

Recent Advances in Targeted Drug Delivery Strategy for Enhancing Oncotherapy

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
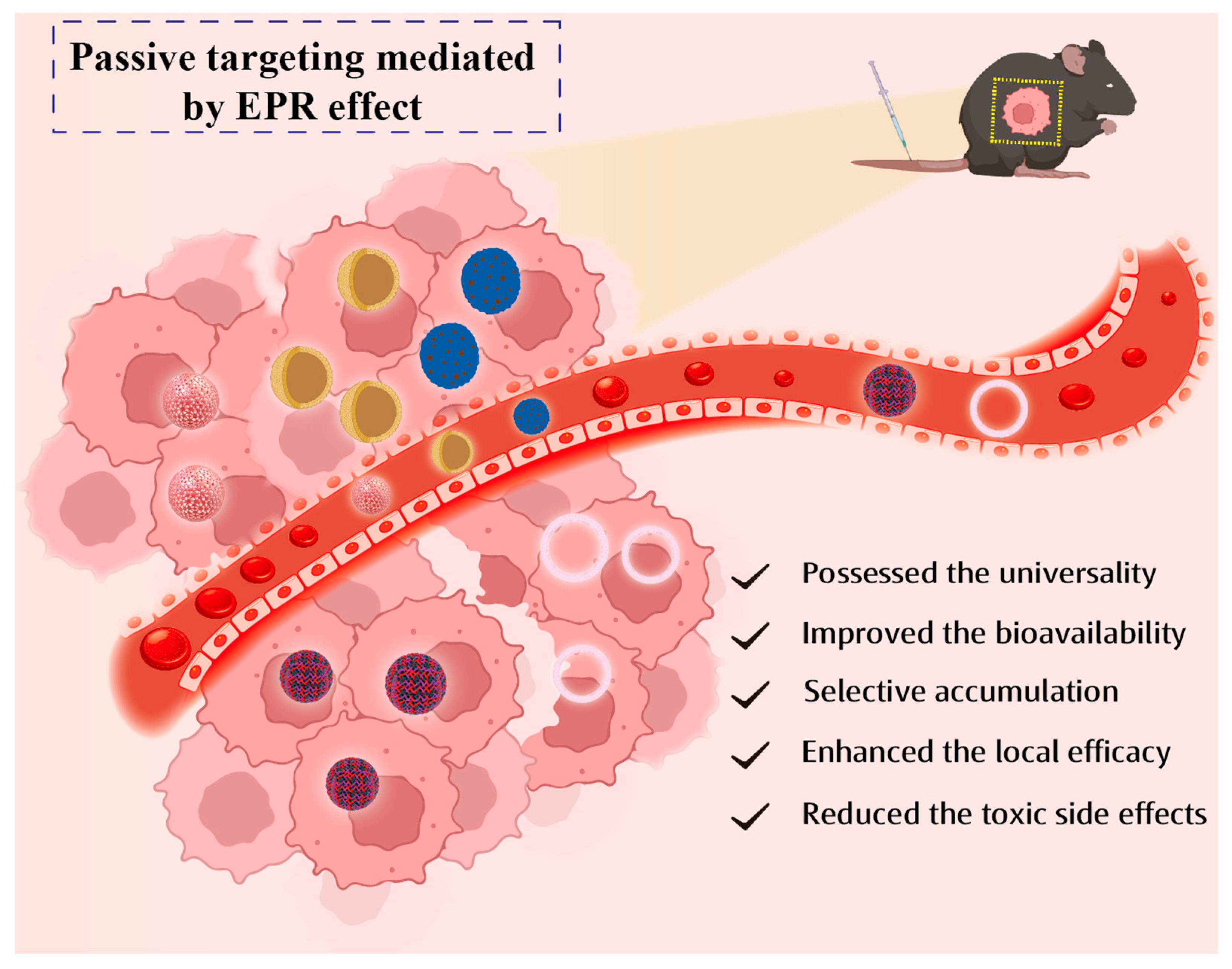
2. Passive Targeting Targeted of Drug Delivery
2.1. Liposomes

2.2. Polymeric Nanoparticles
2.3. Metal Oxide Nanoparticles
2.4. Silicon Dioxide Nanoparticles
2.5. Magnetic Nanoparticles (MNPs)
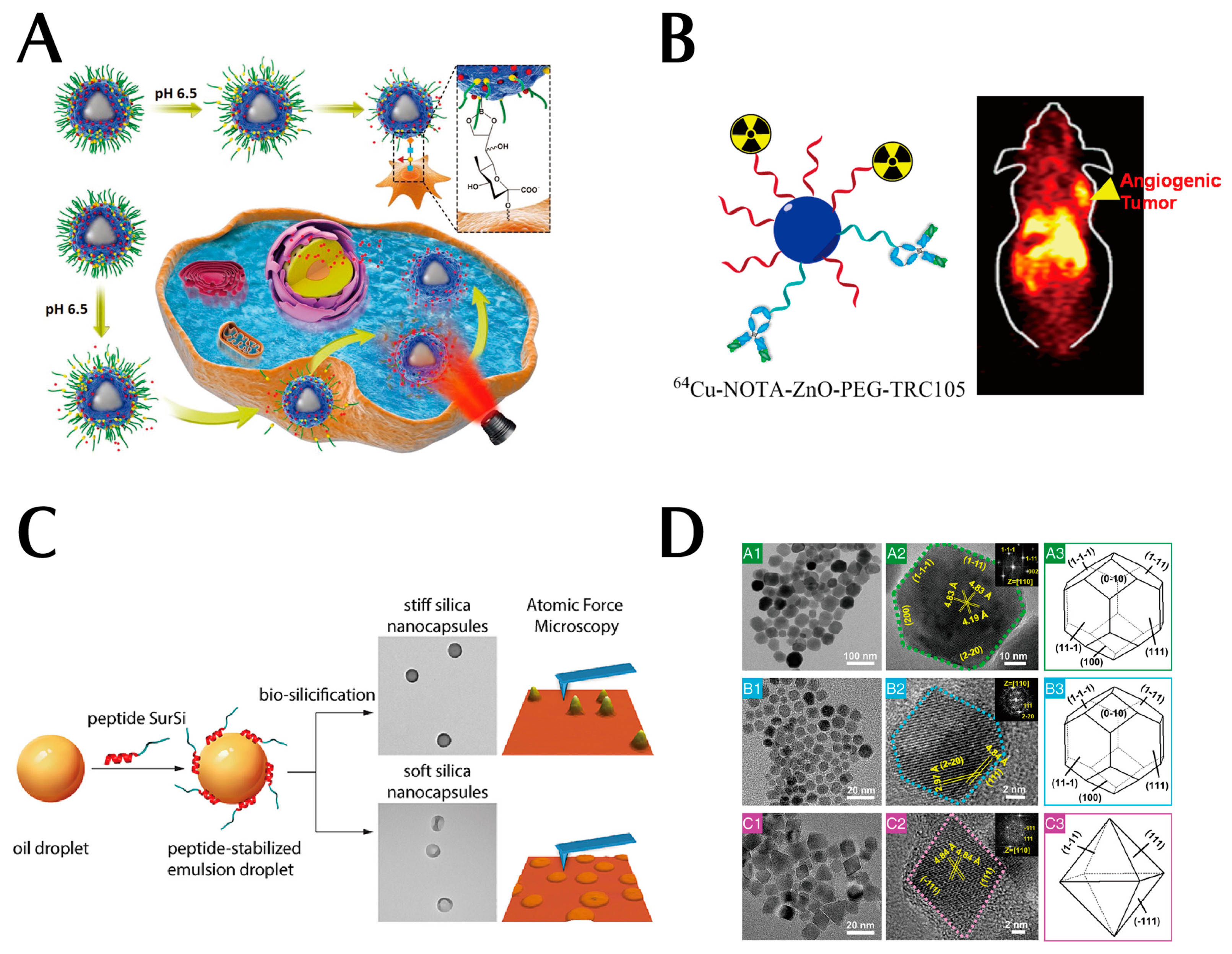
3. Active Targeting of Drug Delivery
3.1. Antibody-Based Targeted Nanoparticles
3.2. Peptide-Based Targeted Nanoparticles

3.3. Aptamer-Based Targeted Nanoparticles
3.4. Small-Molecule-Based Targeted Nanoparticles
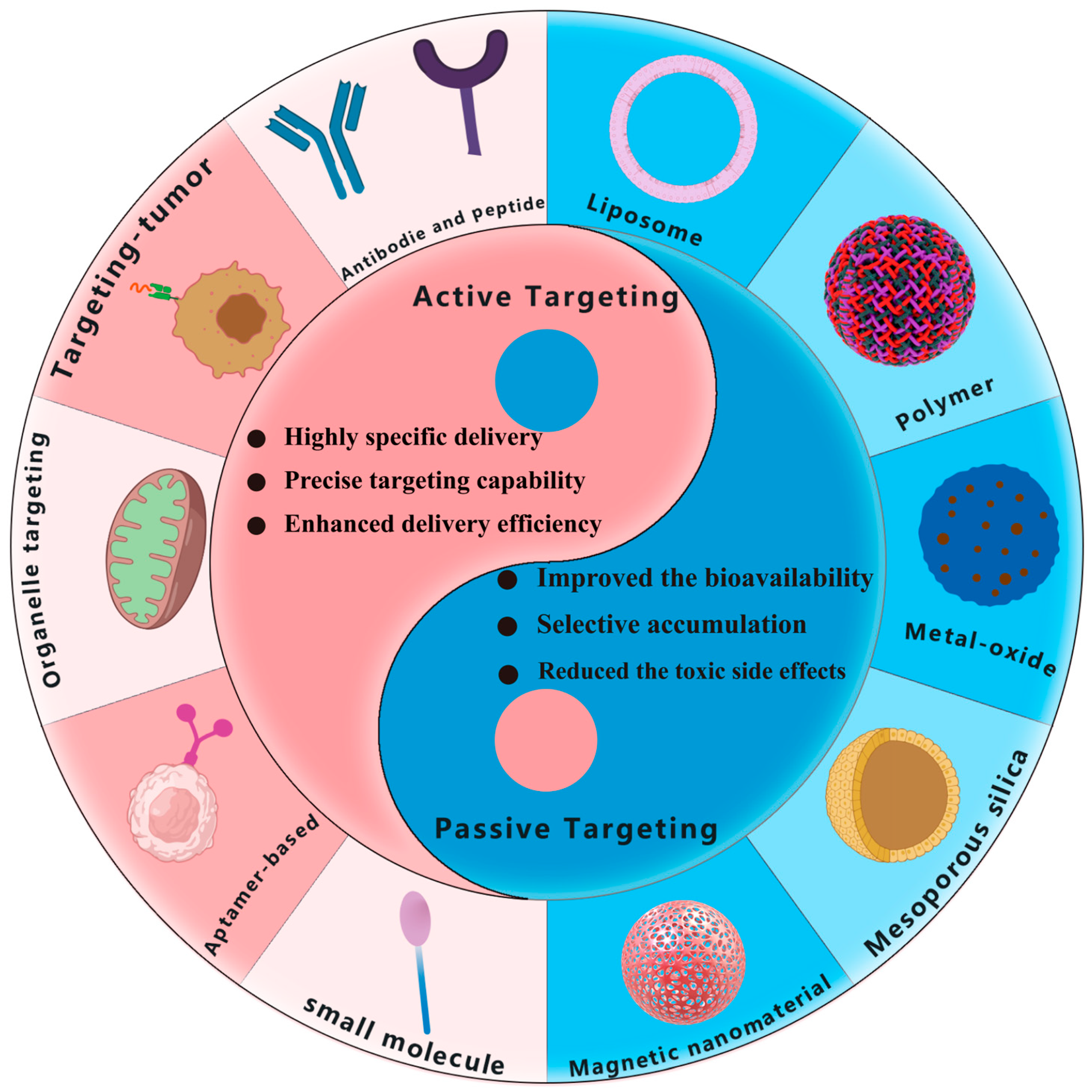
4. Comparison and Combination of Passive and Active Targeting
4.1. Comparison of Passive and Active Targeting
4.2. Combination of Passive and Active Targeting
5. Future Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADC | antibody-drug couples |
| ADCC | antibody-dependent cell-mediated cytotoxic effects |
| ADCP | antibody-dependent cytophagy |
| ALPPL2 | alkaline phosphatase placenta-like 2 |
| C5 | component 5 |
| CAF | cancer-associated fibroblasts |
| CCKBR | cholecystokinin B receptor |
| CDC | cytotoxic effects |
| CRC | colorectal cancer |
| CRP | complement regulatory protein |
| DC | dendritic cells |
| DDS | drug delivery systems |
| DLPC | 1,2-Dilauroyl-sn-glycero-3-phosphorylcholine |
| DMPC | 1,2-dimyristoyl-sn-glycero-3-phosphocholine |
| DOCP | 2-((2,3-Bis(oleoyloxy)propyl)dimethylammonio)ethyl hydrogen Phosphate |
| DOCPe | 2-((2,3-bis(oleoyloxy)propyl)-dimethylammonio)ethyl ethyl phosphate |
| DOX | doxorubicin |
| DPPC | 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine |
| DSPE-mPEG | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine with conjugated methoxyl PEG |
| ECM | extracellular matrix |
| EGFR | growth factor receptor |
| EPR | enhanced permeability and retention |
| ES | Estrone |
| FA-PEG | folic acid and poly(ethylene glycol) |
| FGNP | core–shell Fe3O4/Gd2O3 heterogeneous nanoparticles |
| GD2 | glycosides deaminate |
| GEM | Gemcitabine |
| GM-CSF | granulocyte-macrophage colony stimulating factor |
| H2O2 | hydrogen peroxide |
| ICG | indocyanine green |
| LPO | lipid peroxides |
| NE | nuclear envelope |
| NK | natural killer cell |
| NL | NGR peptide |
| NLS | nuclear targeting molecules |
| NPC | nuclear pore complex |
| NSCLC | non-small cell lung cancer |
| mAb | monoclonal antibody |
| MAC | membrane attack complex |
| mtDNA | mitochondrial DNA |
| MRI | magnetic resonance imaging |
| PAC1 | phosphatase 1 |
| PAMAM | Polyamidoamine |
| PBA | phenylboronic acid |
| PC | Phosphatidylcholine |
| PDAC | pancreatic ductal adenocarcinoma |
| PD-L1 | programmed death ligand 1 |
| PEG | polyethylene glycol |
| PET | positron emission tomography |
| PRR | pattern recognition receptor |
| PTX | paclitaxel-loaded |
| Que | quercetin |
| RGD | arginine–glycine–aspartate |
| ROS | reactive oxygen species |
| RPM | RGD modified paclitaxel (PTX) |
| SELEX | Systematic Evolution of Ligands by Exponential Enrichment |
| SFN | sorafenib |
| SPC | soy phosphatidylcholine |
| STCT | Subcellular targeting cancer therapy |
| TAA | tumor-associated antigens |
| TfR-BP | transferrin receptor binding peptide |
| TGF-α | Transforming growth factor α |
| TME | tumor microenvironment |
| TNBC | triple-negative breast cancer |
| TNF | tumor necrosis factor |
| TRC105 | a monoclonal antibody against CD105 |
| TSA | tumor-specific antigens |
| VEGF | vascular endothelial growth factor |
| ZH-1 | a DNA aptamer |
| ZnO | zinc oxide |
References
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Tam, K.Y.; Leung, K.C.F.; Wang, Y.X.J. Chemoembolization Agents Cancer Treatment. Eur. J. Pharm. Sci. 2011, 44, 1–10. [Google Scholar] [CrossRef]
- Haque, M.; Shakil, M.S.; Mahmud, K.M. The Promise of Nanoparticles-Based Radiotherapy in Cancer Treatment. Cancers 2023, 15, 1892. [Google Scholar] [CrossRef]
