
Praziquantel Fifty Years on: A Comprehensive Overview of Its Solid State †



Abstract
:
1. Introduction
2. (RS)-Praziquantel ((RS)-PZQ) (Form A)
2.1. Physical and Chemical Properties
2.2. Crystal Structure of ((RS)-PZQ) (Form A)
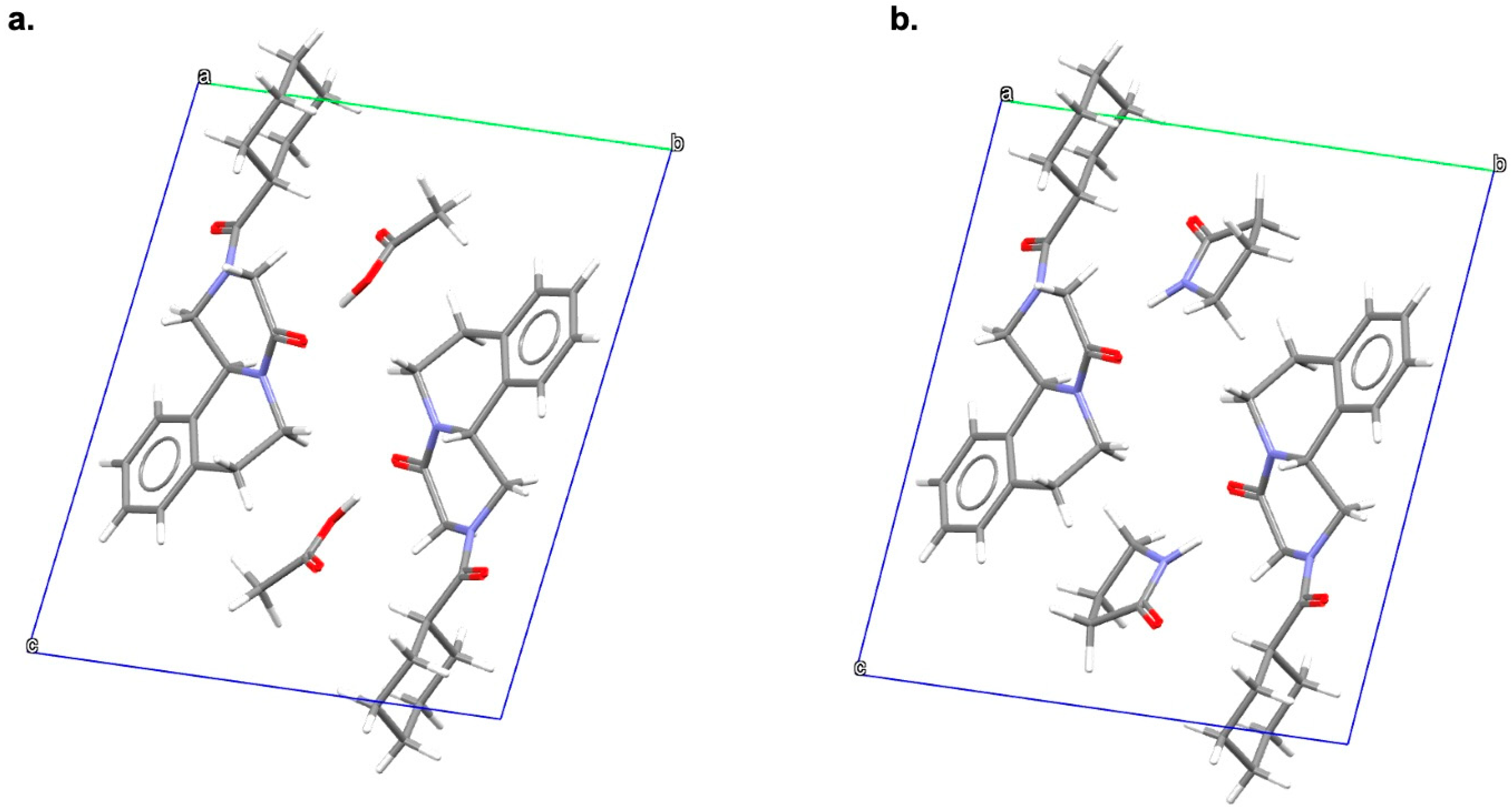
2.3. Solid-State Characterization Analyses
2.3.1. FT-IR Spectrum
2.3.2. SSNMR Analysis
3. PZQ Enantiomers
4. PZQ Crystalline Polymorphs
5. PZQ Amorphous Forms
6. PZQ Multicomponent Solid Forms
6.1. PZQ Hydrates
6.2. PZQ Solvates
6.3. PZQ Cocrystals
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AA | acetic acid |
| CAN | Acetonitrile |
| AcT | Acetone |
| AIM | atoms in molecules |
| API | active pharmaceutical ingredient |
| ASU | asymmetric unit |
| AUC | area under the curve |
| BCS | Biopharmaceutics Classification System |
| CCDC | Cambridge Crystallographic Data Centre |
| CHF | chloroform |
| CSD | Cambridge Structural Database |
| CyHXN | cyclohexanone |
| DFT | density functional theory |
| DMA | dimethylacetamide |
| DMSO | dimethyl sulfoxide |
| DSC | differential scanning calorimetry |
| EA | ethyl acetate |
| EDA | energy decomposition analysis |
| EDD | electron density difference |
| 1,2-EtdOH | 1,2-ethanediol |
| EtOH | ethanol |
| FE-SEM | field-emission SEM |
| FT-IR | Fourier Transform Infrared spectroscopy |
| GRAS | generally recognized as safe |
| H-bonds | hydrogen bonds |
| HPC | hydroxy propyl cellulose |
| HSM | hot-stage microscopy |
| n-HXN | n-hexane |
| H2O | water |
| IDR | intrinsic dissolution rate |
| IPOH | isopropanol |
| LAG | liquid-assisted grinding |
| MeOH | methanol |
| MEPS | molecular electrostatic potential surfaces |
| MTDSC | modulated differential scanning calorimetry |
| NG | neat grinding |
| PM | physical mixture |
| 2-PROH | 2-propanol |
| PXRD | powder X-ray diffraction |
| PZQ | Praziquantel |
| 2-pyr | 2-pyrrolidone |
