 2021, 29, 527–540. [Google Scholar] [CrossRef]
- Brune, K.; Renner, B.; Tiegs, G. Acetaminophen/paracetamol: A history of errors, failures and false decisions. Eur. J. Pain 2015, 19, 953–965. [Google Scholar] [CrossRef]
- Saragiotto, B.T.; Abdel Shaheed, C.; Maher, C.G. Paracetamol for pain in adults. BMJ 2019, 367, l6693. [Google Scholar] [CrossRef] [PubMed]
- Derry, S.; Moore, R.A. Single dose oral aspirin for acute postoperative pain in adults. Cochrane Database Syst. Rev. 2012, 4, CD002067. [Google Scholar] [CrossRef]
- Parri, N.; Lazzeri, S. Efficacy of ibuprofen in musculoskeletal post-traumatic pain in children: A systematic review. PLoS ONE 2020, 15, e0243314. [Google Scholar] [CrossRef]
- Weisman, S. Naproxen for Post-Operative Pain. J. Pharm. Pharm. Sci. 2021, 24, 62–70. [Google Scholar] [CrossRef]
- Brutzkus, J.C.; Shahrokhi, M.; Varacallo, M. Naproxen. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Wan, R.; Li, P.; Jiang, H. The efficacy of celecoxib for pain management of arthroscopy: A meta-analysis of randomized controlled trials. Medicine 2019, 98, e17808. [Google Scholar] [CrossRef]
- Bedaiwi, M.K.; Sari, I.; Wallis, D.; O’shea, F.D.; Salonen, D.; Haroon, N.; Omar, A.; Inman, R.D. Clinical Efficacy of Celecoxib Compared to Acetaminophen in Chronic Nonspecific Lower back Pain: Results of a Randomized Controlled Trial. Arthritis Care Res. 2016, 68, 845–852. [Google Scholar] [CrossRef] [Green Version]
- Alchin, J.; Dhar, A.; Siddiqui, K.; Christo, P.J. Why paracetamol (acetaminophen) is a suitable first choice for treating mild to moderate acute pain in adults with liver, kidney or cardiovascular disease, gastrointestinal disorders, asthma, or who are older. Curr. Med. Res. Opin. 2022, 38, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Chou, D.; Abalos, E.; Gyte, G.M.; Gülmezoglu, A.M. Paracetamol/acetaminophen (single administration) for perineal pain in the early postpartum period. Cochrane Database Syst. Rev. 2013, 1, CD008407. [Google Scholar] [CrossRef]
- García-Perdomo, H.A.; Echeverría-García, F.; López, H.; Fernández, N.; Manzano-Nunez, R. Pharmacologic interventions to treat renal colic pain in acute stone episodes: Systematic review and meta-analysis. Prog. Urol. 2017, 27, 654–665. [Google Scholar] [CrossRef]
- Wiffen, P.J.; Derry, S.; Moore, R.A. Tramadol with or without paracetamol (acetaminophen) for cancer pain. Cochrane Database Syst. Rev. 2017, 5, CD012508. [Google Scholar] [CrossRef]
- Nafziger, A.N.; Barkin, R.L. Opioid Therapy in Acute and Chronic Pain. J. Clin. Pharmacol. 2018, 58, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Trescot, A.M.; Datta, S.; Lee, M.; Hansen, H. Opioid pharmacology. Pain Physician 2008, 11, S133–S153. [Google Scholar] [CrossRef]
- Childers, S.R.; Fleming, L.; Konkoy, C.; Marckel, D.; Pacheco, M.; Sexton, T.; Ward, S. Opioid and cannabinoid receptor inhibition of adenylyl cyclase in brain. Ann. N. Y. Acad. Sci. 1992, 654, 33–51. [Google Scholar] [CrossRef]
- McCleane, G.; Smith, H. Opioids for persistent noncancer pain. Med. Clin. N. Am. 2007, 91, 177–197. [Google Scholar] [CrossRef]
- Stein, C. Opioid Receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, S.; Lu, X.; Tao, F. Role of Descending Dopaminergic Pathways in Pain Modulation. Curr. Neuropharmacol. 2019, 17, 1176–1182. [Google Scholar] [CrossRef]
- Mesgarpour, B.; Griebler, U.; Glechner, A.; Kien, C.; Strobelberger, M.; Van Noord, M.G.; Michalek-Sauberer, A. Extended-release opioids in the management of cancer pain: A systematic review of efficacy and safety. Eur. J. Pain 2014, 18, 605–616. [Google Scholar] [CrossRef]
- Drewes, A.M.; Jensen, R.D.; Nielsen, L.M.; Droney, J.; Christrup, L.L.; Arendt-Nielsen, L.; Riley, J.; Dahan, A. Differences between opioids: Pharmacological, experimental, clinical and economical perspectives. Br. J. Clin. Pharmacol. 2013, 75, 60–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.M.; Christie, M.J. Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br. J. Pharmacol. 2011, 164, 1322–1334. [Google Scholar] [CrossRef] [Green Version]