- Glasgow, M.D.; Chougule, M.B. Recent Developments in Active Tumor Targeted Multifunctional Nanoparticles for Combination Chemotherapy in Cancer Treatment and Imaging. J. Biomed. Nanotechnol. 2015, 11, 1859–1898. [Google Scholar] [CrossRef]
- Dang, Y.; Guan, J. Nanoparticle-Based Drug Delivery Systems for Cancer Therapy. Smart Mater. Med. 2020, 1, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A. Nanotechnology in Targeted Drug Delivery. IJMS 2023, 24, 8194. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Barick, K.C.; Bahadur, D. Oxide and Hybrid Nanostructures for Therapeutic Applications. Adv. Drug Deliv. Rev. 2011, 63, 1267–1281. [Google Scholar] [CrossRef]
- Chamundeeswari, M.; Jeslin, J.; Verma, M.L. Nanocarriers for Drug Delivery Applications. Environ. Chem. Lett. 2019, 17, 849–865. [Google Scholar] [CrossRef]
- Pushpalatha, R.; Selvamuthukumar, S.; Kilimozhi, D. Nanocarrier Mediated Combination Drug Delivery for Chemotherapy—A Review. J. Drug Deliv. Sci. Technol. 2017, 39, 362–371. [Google Scholar] [CrossRef]
- Khizar, S.; Alrushaid, N.; Alam Khan, F.; Zine, N.; Jaffrezic-Renault, N.; Errachid, A.; Elaissari, A. Nanocarriers Based Novel and Effective Drug Delivery System. Int. J. Pharm. 2023, 632, 122570. [Google Scholar] [CrossRef]
- Shah, A.; Aftab, S.; Nisar, J.; Ashiq, M.N.; Iftikhar, F.J. Nanocarriers for Targeted Drug Delivery. J. Drug Deliv. Sci. Technol. 2021, 62, 102426. [Google Scholar] [CrossRef]
- Bazak, R.; Houri, M.; Achy, S.E.; Hussein, W.; Refaat, T. Passive Targeting of Nanoparticles to Cancer: A Comprehensive Review of the Literature. Mol. Clin. Oncol. 2014, 2, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tu-moritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 2017, 46, 6387–6392. [Google Scholar]
- Maeda, H.; Bharate, G.; Daruwalla, J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar]
- Feliu, N.; Docter, D.; Heine, M.; Del Pino, P.; Ashraf, S.; Kolosnjaj-Tabi, J.; Macchiarini, P.; Nielsen, P.; Alloyeau, D.; Gazeau, F.; et al. In Vivo Degeneration and the Fate of Inorganic Nanoparticles. Chem. Soc. Rev. 2016, 45, 2440–2457. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Hu, C.-M.J.; Fang, R.H.; Zhang, L. Liposome-like Nanostructures for Drug Delivery. J. Mater. Chem. B 2013, 1, 6569. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. Interfacing Zwitterionic Liposomes with Inorganic Nanomaterials: Surface Forces, Membrane Integrity, and Applications. Langmuir 2016, 32, 4393–4404. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Li, X.; Guo, Y.; Zhang, Z. Lipid-Enveloped Hybrid Nanoparticles for Drug Delivery. Nanoscale 2013, 5, 860. [Google Scholar] [CrossRef]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumor sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef]
- Kibria, G.; Hatakeyama, H.; Ohga, N.; Hida, K.; Harashima, H. The Effect of Liposomal Size on the Targeted Delivery of Doxorubicin to Integrin Avβ3-Expressing Tumor Endothelial Cells. Biomaterials 2013, 34, 5617–5627. [Google Scholar] [CrossRef]
- Deshpande, P.P.; Biswas, S.; Torchilin, V.P. Current Trends in the Use of Liposomes for Tumor Targeting. Nanomedicine 2013, 8, 1509–1528. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; He, Y.; Liang, J.; Cheng, Z.; Zhang, M.; Zhu, Y.; Hong, C.; Qin, J.; Xu, X.; Wang, J. Role of Liposome Size, Surface Charge, and PEGylation on Rheumatoid Arthritis Targeting Therapy. ACS Appl. Mater. Interfaces 2019, 11, 20304–20315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, C.; Wang, C.; Jankovic, K.E.; Dong, Y. Lipids and Lipid Derivatives for RNA Delivery. Chem. Rev. 2021, 121, 12181–12277. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, F.; Li, X.; Chen, J.; Zhang, X.; Wang, Y.; Liu, J. Dipole Orientation Matters: Longer-Circulating Choline Phosphate than Phosphocholine Liposomes for Enhanced Tumor Targeting. ACS Appl. Mater. Interfaces 2017, 9, 17736–17744. [Google Scholar] [CrossRef]