| RH % | relative humidity percentage |
| SEE | solvent evaporation experiment |
| SEM | scanning electron microscopy |
| SI | saturation index |
| SPE | (Single Pulse Excitation) |
| SSNMR | solid-state NMR |
| STM | saturation temperature measurements |
| SXRD | single-crystal X-ray diffraction |
| TEA | triethylamine |
| TGA | thermogravimetric analysis |
| THF | tetrahydrofuran |
| WHO | World Health Organization |
| WBR | worm burden reduction |
References
- Schistosomiasis. Available online: https://www.who.int/news-room/fact-sheets/detail/schistosomiasis (accessed on 20 November 2023).
- Waechtler, A.; Cezanne, B.; Maillard, D.; Sun, R.; Wang, S.; Wang, J.; Harder, A. Praziquantel–50 Years of Research. ChemMedChem 2023, 18, e202300154. [Google Scholar] [CrossRef] [PubMed]
- SIMET (Ed.) Schistosomiasi: Raccomandazioni per la gestione clinica in Italia. In Quaderni della Società Italiana di Medicina Tropicale e Salute Globale (SIMET) n°7; SIMET: Roma, Italy, 2023; ISBN 978-88-900025-8-8. [Google Scholar]
- McManus, D.P.; Dunne, D.W.; Sacko, M.; Utzinger, J.; Vennervald, B.J.; Zhou, X.-N. Schistosomiasis. Nat. Rev. Dis. Primers 2018, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human Schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef] [PubMed]
- Gryseels, B.; Polman, K.; Clerinx, J.; Kestens, L. Human Schistosomiasis. Lancet 2006, 368, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Cioli, D.; Pica-Mattoccia, L.; Basso, A.; Guidi, A. Schistosomiasis Control: Praziquantel Forever? Mol. Biochem. Parasitol. 2014, 195, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Cioli, D.; Pica-Mattoccia, L. Praziquantel. Parasitol. Res. 2003, 90, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- WHO Model List of Essential Medicines-22nd List. 2021. Available online: https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2021.02 (accessed on 18 September 2023).