- Mercadante, S.; Arcuri, E.; Santoni, A. Opioid-Induced Tolerance and Hyperalgesia. CNS Drugs 2019, 33, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Leppert, W.; Majkowicz, M. The impact of tramadol and dihydrocodeine treatment on quality of life of patients with cancer pain. Int. J. Clin. Pract. 2010, 64, 1681–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, L.E.; Madadi, P. Is there a role for therapeutic drug monitoring with codeine? Ther. Drug Monit. 2012, 34, 249–256. [Google Scholar] [CrossRef]
- Peechakara, B.V.; Tharp, J.G.; Gupta, M. Codeine. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Franceschi, F.; Iacomini, P.; Marsiliani, D.; Cordischi, C.; Antonini, E.F.; Alesi, A.; Giacobelli, D.; Zuccalà, G. Safety and efficacy of the combination acetaminophen-codeine in the treatment of pain of different origin. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2129–2135. [Google Scholar] [PubMed]
- Bvaarakat, A. Revisiting Tramadol: A Multi-Modal Agent for Pain Management. CNS Drugs 2019, 33, 481–501. [Google Scholar] [CrossRef]
- Grond, S.; Sablotzki, A. Clinical pharmacology of tramadol. Clin. Pharmacokinet. 2004, 43, 879–923. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Martucci, C.; Ferrario, P.; Franchi, S.; Sacerdote, P. Increased tumor necrosis factor-alpha and prostaglandin E2 concentrations in the cerebrospinal fluid of rats with inflammatory hyperalgesia: The effects of analgesic drugs. Anesth. Analg. 2007, 104, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Beakley, B.D.; Kaye, A.M.; Kaye, A.D. Tramadol, Pharmacology, Side Effects, and Serotonin Syndrome: A Review. Pain Physician 2015, 18, 395–400. [Google Scholar]
- Vazzana, M.; Andreani, T.; Fangueiro, J.; Faggio, C.; Silva, C.; Santini, A.; Garcia, M.L.; Silva, A.M.; Souto, E.B. Tramadol hydrochloride: Pharmacokinetics, pharmacodynamics, adverse side effects, co-administration of drugs and new drug delivery systems. Biomed. Pharmacother. 2015, 70, 234–238. [Google Scholar] [CrossRef]
- Hollingshead, J.; Duehmke, R.M.; Cornblath, D.R. Tramadol for neuropathic pain. Cochrane Database Syst. Rev. 2006, 3, CD003726. [Google Scholar] [CrossRef]
- Cepeda, M.S.; Camargo, F.; Zea, C.; Valencia, L. Tramadol for osteoarthritis. Cochrane Database Syst. Rev. 2006, 3, CD005522. [Google Scholar] [CrossRef] [PubMed]
- Chaparro, L.E.; Furlan, A.D.; Deshpande, A.; Mailis-Gagnon, A.; Atlas, S.; Turk, D.C. Opioids compared to placebo or other treatments for chronic low-back pain. Cochrane Database Syst. Rev. 2013, 8, CD004959. [Google Scholar] [CrossRef] [Green Version]
- Sonis, J. Tramadol for acute pain: A review of the evidence. Am. Fam. Physician 2005, 72, 1964. [Google Scholar]
- Kubota, R.; Komiyama, T.; Miwa, Y.; Ide, T.; Toyoda, H.; Asanuma, F.; Yamada, Y. Pharmacokinetics and postoperative analgesia of epidural tramadol: A prospective, pilot study. Curr. Ther. Res. Clin. Exp. 2008, 69, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Brook, K.; Bennett, J.; Desai, S.P. The Chemical History of Morphine: An 8000-year Journey, from Resin to de-novo Synthesis. J. Anesth. Hist. 2017, 3, 50–55. [Google Scholar] [CrossRef]
- Sverrisdóttir, E.; Lund, T.M.; Olesen, A.E.; Drewes, A.M.; Christrup, L.L.; Kreilgaard, M. A review of morphine and morphine-6-glucuronide’s pharmacokinetic-pharmacodynamic relationships in experimental and clinical pain. Eur. J. Pharm. Sci. 2015, 74, 45–62. [Google Scholar] [CrossRef] [PubMed]
- De Gregori, S.; De Gregori, M.; Ranzani, G.N.; Allegri, M.; Minella, C.; Regazzi, M. Morphine metabolism, transport and brain disposition. Metab. Brain Dis. 2012, 27, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.T. Neuroexcitatory effects of morphine and hydromorphone: Evidence implicating the 3-glucuronide metabolites. Clin. Exp. Pharmacol. Physiol. 2000, 27, 524–528. [Google Scholar] [CrossRef]