- Takechi-Haraya, Y.; Goda, Y.; Sakai-Kato, K. Control of Liposomal Penetration into Three-Dimensional Multicellular Tumor Spheroids by Modulating Liposomal Membrane Rigidity. Mol. Pharm. 2017, 14, 2158–2165. [Google Scholar] [CrossRef]
- Dai, Z.; Yu, M.; Yi, X.; Wu, Z.; Tian, F.; Miao, Y.; Song, W.; He, S.; Ahmad, E.; Guo, S.; et al. Chain-Length- and Saturation-Tuned Mechanics of Fluid Nanovesicles Direct Tumor Delivery. ACS Nano 2019, 13, 7676–7689. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.R.; Regen, S.L. The Structural Role of Cholesterol in Cell Membranes: From Condensed Bilayers to Lipid Rafts. Acc. Chem. Res. 2014, 47, 3512–3521. [Google Scholar] [CrossRef]
- Simons, K.; Vaz, W.L.C. Model Systems, Lipid Rafts, and Cell Membranes. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 269–295. [Google Scholar] [CrossRef]
- Abumanhal-Masarweh, H.; Da Silva, D.; Poley, M.; Zinger, A.; Goldman, E.; Krinsky, N.; Kleiner, R.; Shenbach, G.; Schroeder, J.E.; Shklover, J.; et al. Tailoring the Lipid Composition of Nanoparticles Modulates Their Cellular Uptake and Affects the Viability of Triple Negative Breast Cancer Cells. J. Control. Release 2019, 307, 331–341. [Google Scholar] [CrossRef]
- Tang, H.; Zhang, Z.; Zhu, M.; Xie, Y.; Lv, Z.; Liu, R.; Shen, Y.; Pei, J. Efficient Delivery of Gemcitabine by Estrogen Receptor-Targeted PEGylated Liposome and Its Anti-Lung Cancer Activity In Vivo and In Vitro. Pharmaceutics 2023, 15, 988. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR Effect: Unique Features of Tumor Blood Vessels for Drug Delivery, Factors Involved, and Limitations and Augmentation of the Effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Cabral, H.; Kataoka, K. Progress of Drug-Loaded Polymeric Micelles into Clinical Studies. J. Control. Release 2014, 190, 465–476. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Nixon, L.S.; Hedstrand, D.M. The Role of Branch Cell Symmetry and Other Critical Nanoscale Design Parameters in the Determination of Dendrimer Encapsulation Properties. Biomolecules 2020, 10, 642. [Google Scholar] [CrossRef] [PubMed]
- Kaminskas, L.M.; McLeod, V.M.; Kelly, B.D.; Sberna, G.; Boyd, B.J.; Williamson, M.; Owen, D.J.; Porter, C.J.H. A Comparison of Changes to Doxorubicin Pharmacokinetics, Antitumor Activity, and Toxicity Mediated by PEGylated Dendrimer and PEGylated Liposome Drug Delivery Systems. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 103–111. [Google Scholar] [CrossRef]
- Kojima, C.; Regino, C.; Umeda, Y.; Kobayashi, H.; Kono, K. Influence of Dendrimer Generation and Polyethylene Glycol Length on the Biodistribution of PEGylated Dendrimers. Int. J. Pharm. 2010, 383, 293–296. [Google Scholar] [CrossRef]
- Liu, S.; Pan, J.; Liu, J.; Ma, Y.; Qiu, F.; Mei, L.; Zeng, X.; Pan, G. Dynamically PEGylated and Borate-Coordination-Polymer-Coated Polydopamine Nanoparticles for Synergetic Tumor-Targeted, Chemo-Photothermal Combination Therapy. Small 2018, 14, 1703968. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Bi, S.; Fan, Z.; Fu, Z.; Meng, Z.; Hou, Z.; Zhang, F. A Metal-Free Approach to Bipyridinium Salt-Based Conjugated Porous Polymers with Olefin Linkages. Polym. Chem. 2021, 12, 1661–1667. [Google Scholar] [CrossRef]
- Xiong, H.-M. ZnO Nanoparticles Applied to Bioimaging and Drug Delivery. Adv. Mater. 2013, 25, 5329–5335. [Google Scholar] [CrossRef]
- Ko, W.; Jung, N.; Lee, M.; Yun, M.; Jeon, S. Electronic Nose Based on Multipatterns of ZnO Nanorods on a Quartz Resonator with Remote Electrodes. ACS Nano 2013, 7, 6685–6690. [Google Scholar] [CrossRef]