- WHO Model List of Essential Medicines for Children-8th List. 2021. Available online: https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2021.03 (accessed on 18 September 2023).
- Sousa-Figueiredo, J.C.; Betson, M.; Stothard, J.R. Treatment of Schistosomiasis in African Infants and Preschool-Aged Children: Downward Extension and Biometric Optimization of the Current Praziquantel Dose Pole. Int. Health 2012, 4, 95–102. [Google Scholar] [CrossRef]
- Lindenberg, M.; Kopp, S.; Dressman, J.B. Classification of Orally Administered Drugs on the World Health Organization Model List of Essential Medicines According to the Biopharmaceutics Classification System. Eur. J. Pharm. Biopharm. 2004, 58, 265–278. [Google Scholar] [CrossRef]
- El-Subbagh, H.I.; Al-Badr, A.A. Praziquantel. In Analytical Profiles of Drug Substances and Excipients; Harry, B., Ed.; Academic Press: Cambridge, MA, USA, 1998; Volume 25, pp. 463–500. [Google Scholar]
- Benet, L.Z.; Broccatelli, F.; Oprea, T.I. BDDCS Applied to Over 900 Drugs. AAPS J. 2011, 13, 519–547. [Google Scholar] [CrossRef]
- González-Esquivel, D.; Rivera, J.; Castro, N.; Yepez-Mulia, L.; Helgi Jung, C. In Vitro Characterization of Some Biopharmaceutical Properties of Praziquantel. Int. J. Pharm. 2005, 295, 93–99. [Google Scholar] [CrossRef]
- Jung-Cook, H. Pharmacokinetic Variability of Anthelmintics: Implications for the Treatment of Neurocysticercosis. Expert. Rev. Clin. Pharmacol. 2012, 5, 21–30. [Google Scholar] [CrossRef]
- El-Arini, S.K.; Leuenberger, H. Dissolution Properties of Praziquantel–PVP Systems. Pharm. Acta Helv. 1998, 73, 89–94. [Google Scholar] [CrossRef]
- FDA Biltricide (Praziquantel) Tablets Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/018714s012lbl.pdf (accessed on 4 November 2023).
- Cedillo-Cruz, A.; Aguilar, M.I.; Flores-Alamo, M.; Palomares-Alonso, F.; Jung-Cook, H. A Straightforward and Efficient Synthesis of Praziquantel Enantiomers and Their 4′-Hydroxy Derivatives. Tetrahedron Asymmetry 2014, 25, 133–140. [Google Scholar] [CrossRef]
- Meister, I.; Ingram-Sieber, K.; Cowan, N.; Todd, M.; Robertson, M.N.; Meli, C.; Patra, M.; Gasser, G.; Keiser, J. Activity of Praziquantel Enantiomers and Main Metabolites against Schistosoma mansoni. Antimicrob. Agents Chemother. 2014, 58, 5466–5472. [Google Scholar] [CrossRef]
- Kovač, J.; Vargas, M.; Keiser, J. In Vitro and in Vivo Activity of R- and S- Praziquantel Enantiomers and the Main Human Metabolite Trans-4-Hydroxy-Praziquantel against Schistosoma haematobium. Parasit. Vectors 2017, 10, 365. [Google Scholar] [CrossRef]
- Bagchus, W.M.; Bezuidenhout, D.; Harrison-Moench, E.; Kourany-Lefoll, E.; Wolna, P.; Yalkinoglu, O. Relative Bioavailability of Orally Dispersible Tablet Formulations of Levo- and Racemic Praziquantel: Two Phase I Studies. Clin. Transl. Sci. 2019, 12, 66–76. [Google Scholar] [CrossRef]
- Shu-Hua, X.; Catto, B.A. Comparative in Vitro and in Vivo Activity of Racemic Praziquantel and Its Levorotated Isomer on Schistosoma mansoni. J. Infect. Dis. 1989, 159, 589–592. [Google Scholar] [CrossRef]