- Klimas, R.; Mikus, G. Morphine-6-glucuronide is responsible for the analgesic effect after morphine administration: A quantitative review of morphine, morphine-6-glucuronide, and morphine-3-glucuronide. Br. J. Anaesth. 2014, 113, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Thigpen, J.C.; Odle, B.L.; Harirforoosh, S. Opioids: A Review of Pharmacokinetics and Pharmacodynamics in Neonates, Infants, and Children. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 591–609. [Google Scholar] [CrossRef]
- Deer, T.R.; Pope, J.E.; Hanes, M.C.; McDowell, G.C. Intrathecal Therapy for Chronic Pain: A Review of Morphine and Ziconotide as Firstline Options. Pain Med. 2019, 20, 784–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayek, S.M.; Hanes, M.C. Intrathecal therapy for chronic pain: Current trends and future needs. Curr. Pain Headache Rep. 2014, 18, 388. [Google Scholar] [CrossRef]
- Martinez-Velez, A.; Singh, P. Epidural Morphine. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK541073/ (accessed on 22 March 2023).
- Mugabure Bujedo, B. A clinical approach to neuraxial morphine for the treatment of postoperative pain. Pain Res. Treat. 2012, 2012, 612145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundeberg, S.; Hatava, P.; Lagerkranser, M.; Olsson, G.L. Perception of pain following rectal administration of morphine in children: A comparison of a gel and a solution. Paediatr. Anaesth. 2006, 16, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Campbell, W.I. Rectal controlled-release morphine: Plasma levels of morphine and its metabolites following the rectal administration of MST Continus 100 mg. J. Clin. Pharm. Ther. 1996, 21, 65–71. [Google Scholar] [CrossRef]
- Elsner, F.; Radbruch, L.; Loick, G.; Gaertner, J.; Sabatowski, R. Intravenous versus subcutaneous morphine titration in patients with persisting exacerbation of cancer pain. J. Palliat. Med. 2005, 8, 743–750. [Google Scholar] [CrossRef]
- Sun, Y.; Wei, H.; Yu, M.; Zheng, R.; Li, J.; Fu, Y.; Zheng, Y.; Zhang, X.; Shou, F.; Zhou, J.; et al. Rapid titration with oral sustained-release morphine plus subcutaneous morphine in a multi-center, randomized control study of cancer patients with moderate to severe cancer pain. Jpn. J. Clin. Oncol. 2022, 52, 1303–1310. [Google Scholar] [CrossRef]
- Cooper, T.E.; Chen, J.; Wiffen, P.J.; Derry, S.; Carr, D.B.; Aldington, D.; Cole, P.; Moore, R.A. Morphine for chronic neuropathic pain in adults. Cochrane Database Syst. Rev. 2017, 5, CD011669. [Google Scholar] [CrossRef]
- Bechakra, M.; Moerdijk, F.; van Rosmalen, J.; Koch, B.C.P.; van der Rijt, C.C.D.; Sillevis Smitt, P.A.E.; Jongen, J.L.M. Opioid responsiveness of nociceptive versus mixed pain in clinical cancer patients. Eur. J. Cancer 2018, 105, 79–87. [Google Scholar] [CrossRef]
- Preuss, C.V.; Kalava, A.; King, K.C. Prescription of Controlled Substances: Benefits and Risks. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Venkatraman, R.; Pushparani, A.; Balaji, R.; Nandhini, P. Comparison of low dose intravenous fentanyl and morphine infusion for postoperative analgesia in spine fusion surgeries—A randomized control trial. Braz. J. Anesthesiol. 2021, 71, 339–344. [Google Scholar] [CrossRef]