- Zhang, Y.; Nayak, T.; Hong, H.; Cai, W. Biomedical Applications of Zinc Oxide Nanomaterials. CMM 2013, 13, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, W.; Qiu, Y.; Yang, S.; Xiao, S.; Wang, Q.-Q.; Ding, L.; Wang, J. Surface Functionalization of ZnO Nanotetrapods with Photoactive and Electroactive Organic Monolayers. Langmuir 2008, 24, 5052–5059. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Wang, F.; Zhang, Y.; Graves, S.A.; Eddine, S.B.Z.; Yang, Y.; Theuer, C.P.; Nickles, R.J.; Wang, X.; Cai, W. Red Fluorescent Zinc Oxide Nanoparticle: A Novel Platform for Cancer Targeting. ACS Appl. Mater. Interfaces 2015, 7, 3373–3381. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer Nanotechnology: The Impact of Passive and Active Targeting in the Era of Modern Cancer Biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Impact of Particle Elasticity on Particle-Based Drug Delivery Systems. Adv. Drug Deliv. Rev. 2017, 108, 51–67. [Google Scholar] [CrossRef]
- Guo, D.; Xie, G.; Luo, J. Mechanical Properties of Nanoparticles: Basics and Applications. J. Phys. D Appl. Phys. 2014, 47, 013001. [Google Scholar] [CrossRef]
- Yi, X.; Shi, X.; Gao, H. Cellular Uptake of Elastic Nanoparticles. Phys. Rev. Lett. 2011, 107, 098101. [Google Scholar] [CrossRef]
- Best, J.P.; Yan, Y.; Caruso, F. The Role of Particle Geometry and Mechanics in the Biological Domain. Adv. Healthc. Mater. 2012, 1, 35–47. [Google Scholar] [CrossRef]
- Hui, Y.; Wibowo, D.; Liu, Y.; Ran, R.; Wang, H.-F.; Seth, A.; Middelberg, A.P.J.; Zhao, C.-X. Understanding the Effects of Nanocapsular Mechanical Property on Passive and Active Tumor Targeting. ACS Nano 2018, 12, 2846–2857. [Google Scholar] [CrossRef]
- Ma, K.; Xu, S.; Tao, T.; Qian, J.; Cui, Q.; Rehman, S.U.; Zhu, X.; Chen, R.; Zhao, H.; Wang, C.; et al. Magnetosome-Inspired Synthesis of Soft Ferrimagnetic Nanoparticles for Magnetic Tumor Targeting. Proc. Natl. Acad. Sci. USA 2022, 119, e2211228119. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Song, J.; Yung, B.C.; Zhou, Z.; Wu, A.; Chen, X. Emerging Strategies of Cancer Therapy Based on Ferroptosis. Adv. Mater. 2018, 30, 1704007. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and Function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lu, X.; Du, C.; Zhou, Z.; Feng, J.; Liang, Z.; Xu, Y.; Qiu, X.; Shen, Z. Cycloacceleration of Reactive Oxygen Species Generation Based on Exceedingly Small Magnetic Iron Oxide Nanoparticles for Tumor Ferroptosis Therapy. Small 2022, 18, 2202705. [Google Scholar] [CrossRef]
- Biffi, S.; Voltan, R.; Bortot, B.; Zauli, G.; Secchiero, P. Actively Targeted Nanocarriers for Drug Delivery to Cancer Cells. Expert. Opin. Drug Deliv. 2019, 16, 481–496. [Google Scholar] [CrossRef]
- Roex, M.C.J.; Hageman, L.; Veld, S.A.J.; van Egmond, E.; Hoogstraten, C.; Stemberger, C.; Germeroth, L.; Einsele, H.; Falkenburg, J.H.F.; Jedema, I. A Minority of T Cells Recognizing Tumor-Associated Antigens Presented in Self-HLA Can Provoke Antitumor Reactivity. Blood 2020, 136, 455–467. [Google Scholar] [CrossRef]
- Greiner, J.; Schmitt, M.; Li, L.; Giannopoulos, K.; Bosch, K.; Schmitt, A.; Dohner, K.; Schlenk, R.F.; Pollack, J.R.; Dohner, H.; et al. Expression of Tumor-Associated Antigens in Acute Myeloid Leukemia: Implications for Specific Immunotherapeutic Approaches. Blood 2006, 108, 4109–4117. [Google Scholar] [CrossRef]
- Seeger, R.C. Immunology and Immunotherapy of Neuroblastoma. Semin. Cancer Biol. 2011, 21, 229–237. [Google Scholar] [CrossRef]
- Su, Y.; Zhang, X.; Bidlingmaier, S.; Behrens, C.R.; Lee, N.-K.; Liu, B. ALPPL2 Is a Highly Specific and Targetable Tumor Cell Surface Antigen. Cancer Res. 2020, 80, 4552–4564. [Google Scholar] [CrossRef]
- Capece, D.; Verzella, D.; Fischietti, M.; Zazzeroni, F.; Alesse, E. Targeting Costimulatory Molecules to Improve Antitumor Immunity. BioMed Res. Int. 2012, 2012, e926321. [Google Scholar] [CrossRef]
- Schwartz, R.H. T Cell Anergy. Annu. Rev. Immunol. 2003, 21, 305–334. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Liu, Y. Immunotherapy of Melanoma with the Immune Costimulatory Monoclonal Antibodies Targeting CD137. CPAA 2013, 5, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.-B.; He, T.-P.; Li, H.-B.; Tang, H.-W.; Xia, E.-Q. The Structure-Activity Relationship of the Antioxidant Peptides from Natural Proteins. Molecules 2016, 21, 72. [Google Scholar] [CrossRef]