- Liu, Y.H.; Qian, M.X.; Wang, X.G.; Quan, Y.Z.; Yan, S.H.; Chen, B.Y.; Li, J.S.; Qiu, Z.Y. Levo-Praziquantel versus Praziquantel in Experimental and Clinical Treatment of Schistosomiasis japonica. Chin. Med. J. 1993, 106, 593–596. [Google Scholar]
- Liu, Y.; Wang, X.; Wang, J.-K.; Ching, C.B. Investigation of the Phase Diagrams of Chiral Praziquantel. Chirality 2006, 18, 259–264. [Google Scholar] [CrossRef]
- Meyer, T.; Sekljic, H.; Fuchs, S.; Bothe, H.; Schollmeyer, D.; Miculka, C. Taste, A New Incentive to Switch to (R)-Praziquantel in Schistosomiasis Treatment. PLoS Negl. Trop. Dis. 2009, 3, 357. [Google Scholar] [CrossRef]
- Olliaro, P.; Delgado-Romero, P.; Keiser, J. The Little We Know about the Pharmacokinetics and Pharmacodynamics of Praziquantel (Racemate and R-Enantiomer). J. Antimicrob. Chemother. 2014, 69, 863–870. [Google Scholar] [CrossRef]
- Liu, Y.H.; Qian, M.X.; Wang, X.G.; Jia, J.; Wang, Q.N.; Jiang, Y.F.; Wang, R.Q.; Yan, S.H.; Chen, B.Y.; Li, J.S. Comparative efficacy of Praziquantel and its optic isomers in experimental therapy of Schistosomiasis japonica in rabbits. Chin. Med. J. 1986, 99, 935–940. [Google Scholar]
- De La Torre, P.; Torrado, S.; Torrado, S. Preparation, Dissolution and Characterization of Praziquantel Solid Dispersions. Chem. Pharm. Bull. 1999, 47, 1629–1633. [Google Scholar] [CrossRef]
- Costa, E.D.; Priotti, J.; Orlandi, S.; Leonardi, D.; Lamas, M.C.; Nunes, T.G.; Diogo, H.P.; Salomon, C.J.; Ferreira, M.J. Unexpected Solvent Impact in the Crystallinity of Praziquantel/Poly (Vinylpyrrolidone) Formulations. A Solubility, DSC and Solid-State NMR Study. Int. J. Pharm. 2016, 511, 983–993. [Google Scholar] [CrossRef]
- El-Lakkany, N.; Seif el-Din, S.H.; Heikal, L. Bioavailability and in Vivo Efficacy of a Praziquantel–Polyvinylpyrrolidone Solid Dispersion in Schistosoma mansoni-Infected Mice. Eur. J. Drug Metab. Pharmacokinet. 2012, 37, 289–299. [Google Scholar] [CrossRef]
- Chaud, M.; Lima, A.; Vila, M.; Paganelli, M.; Paula, F.; Pedreiro, L.; Gremião, M. Development and Evaluation of Praziquantel Solid Dispersions in Sodium Starch Glycolate. Trop. J. Pharm. Res. 2013, 12, 163–168. [Google Scholar] [CrossRef]
- Šagud, I.; Zanolla, D.; Zingone, G.; Perissutti, B.; Škorić, I. Impact of Mesoporous Silica on the Chemical Degradation of Praziquantel upon Grinding. Comptes Rendus Chim. 2021, 24, 233–242. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Carazo, E.; Albertini, B.; Passerini, N.; Perissutti, B.; Cerezo, P.; Viseras, C.; Hernández-Laguna, A.; Aguzzi, C.; Sainz-Díaz, I. Conformational Polymorphic Changes in the Crystal Structure of the Chiral Antiparasitic Drug Praziquantel and Interactions with Calcium Carbonate. Eur. J. Pharm. 2018, 132, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Di Marzio, L.; Borrego-Sánchez, A.; Felaco, M.; Pacinelli, M.; Gómez-Morales, J.; d’Avanzo, N.; Sainz-Díaz, C.I.; Celia, C.; Viseras, C. Praziquantel-Loaded Calcite Crystals: Synthesis, Physicochemical Characterization, and Biopharmaceutical Properties of Inorganic Biomaterials for Drug Delivery. J. Drug Deliv. Sci. Technol. 2022, 68, 103021. [Google Scholar] [CrossRef]