- Tuerxun, H.; Cui, J. The dual effect of morphine on tumor development. Clin. Transl. Oncol. 2019, 21, 695–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Viswanath, O.; Saadabadi, A. Buprenorphine. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Coe, M.A.; Lofwall, M.R.; Walsh, S.L. Buprenorphine Pharmacology Review: Update on Transmucosal and Long-acting Formulations. J. Addict. Med. 2019, 13, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Volpe, D.A.; Tobin, G.A.M.; Mellon, R.D.; Katki, A.G.; Parker, R.J.; Colatsky, T.; Kropp, T.J.; Verbois, S.L. Uniform assessment and ranking of opioid µ receptor binding constants for selected opioid drugs. Regul. Toxicol. Pharmacol. 2011, 59, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Shulman, M.; Wai, J.M.; Nunes, E.V. Buprenorphine Treatment for Opioid Use Disorder: An Overview. CNS Drugs 2019, 33, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.M.; Holtzman, M.; Kim, T.; Kharasch, E.D. Buprenorphine metabolites, buprenorphine-3-glucuronide and norbuprenorphine-3-glucuronide, are biologically active. Anesthesiology 2011, 115, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, A.S.; Mantha, S.S.P.; Kumar, K.P.; Rayani, B.K. Sublingual Buprenorphine: A Feasible Alternative for Treating Breakthrough Chronic Pain. Indian J. Palliat. Care 2019, 25, 595–596. [Google Scholar] [CrossRef]
- Davis, M.P.; Pasternak, G.; Behm, B. Treating Chronic Pain: An Overview of Clinical Studies Centered on the Buprenorphine Option. Drugs 2018, 78, 1211–1228. [Google Scholar] [CrossRef]
- Chen, K.Y.; Chen, L.; Mao, J. Buprenorphine-naloxone therapy in pain management. Anesthesiology 2014, 120, 1262–1274. [Google Scholar] [CrossRef] [Green Version]
- Comer, S.D.; Cahill, C.M. Fentanyl: Receptor pharmacology, abuse potential, and implications for treatment. Neurosci. Biobehav. Rev. 2019, 106, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Lötsch, J.; Walter, C.; Parnham, M.J.; Oertel, B.G.; Geisslinger, G. Pharmacokinetics of non-intravenous formulations of fentanyl. Clin. Pharmacokinet. 2013, 52, 23–36. [Google Scholar] [CrossRef]
- Stanley, T.H. The fentanyl story. J. Pain 2014, 15, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Kalso, E. Oxycodone. J. Pain Symptom Manag. 2005, 29, S47–S56. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, M.; Piirainen, P.; Kokki, H.; Lammi, P.; Kokki, M. Updated Clinical Pharmacokinetics and Pharmacodynamics of Oxycodone. Clin. Pharmacokinet. 2019, 58, 705–725. [Google Scholar] [CrossRef] [Green Version]
- Umukoro, N.N.; Aruldhas, B.W.; Rossos, R.; Pawale, D.; Renschler, J.S.; Sadhasivam, S. Pharmacogenomics of oxycodone: A narrative literature review. Pharmacogenomics 2021, 22, 275–290. [Google Scholar] [CrossRef]
- Pergolizzi, J.V., Jr.; Seow-Choen, F.; Wexner, S.D.; Zampogna, G.; Raffa, R.B.; Taylor, R., Jr. Perspectives on Intravenous Oxycodone for Control of Postoperative Pain. Pain Pract. 2016, 16, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Di, J.; Zhang, Y.; Xing, E. Oxycodone-paracetamol tablet exhibits increased analgesic efficacy for acute postoperative pain, higher satisfaction and comparable safety profiles compared with celecoxib in patients underwent arthroscopic knee surgery. Inflammopharmacology 2021, 29, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- De Santis, S.; Simone, M.D.; Mercadante, S.; Mediati, R.D.; Vellucci, R.; Marchetti, P.; Tonini, G.; Cuomo, A.; Caraceni, A.; Natoli, S.; et al. Oxycodone/Acetaminophen: The Tailoring Combination Treatment for Specific Clinical Profile of Opioid Well-Responsive Cancer Pain. Cancer Manag. Res. 2021, 13, 1747–1756. [Google Scholar] [CrossRef]
- Polati, E.; Canonico, P.L.; Schweiger, V.; Collino, M. Tapentadol: An overview of the safety profile. J. Pain Res. 2019, 12, 1569–1576. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, J.; Faria, J.; Queirós, O.; Moreira, R.; Carvalho, F.; Dinis-Oliveira, R.J. Comparative metabolism of tramadol and tapentadol: A toxicological perspective. Drug Metab. Rev. 2016, 48, 577–592. [Google Scholar] [CrossRef]
- Faria, J.; Barbosa, J.; Moreira, R.; Queirós, O.; Carvalho, F.; Dinis-Oliveira, R.J. Comparative pharmacology and toxicology of tramadol and tapentadol. Eur. J. Pain 2018, 22, 827–844. [Google Scholar] [CrossRef]