- Dmitrieva, M.D.; Voitova, A.A.; Dymova, M.A.; Richter, V.A.; Kuligina, E.V. Tumor-Targeting Peptides Search Strategy for the Delivery of Therapeutic and Diagnostic Molecules to Tumor Cells. Int. J. Mol. Sci. 2021, 22, 314. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Qiu, P.; Zhu, Y.; Yang, M.; Mao, C. Guiding Nanomaterials to Tumors for Breast Cancer Precision Medicine: From Tumor-Targeting Small-Molecule Discovery to Targeted Nanodrug Delivery. NPG Asia Mater. 2017, 9, e452. [Google Scholar] [CrossRef]
- Ma, C.; Yin, G.; Yan, D.; He, X.; Zhang, L.; Wei, Y.; Huang, Z. A Novel Peptide Specifically Targeting Ovarian Cancer Identified by in Vivo Phage Display. J. Pept. Sci. 2013, 19, 730–736. [Google Scholar] [CrossRef]
- Zhang, L.; Yin, G.; Yan, D.; Wei, Y.; Ma, C.; Huang, Z.; Liao, X.; Yao, Y.; Chen, X.; Hao, B. In Vitro Screening of Ovarian Tumor Specific Peptides from a Phage Display Peptide Library. Biotechnol. Lett. 2011, 33, 1729–1735. [Google Scholar] [CrossRef]
- Saw, P.E.; Song, E.-W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 11, 787–807. [Google Scholar] [CrossRef]
- Scodeller, P.; Simón-Gracia, L.; Kopanchuk, S.; Tobi, A.; Kilk, K.; Säälik, P.; Kurm, K.; Squadrito, M.L.; Kotamraju, V.R.; Rinken, A.; et al. Precision Targeting of Tumor Macrophages with a CD206 Binding Peptide. Sci. Rep. 2017, 7, 14655. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, W.; Li, R.; Qiu, W.; Zhuang, Z.; Cheng, S.; Zhang, X. Peptide-Based Multifunctional Nanomaterials for Tumor Imaging and Therapy. Adv. Funct. Mater. 2018, 28, 1804492. [Google Scholar] [CrossRef]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.-J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the Treatment of Colorectal Cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Peeters, M.; Siena, S.; Humblet, Y.; Hendlisz, A.; Neyns, B.; Canon, J.-L.; Van Laethem, J.-L.; Maurel, J.; Richardson, G.; et al. Open-Label Phase III Trial of Panitumumab Plus Best Supportive Care Compared with Best Supportive Care Alone in Patients With Chemotherapy-Refractory Metastatic Colorectal Cancer. JCO 2007, 25, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab Monotherapy and Cetuximab plus Irinotecan in Irinotecan-Refractory Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Erratum: Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer 2010, 10, 890. [Google Scholar] [CrossRef]
- Conibear, A.C.; Hager, S.; Mayr, J.; Klose, M.H.M.; Keppler, B.K.; Kowol, C.R.; Heffeter, P.; Becker, C.F.W. Multifunctional α v β 6 Integrin-Specific Peptide–Pt(IV) Conjugates for Cancer Cell Targeting. Bioconjugate Chem. 2017, 28, 2429–2439. [Google Scholar] [CrossRef]
- Li, S.; Gray, B.P.; McGuire, M.J.; Brown, K.C. Synthesis and Biological Evaluation of a Peptide–Paclitaxel Conjugate Which Targets the Integrin Avβ6. Bioorganic Med. Chem. 2011, 19, 5480–5489. [Google Scholar] [CrossRef]
- Murray, E.R.; Brown, N.F.; Howard, P.; Masterson, L.; Zammarchi, F.; van Berkel, P.H.; Marshall, J.F. Marshall. Abstract 958: Effective targeting of pancreatic ductal adenocarcinoma metastases with an integrin αvβ6-targeting pep-tide-drug conjugate. Cancer Res. 2021, 81, 958. [Google Scholar] [CrossRef]
- Li, K.; Liu, C.-J.; Zhang, X.-Z. Multifunctional Peptides for Tumor Therapy. Adv. Drug Deliv. Rev. 2020, 160, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Zhao, Y.; Ding, Y.; Nie, G. Using Functional Nanomaterials to Target and Regulate the Tumor Microenvironment: Diagnostic and Therapeutic Applications. Adv. Mater. 2013, 25, 3508–3525. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- De La Granja, P.P.; Alejos, A.D.; Serrano, J.A.C.; Bareas, C.P. Archaeometric Analysis of the Pottery from the Chalcolithic Site of El Cortijo de Montiel Bajo (Santo Tomé de La Vega, Jaén, Spain). Archaeol. Anthr. Sci. 2022, 14, 194. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef]
- Perez-Penco, M.; Weis-Banke, S.E.; Schina, A.; Siersbæk, M.; Hübbe, M.L.; Jørgensen, M.A.; Lecoq, I.; de la Torre, L.L.; Bendtsen, S.K.; Martinenaite, E.; et al. TGFβ-derived immune modulatory vaccine: Targeting the immunosuppressive and fibrotic tumor microenvironment in a murine model of pancreatic cancer. J. Immunother. Cancer 2022, 10, e005491. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Liu, C.; Hou, Y.; Liu, Y.; Zhang, Z.; Zhao, H.; Xin, X.; Liu, W.; Zhang, X.; Chen, L.; et al. Sequential Delivery of Quercetin and Paclitaxel for the Fibrotic Tumor Microenvironment Remodeling and Chemotherapy Potentiation via a Dual-Targeting Hybrid Micelle-in-Liposome System. ACS Appl. Mater. Interfaces 2022, 14, 10102–10116. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Q.; Zhou, M.; Li, X.; Huang, Y.; Yang, N.; Zhou, Z. Concurrent impairment of nucleus and mito-chondria for synergistic inhibition of cancer metastasis. Int. J. Pharm. 2021, 608, 0378–5173. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.; Wagstaff, K.; Roth, D.; Moseley, G.; Jans, D. Targeted Delivery to the Nucleus☆. Adv. Drug Deliv. Rev. 2007, 59, 698–717. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.-W.; Li, B.; Li, C.-X.; Xu, L.; Fan, J.-X.; Lei, Q.; Zhang, X.-Z. Photocatalyzing CO2 to CO for Enhanced Cancer Therapy. Adv. Mater. 2017, 29, 1703822. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Liu, L.-H.; Jia, H.-Z.; Qiu, W.-X.; Rong, L.; Cheng, H.; Zhang, X.-Z. A PH-Responsive Prodrug for Real-Time Drug Release Monitoring and Targeted Cancer Therapy. Chem. Commun. 2014, 50, 11852–11855. [Google Scholar] [CrossRef] [PubMed]
- Şahin, A.; Eke, G.; Buyuksungur, A.; Hasirci, N.; Hasirci, V. Nuclear Targeting Peptide-Modified, DOX-Loaded, PHBV Nanoparticles Enhance Drug Efficacy by Targeting to Saos-2 Cell Nuclear Membranes. J. Biomater. Sci. Polym. Ed. 2018, 29, 507–519. [Google Scholar] [CrossRef]
- Chiu, H.Y.; Tay, E.X.Y.; Ong, D.S.T.; Taneja, R. Mitochondrial Dysfunction at the Center of Cancer Therapy. Antioxid. Redox Signal. 2020, 32, 309–330. [Google Scholar] [CrossRef]
- Deng, J. How Unleash Mitochondrial Apoptotic Blockades Kill Cancers? Acta Pharm. Sin. B 2017, 7, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, P.P.; Naik, P.P.; Panigrahi, D.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Sethi, G.; Bhutia, S.K. Intricate Role of Mitochondrial Lipid in Mitophagy and Mitochondrial Apoptosis: Its Implication in Cancer Therapeutics. Cell. Mol. Life Sci. 2019, 76, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, M.E.; Moreno-Loshuertos, R.; López, C.; Casqueiro, M.; Silva, J.; Bonilla, F.; de Córdoba, S.R.; Enríquez, J.A. m.6267G>A: A recurrent mutation in the human mitochondrial DNA that reduces cytochrome c oxidase activity and is asso-ciated with tumors. Hum. Mutat. 2006, 27, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Dakubo, G.D.; Parr, R.L.; Costello, L.C.; Franklin, R.B.; E Thayer, R. Altered metabolism and mitochondrial genome in prostate cancer. J. Clin. Pathol. 2006, 59, 10–16. [Google Scholar] [CrossRef]
- Morales-Valencia, J.; David, G. The origins of cancer cell dormancy. Curr. Opin. Genet. Dev. 2022, 74, 101914. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial Dysfunction and Mitochondrial Dynamics-The Cancer Connection. Biochim. Biophys. Acta (BBA)—Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Jiang, L.; Zhou, S.; Zhang, X.; Li, C.; Ji, S.; Mao, H.; Jiang, X. Mitochondrion-Specific Dendritic Lipopeptide Liposomes for Targeted Sub-Cellular Delivery. Nat. Commun. 2021, 12, 2390. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Nezlin, R. Aptamers in Immunological Research. Immunol. Lett. 2014, 162, 252–255. [Google Scholar] [CrossRef]
- Cerchia, L.; de Franciscis, V. Targeting Cancer Cells with Nucleic Acid Aptamers. Trends Biotechnol. 2010, 28, 517–525. [Google Scholar] [CrossRef]