- Deeks, E.D. Tapentadol Prolonged Release: A Review in Pain Management. Drugs 2019, 79, 589. [Google Scholar] [CrossRef] [Green Version]
- Freo, U.; Romualdi, P.; Kress, H.G. Tapentadol for neuropathic pain: A review of clinical studies. J. Pain Res. 2019, 12, 1537–1551. [Google Scholar] [CrossRef] [Green Version]
- Hoskin, P.J.; Hanks, G.W. Morphine: Pharmacokinetics and clinical practice. Br. J. Cancer 1990, 62, 705–707. [Google Scholar] [CrossRef] [Green Version]
- Stuart-Harris, R.; Joel, S.P.; McDonald, P.; Currow, D.; Slevin, M.L. The pharmacokinetics of morphine and morphine glucuronide metabolites after subcutaneous bolus injection and subcutaneous infusion of morphine. Br. J. Clin. Pharmacol. 2000, 49, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Avdeef, A.; Barrett, D.A.; Shaw, P.N.; Knaggs, R.D.; Davis, S.S. Octanol-, chloroform-, and propylene glycol dipelargonat-water partitioning of morphine-6-glucuronide and other related opiates. J. Med. Chem. 1996, 39, 4377–4381. [Google Scholar] [CrossRef]
- Saari, T.I.; Ihmsen, H.; Neuvonen, P.J.; Olkkola, K.T.; Schwilden, H. Oxycodone clearance is markedly reduced with advancing age: A population pharmacokinetic study. Br. J. Anaesth. 2012, 108, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Ordóñez Gallego, A.; González Barón, M.; Espinosa Arranz, E. Oxycodone: A pharmacological and clinical review. Clin. Transl. Oncol. 2007, 9, 298–307. [Google Scholar] [CrossRef]
- Takala, A.; Kaasalainen, V.; Seppälä, T.; Kalso, E.; Olkkola, K.T. Pharmacokinetic comparison of intravenous and intranasal administration of oxycodone. Acta Anaesthesiol. Scand. 1997, 41, 309–312. [Google Scholar] [CrossRef]
- Concheiro, M.; Chesser, R.; Pardi, J.; Cooper, G. Postmortem Toxicology of New Synthetic Opioids. Front. Pharmacol. 2018, 9, 1210. [Google Scholar] [CrossRef]
- Elkader, A.; Sproule, B. Buprenorphine: Clinical pharmacokinetics in the treatment of opioid dependence. Clin. Pharmacokinet. 2005, 44, 661–680. [Google Scholar] [CrossRef]
- Ellis, C.R.; Kruhlak, N.L.; Kim, M.T.; Hawkins, E.G.; Stavitskaya, L. Predicting opioid receptor binding affinity of pharmacologically unclassified designer substances using molecular docking. PLoS ONE 2018, 13, e0197734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, J.; Steinfath, M.; Schulz, M. Clinical pharmacokinetics of alfentanil, fentanyl and sufentanil. An update. Clin. Pharmacokinet. 1996, 31, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Bista, S.R.; Haywood, A.; Hardy, J.; Lobb, M.; Tapuni, A.; Norris, R. Protein binding of fentanyl and its metabolite nor-fentanyl in human plasma, albumin and α-1 acid glycoprotein. Xenobiotica 2015, 45, 207–212. [Google Scholar] [CrossRef]
- Kuip, E.J.; Zandvliet, M.L.; Koolen, S.L.; Mathijssen, R.H.; van der Rijt, C.C. A review of factors explaining variability in fentanyl pharmacokinetics; focus on implications for cancer patients. Br. J. Clin. Pharmacol. 2017, 83, 294–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.R.; Nag, K.; Shetti, A.N.; Krishnaveni, N. Tapentadol hydrochloride: A novel analgesic. Saudi J. Anaesth. 2013, 7, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Fidman, B.; Nogid, A. Role of Tapentadol Immediate Release (Nucynta) in the Management of Moderate-to-Severe Pain. Pharm. Ther. 2010, 35, 330–333, 357. [Google Scholar]
- Jain, D.; Basniwal, P.K. Tapentadol, a novel analgesic: Review of recent trends in synthesis, related substances, analytical methods, pharmacodynamics and pharmacokinetics. Bull. Fac. Pharm. Cairo Univ. 2013, 51, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Raffa, R.B.; Elling, C.; Tzschentke, T.M. Does ‘Strong Analgesic’ Equal ‘Strong Opioid’? Tapentadol and the Concept of ‘µ-Load’. Adv. Ther. 2018, 35, 1471–1484. [Google Scholar] [CrossRef] [Green Version]
- Owusu Obeng, A.; Hamadeh, I.; Smith, M. Review of Opioid Pharmacogenetics and Considerations for Pain Management. Pharmacotherapy 2017, 37, 1105–1121. [Google Scholar] [CrossRef]
- Vieira, C.M.P.; Fragoso, R.M.; Pereira, D.; Medeiros, R. Pain polymorphisms and opioids: An evidence based review. Mol. Med. Rep. 2019, 19, 1423–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relling, M.V.; Evans, W.E. Pharmacogenomics in the clinic. Nature 2015, 526, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, R.A.; Hotham, E.; Hall, C.; Williams, M.; Suppiah, V. Pharmacogenomics and Patient Treatment Parameters to Opioid Treatment in Chronic Pain: A Focus on Morphine, Oxycodone, Tramadol, and Fentanyl. Pain Med. 2017, 18, 2369–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.F. Polimorphism of human cytochrome P450 2D6 and its clinical significance: Part I. Clin. Pharmacokinet. 2009, 48, 689–723. [Google Scholar] [CrossRef]
- Gong, X.D.; Wang, J.Y.; Liu, F.; Yuan, H.H.; Zhang, W.Y.; Guo, Y.H.; Jiang, B. Gene polymorphisms of OPRM1 A118G and ABCB1 C3435T may influence opioid requirements in Chinese patients with cancer pain. Asian Pac. J. Cancer Prev. 2013, 14, 2937–2943. [Google Scholar] [CrossRef] [Green Version]
- Crews, K.R.; Monte, A.A.; Huddart, R.; Caudle, K.E.; Kharasch, E.D.; Gaedigk, A.; Dunnenberger, H.M.; Leeder, J.S.; Callaghan, J.T.; Samer, C.F.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for CYP2D6, OPRM1, and COMT Genotypes and Select Opioid Therapy. Clin. Pharmacol. Ther. 2021, 110, 888–896. [Google Scholar] [CrossRef]
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid complications and side effects. Pain Physician 2008, 11, S105–S120. [Google Scholar] [CrossRef]
- Zha, W.; Shum, L. Simultaneous determination of oxymorphone and its active metabolite 6-OH-oxymorphone in human plasma by high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 902, 116–121. [Google Scholar] [CrossRef]
- Fda.gov. Available online: https://www.fda.gov/news-events/press-announcements/califf-fda-top-officials-call-sweeping-review-agency-opioids-policies (accessed on 17 April 2023).
- EMCDDA.europa.eu. Available online: https://www.emcdda.europa.eu/publications/mini-guides/opioids-health-and-social-responses_en (accessed on 17 April 2023).
- Volkow, N.D.; McLellan, A.T. Opioid Abuse in Chronic Pain--Misconceptions and Mitigation Strategies. N. Engl. J. Med. 2016, 374, 1253–1263. [Google Scholar] [CrossRef]
- CDC.gov. Available online: https://www.cdc.gov/opioids/basics/epidemic.html (accessed on 17 April 2023).
- Butler, S.F.; Black, R.A.; Cassidy, T.A.; Dailey, T.M.; Budman, S.H. Abuse risks and routes of administration of different prescription opioid compounds and formulations. Harm Reduct. J. 2011, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Pruskowski, J.; Arnold, R.M. Tramadol in palliative care #290. J. Palliat. Med. 2015, 18, 461–462. [Google Scholar] [CrossRef]
- Walker, D.J.; Zacny, J.P. Subjective, psychomotor, and analgesic effects of oral codeine and morphine in healthy volunteers. Psychopharmacology 1998, 140, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W. When it comes to opiates, just say NO. J. Clin. Investig. 2007, 117, 3185–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, G.; Chen, L.; Mao, J. Opioid tolerance and hyperalgesia. Med. Clin. N. Am. 2007, 91, 199–211. [Google Scholar] [CrossRef] [PubMed]
- DuPen, A.; Shen, D.; Ersek, M. Mechanisms of opioid-induced tolerance and hyperalgesia. Pain Manag. Nurs. 2007, 8, 113–121. [Google Scholar] [CrossRef]
- Duttaroy, A.; Yoburn, B.C. The effect of intrinsic efficacy on opioid tolerance. Anesthesiology 1995, 82, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Abram, S.E.; Mampilly, G.A.; Milosavljevic, D. Assessment of the potency and intrinsic activity of systemic versus intrathecal opioids in rats. Anesthesiology 1997, 87, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Stoicea, N.; Soghomonyan, S.; Bergese, S.D. Remifentanil-acute opioid tolerance and opioid-induced hyperalgesia: A systematic review. Am. J. Ther. 2015, 22, e62–e74. [Google Scholar] [CrossRef]