- Banerjee, J. Antibodies Are Challenged. Indian J. Med. Sci. 2010, 64, 144–147. [Google Scholar]
- Jayasena, S.D. Aptamers: An Emerging Class of Molecules That Rival Antibodies in Diagnostics. Clin. Chem. 1999, 45, 1628–1650. [Google Scholar] [CrossRef]
- Huang, G.N. Biotinylation of Cell Surface Proteins. Bio Protoc. 2012, 2, e170. [Google Scholar] [CrossRef]
- Zhu, H.; Wu, E.; Pan, Z.; Zhang, C.; Zhang, Y.; Liao, Q.; Wang, Y.; Sun, Y.; Ye, M.; Wu, W. Development of an Aptamer-Based Molecular Tool for Specifically Targeting Microglia via the CD64 Protein. Anal. Chem. 2023, 95, 3238–3246. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Qiao, L.; Zhou, S.-F.; Xiang, D.; Wang, T.; Li, Y.; Lim, L.Y.; Kong, L.; Li, L.; Duan, W. RNA Aptamers Targeting Cancer Stem Cell Marker CD133. Cancer Lett. 2013, 330, 84–95. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V. The Role of the Complement System in Cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, Q.; Li, T.; Liao, Q.; Zhao, Y. Role of the Complement System in the Tumor Microenvironment. Cancer Cell Int. 2019, 19, 300. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A Key System for Immune Surveillance and Homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Fishelson, Z.; Donin, N.; Zell, S.; Schultz, S.; Kirschfink, M. Obstacles to Cancer Immunotherapy: Expression of Membrane Complement Regulatory Proteins (MCRPs) in Tumors. Mol. Immunol. 2003, 40, 109–123. [Google Scholar] [CrossRef]
- Kolev, M.; Le Friec, G.; Kemper, C. Complement—Tapping into New Sites and Effector Systems. Nat. Rev. Immunol. 2014, 14, 811–820. [Google Scholar] [CrossRef]
- Stecker, J.R.; Savage, A.A.; Bruno, J.G.; García, D.M.; Koke, J.R. Dynamics and Visualization of MCF7 Adenocarcinoma Cell Death by Aptamer-C1q-Mediated Membrane Attack. Nucleic Acid Ther. 2012, 22, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.-P. Recent Advances in Aptamers Targeting Immune System. Inflammation 2017, 40, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Michaud, D.S.; Houseman, E.A.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. Understanding the Role of the Immune System in the Development of Cancer: New Opportunities for Population-Based Research. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Gerada, C.; Ryan, K.M. The innate immune response and cancer. Molecular Oncology. Autophagy 2020, 14, 1913–1929. [Google Scholar]
- Masters, S.L.; De Nardo, D. Innate Immunity. Curr. Opin. Immunol. 2014, 26, v–vi. [Google Scholar] [CrossRef]
- Shuai, Z.; Leung, M.W.; He, X.; Zhang, W.; Yang, G.; Leung, P.S.; Gershwin, M.E. Adaptive immunity in the liver. Cell Mol. Immunol. 2016, 13, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Jiang, Z.; Yang, Z.; Liu, L.; Zhu, Z.; Jin, Y.; Yin, Y. Development of a Modularized Aptamer Targeting the Nuclear T-Cell Suppressor PAC1. Analyst 2023, 148, 2616–2625. [Google Scholar] [CrossRef]
- Clawson, G.A.; Abraham, T.; Pan, W.; Tang, X.; Linton, S.S.; McGovern, C.O.; Loc, W.S.; Smith, J.P.; Butler, P.J.; Kester, M.; et al. A Cholecystokinin B Receptor-Specific DNA Aptamer for Targeting Pancreatic Ductal Adenocarcinoma. Nucleic Acid Ther. 2017, 27, 23–35. [Google Scholar] [CrossRef]
- Chen, Z.; Zeng, Z.; Wan, Q.; Liu, X.; Qi, J.; Zu, Y. Targeted immunotherapy of triple-negative breast cancer by aptamer-engineered NK cells. Biomaterials 2022, 280, 121259. [Google Scholar] [CrossRef]
- Kue, C.S.; Kamkaew, A.; Burgess, K.; Kiew, L.V.; Chung, L.Y.; Lee, H.B. Small Molecules for Active Targeting in Cancer. Med. Res. Rev. 2016, 36, 494–575. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Low, P.S. Folate-Targeted Therapies for Cancer. J. Med. Chem. 2010, 53, 6811–6824. [Google Scholar] [CrossRef] [PubMed]