- De Iaco, F.; Mannaioni, G.; Serra, S.; Finco, G.; Sartori, S.; Gandolfo, E.; Sansone, P.; Marinangeli, F. Equianalgesia, opioid switch and opioid association in different clinical settings: A narrative review. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 2000–2017. [Google Scholar] [CrossRef]
- Quigley, C. Opioid switching to improve pain relief and drug tolerability. Cochrane Database Syst. Rev. 2004, 3, CD004847. [Google Scholar] [CrossRef]
- Juurlink, D.N.; Dhalla, I.A. Dependence and addiction during chronic opioid therapy. J. Med. Toxicol. 2012, 8, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Von Korff, M.; Kolodny, A.; Deyo, R.A.; Chou, R. Long-term opioid therapy reconsidered. Ann. Intern. Med. 2011, 155, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Kaye, A.D.; Jones, M.R.; Kaye, A.M.; Ripoll, J.G.; Galan, V.; Beakley, B.D.; Calixto, F.; Bolden, J.L.; Urman, R.D.; Manchikanti, L. Prescription Opioid Abuse in Chronic Pain: An Updated Review of Opioid Abuse Predictors and Strategies to Curb Opioid Abuse: Part 1. Pain Physician 2017, 20, S93–S109. [Google Scholar] [CrossRef] [PubMed]
- Ates, H.C.; Roberts, J.A.; Lipman, J.; Cass, A.E.G.; Urban, G.A.; Dincer, C. On-Site Therapeutic Drug Monitoring. Trends Biotechnol. 2020, 38, 1262–1277. [Google Scholar] [CrossRef] [PubMed]
- Sankowski, B.; Michorowska, S.; Raćkowska, E.; Sikora, M.; Giebułtowicz, J. Saliva as Blood Alternative in Therapeutic Monitoring of Teriflunomide-Development and Validation of the Novel Analytical Method. Int. J. Mol. Sci. 2022, 23, 9544. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Lee, M.H. Overview of therapeutic drug monitoring. Korean J. Intern. Med. 2009, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shipkova, M.; Svinarov, D. LC-MS/MS as a tool for TDM services: Where are we? Clin. Biochem. 2016, 49, 1009–1023. [Google Scholar] [CrossRef]
- Mensitieri, F.; Coglianese, A.; Giudice, V.; Charlier, B.; De Rosa, F.; Filippelli, A.; Dal Piaz, F.; Izzo, V. Effects of selected preanalytical variables on Dried Blood Spot (DBS) and Volumetric Adsorptive Microsampling (VAMS) based bioanalytical methods for the determination of four β-lactam antibiotics. Biochim. Clin. 2022, 46, 141–153. [Google Scholar]
- D’Urso, A.; Locatelli, M.; Tartaglia, A.; Molteni, L.; D’Ovidio, C.; Savini, F.; Rudge, J.; de Grazia, U. Therapeutic Drug Monitoring of Antiseizure Medications Using Volumetric Absorptive Microsampling: Where Are We? Pharmaceuticals 2021, 14, 627. [Google Scholar] [CrossRef]
- Musshoff, F.; Lachenmeier, K.; Trafkowski, J.; Madea, B.; Nauck, F.; Stamer, U. Determination of opioid analgesics in hair samples using liquid chromatography/tandem mass spectrometry and application to patients under palliative care. Ther. Drug Monit. 2007, 29, 655–661. [Google Scholar] [CrossRef]
- Avataneo, V.; D’Avolio, A.; Cusato, J.; Cantù, M.; De Nicolò, A. LC-MS application for therapeutic drug monitoring in alternative matrices. J. Pharm. Biomed. Anal. 2019, 166, 40–51. [Google Scholar] [CrossRef]
- Manchikanti, L.; Atluri, S.; Trescot, A.M.; Giordano, J. Monitoring opioid adherence in chronic pain patients: Tools, techniques, and utility. Pain Physician 2008, 11, S155–S180. [Google Scholar] [CrossRef]
- van den Beuken-van Everdingen, M.H.J.; van Kuijk, S.M.J.; Janssen, D.J.A.; Joosten, E.A.J. Treatment of Pain in Cancer: Towards Personalised Medicine. Cancers 2018, 10, 502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantano, F.; Brauneis, S.; Forneris, A.; Pacifici, R.; Marinelli, E.; Kyriakou, C.; Pichini, S.; Busardò, F.P. Determination of oxycodone and its major metabolites noroxycodone and oxymorphone by ultra-high-performance liquid chromatography tandem mass spectrometry in plasma and urine: Application to real cases. Clin. Chem. Lab. Med. 2017, 55, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Protti, M.; Catapano, M.C.; Samolsky Dekel, B.G.; Rudge, J.; Gerra, G.; Somaini, L.; Mandrioli, R.; Mercolini, L. Determination of oxycodone and its major metabolites in haematic and urinary matrices: Comparison of traditional and miniaturised sampling approaches. J. Pharm. Biomed. Anal. 2018, 152, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Elarabi, H.; Elrasheed, A.; Ali, A.; Shawky, M.; Hasan, N.; Gawad, T.A.; Adem, A.; Marsden, J. Suboxone Treatment and Recovery Trial (STAR-T): Study Protocol for a Randomised Controlled Trial of Opioid Medication Assisted Treatment with Adjunctive Medication Management Using Therapeutic Drug Monitoring and Contingency Management. J. Addict. 2019, 2019, 2491063. [Google Scholar] [CrossRef] [Green Version]
- Elarabi, H.F.; Hasan, N.; Marsden, J.; Radwan, D.; Adem, A.; Almamari, S.; Elrasheed, A. Therapeutic Drug Monitoring in Buprenorphine/Naloxone Treatment for Opioid Use Disorder: Clinical Feasibility and Optimizing Assay Precision. Pharmacopsychiatry 2020, 53, 115–121. [Google Scholar] [CrossRef]
- Brünen, S.; Vincent, P.D.; Baumann, P.; Hiemke, C.; Havemann-Reinecke, U. Therapeutic drug monitoring for drugs used in the treatment of substance-related disorders: Literature review using a therapeutic drug monitoring appropriateness rating scale. Ther. Drug Monit. 2011, 33, 561–572. [Google Scholar] [CrossRef]
- Mercadante, S. Opioid titration in cancer pain: A critical review. Eur. J. Pain 2007, 11, 823–830. [Google Scholar] [CrossRef]
- Caraceni, A.; Hanks, G.; Kaasa, S.; Bennett, M.I.; Brunelli, C.; Cherny, N.; Dale, O.; de Conno, F.; Fallon, M.; Hanna, M.; et al. Use of opioid analgesics in the treatment of cancer pain: Evidence-based recommendations from the EAPC. Lancet Oncol. 2012, 13, e58–e68. [Google Scholar] [CrossRef]
- Nicholson, A.B. Methadone for cancer pain. Cochrane Database Syst. Rev. 2007, 4, CD003971. [Google Scholar] [CrossRef]
- Hadley, G.; Derry, S.; Moore, R.A.; Wiffen, P.J. Transdermal fentanyl for cancer pain. Cochrane Database Syst. Rev. 2013, 10, CD010270. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Hansen, M.; Bromham, N.; Taubert, M.; Arnold, S.; Hilgart, J.S. Buprenorphine for treating cancer pain. Cochrane Database Syst. Rev. 2015, 3, CD009596. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Hansen, M.; Bennett, M.I.; Arnold, S.; Bromham, N.; Hilgart, J.S. Oxycodone for cancer-related pain. Cochrane Database Syst. Rev. 2015, 2, CD003870. [Google Scholar] [CrossRef] [Green Version]
- Matic, M.; de Wildt, S.N.; Tibboel, D.; van Schaik, R.H.N. Analgesia and Opioids: A Pharmacogenetics Shortlist for Implementation in Clinical Practice. Clin. Chem. 2017, 63, 1204–1213. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Rosa, F.; Giannatiempo, B.; Charlier, B.; Coglianese, A.; Mensitieri, F.; Gaudino, G.; Cozzolino, A.; Filippelli, A.; Piazza, O.; Dal Piaz, F.; et al. Pharmacological Treatments and Therapeutic Drug Monitoring in Patients with Chronic Pain. Pharmaceutics 2023, 15, 2088. https://doi.org/10.3390/pharmaceutics15082088
De Rosa F, Giannatiempo B, Charlier B, Coglianese A, Mensitieri F, Gaudino G, Cozzolino A, Filippelli A, Piazza O, Dal Piaz F, et al. Pharmacological Treatments and Therapeutic Drug Monitoring in Patients with Chronic Pain. Pharmaceutics. 2023; 15(8):2088. https://doi.org/10.3390/pharmaceutics15082088
Chicago/Turabian StyleDe Rosa, Federica, Bruno Giannatiempo, Bruno Charlier, Albino Coglianese, Francesca Mensitieri, Giulia Gaudino, Armando Cozzolino, Amelia Filippelli, Ornella Piazza, Fabrizio Dal Piaz, and et al. 2023. "Pharmacological Treatments and Therapeutic Drug Monitoring in Patients with Chronic Pain" Pharmaceutics 15, no. 8: 2088. https://doi.org/10.3390/pharmaceutics15082088