- Pilch, J.; Potęga, A.; Kowalczyk, A.; Kasprzak, A.; Kowalik, P.; Bujak, P.; Paluszkiewicz, E.; Augustin, E.; Nowicka, A.M. PH-Responsive Drug Delivery Nanoplatforms as Smart Carriers of Unsymmetrical Bisacridines for Targeted Cancer Therapy. Pharmaceutics 2023, 15, 201. [Google Scholar] [CrossRef]
- Torchilin, V. Tumor Delivery of Macromolecular Drugs Based on the EPR Effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef]
- Gilson, P.; Merlin, J.-L.; Harlé, A. Deciphering Tumour Heterogeneity: From Tissue to Liquid Biopsy. Cancers 2022, 14, 1384. [Google Scholar] [CrossRef] [PubMed]
- Dutta, B.; Barick, K.C.; Hassan, P.A. Recent Advances in Active Targeting of Nanomaterials for Anticancer Drug Delivery. Adv. Colloid Interface Sci. 2021, 296, 102509. [Google Scholar] [CrossRef]
- Fan, Z.; Wang, Y.; Xiang, S.; Zuo, W.; Huang, D.; Jiang, B.; Sun, H.; Yin, W.; Xie, L.; Hou, Z. Dual-Self-Recognizing, Stimulus-Responsive and Carrier-Free Methotrexate–Mannose Conjugate Nanoparticles with Highly Synergistic Chemotherapeutic Effects. J. Mater. Chem. B 2020, 8, 1922–1934. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Shi, D.; Zuo, W.; Feng, J.; Ge, D.; Su, G.; Yang, L.; Hou, Z. Trojan-Horse Diameter-Reducible Nanotheranostics for Macroscopic/Microscopic Imaging-Monitored Chemo-Antiangiogenic Therapy. ACS Appl. Mater. Interfaces 2022, 14, 5033–5052. [Google Scholar] [CrossRef]
- Fan, Z.; Jiang, B.; Zhu, Q.; Xiang, S.; Tu, L.; Yang, Y.; Zhao, Q.; Huang, D.; Han, J.; Su, G.; et al. Tumor-Specific Endogenous Fe II -Activated, MRI-Guided Self-Targeting Gadolinium-Coordinated Theranostic Nanoplatforms for Amplification of ROS and Enhanced Chemodynamic Chemotherapy. ACS Appl. Mater. Interfaces 2020, 12, 14884–14904. [Google Scholar] [CrossRef]
- Barenholz, Y. (Chezy) Doxil®—The First FDA-Approved Nano-Drug: Lessons Learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Danhier, F. To Exploit the Tumor Microenvironment: Since the EPR Effect Fails in the Clinic, What Is the Future of Nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef]
- Chawla, S.P.; Bruckner, H.; Morse, M.A.; Assudani, N.; Hall, F.L.; Gordon, E.M. A Phase I-II Study Using Rexin-G Tumor-Targeted Retrovector Encoding a Dominant-Negative Cyclin G1 Inhibitor for Advanced Pancreatic Cancer. Mol. Ther.-Oncolytics 2019, 12, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Matsumura, Y.; Nakanishi, Y.; Muro, K.; Yamada, Y.; Shimada, Y.; Shirao, K.; Niki, H.; Hosokawa, S.; Tagawa, T.; et al. Antitumor Effect of MCC-465, Pegylated Liposomal Doxorubicin Tagged with Newly Developed Monoclonal Antibody GAH, in Colorectal Cancer Xenografts. Cancer Sci. 2004, 95, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.E.; Gritli, I.; Tolcher, A.; Heidel, J.D.; Lim, D.; Morgan, R.; Chmielowski, B.; Ribas, A.; Davis, M.E.; Yen, Y. Correlating Animal and Human Phase Ia/Ib Clinical Data with CALAA-01, a Targeted, Polymer-Based Nanoparticle Containing SiRNA. Proc. Natl. Acad. Sci. USA 2014, 111, 11449–11454. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Wang, Q.; Xia, G.; Adilijiang, N.; Li, Y.; Hou, Z.; Fan, Z.; Li, J. Recent Advances in Targeted Drug Delivery Strategy for Enhancing Oncotherapy. Pharmaceutics 2023, 15, 2233. https://doi.org/10.3390/pharmaceutics15092233
Li J, Wang Q, Xia G, Adilijiang N, Li Y, Hou Z, Fan Z, Li J. Recent Advances in Targeted Drug Delivery Strategy for Enhancing Oncotherapy. Pharmaceutics. 2023; 15(9):2233. https://doi.org/10.3390/pharmaceutics15092233
Chicago/Turabian StyleLi, Jianmin, Qingluo Wang, Guoyu Xia, Nigela Adilijiang, Ying Li, Zhenqing Hou, Zhongxiong Fan, and Jinyao Li. 2023. "Recent Advances in Targeted Drug Delivery Strategy for Enhancing Oncotherapy" Pharmaceutics 15, no. 9: 2233. https://doi.org/10.3390/pharmaceutics15092233
APA StyleLi, J., Wang, Q., Xia, G., Adilijiang, N., Li, Y., Hou, Z., Fan, Z., & Li, J. (2023). Recent Advances in Targeted Drug Delivery Strategy for Enhancing Oncotherapy. Pharmaceutics, 15(9), 2233. https://doi.org/10.3390/pharmaceutics15092233