- Dametto, P.R.; Dametto, A.C.; Polese, L.; Ribeiro, C.A.; Chorilli, M.; de Freitas, O. Development and Physicochemical Characterization of Solid Dispersions Containing Praziquantel for the Treatment of Schistosomiasis. J. Therm. Anal. Calorim. 2017, 127, 1693–1706. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Carazo, E.; Aguzzi, C.; Viseras, C.; Sainz-Díaz, C.I. Biopharmaceutical Improvement of Praziquantel by Interaction with Montmorillonite and Sepiolite. Appl. Clay Sci. 2018, 160, 173–179. [Google Scholar] [CrossRef]
- Becket, G.; Schep, L.J.; Tan, Y. Improvement of the in Vitro Dissolution of Praziquantel by Complexation with α-, β- and γ-Cyclodextrins. Int. J. Pharm. 1999, 179, 65–71. [Google Scholar] [CrossRef]
- Cugovčan, M.; Jablan, J.; Lovrić, J.; Cinčić, D.; Galić, N.; Jug, M. Biopharmaceutical Characterization of Praziquantel Cocrystals and Cyclodextrin Complexes Prepared by Grinding. J. Pharm. Biomed. Anal. 2017, 137, 42–53. [Google Scholar] [CrossRef]
- Rodrigues, S.G.; Chaves, I.d.S.; Melo, N.F.S.; Jesus, M.B.; Fraceto, L.F.; Fernandes, S.A.; Paula, E.; de Freitas, M.P.; Pinto, L.d.M.A. Computational Analysis and Physico-Chemical Characterization of an Inclusion Compound between Praziquantel and Methyl-β-Cyclodextrin for Use as an Alternative in the Treatment of Schistosomiasis. J. Incl. Phenom. Macrocycl. Chem. 2011, 70, 19–28. [Google Scholar] [CrossRef]
- Maragos, S.; Archontaki, H.; Macheras, P.; Valsami, G. Effect of Cyclodextrin Complexation on the Aqueous Solubility and Solubility/Dose Ratio of Praziquantel. AAPS PharmSciTech 2009, 10, 1444. [Google Scholar] [CrossRef]
- Arrúa, E.C.; João, M.; Ferreira, G.; Salomon, C.J.; Nunes, T.G. Elucidating the Guest-Host Interactions and Complex Formation of Praziquantel and Cyclodextrin Derivatives by 13C and 15N Solid-State NMR Spectroscopy. Int. J. Pharm. 2015, 496, 812–821. [Google Scholar] [CrossRef]
- de Jesus, M.B.; de Matos Alves Pinto, L.; Fraceto, L.F.; Takahata, Y.; Lino, A.C.S.; Jaime, C.; de Paula, E. Theoretical and Experimental Study of a Praziquantel and β-Cyclodextrin Inclusion Complex Using Molecular Mechanic Calculations and 1H-Nuclear Magnetic Resonance. J. Pharm. Biomed. Anal. 2006, 41, 1428–1432. [Google Scholar] [CrossRef]
- da Silva Mourão, L.C.; Ribeiro Batista, D.R.M.; Honorato, S.B.; Ayala, A.P.; de Alencar Morais, W.; Barbosa, E.G.; Raffin, F.N.; de Lima e Moura, T.F.A. Effect of Hydroxypropyl Methylcellulose on Beta Cyclodextrin Complexation of Praziquantel in Solution and in Solid State. J. Incl. Phenom. Macrocycl. Chem. 2016, 85, 151–160. [Google Scholar] [CrossRef]
- Mourão, S.C.; Costa, P.I.; Salgado, H.R.N.; Gremião, M.P.D. Improvement of Antischistosomal Activity of Praziquantel by Incorporation into Phosphatidylcholine-Containing Liposomes. Int. J. Pharm. 2005, 295, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Partridge, G.J.; Rao, S.; Woolley, L.D.; Pilmer, L.; Lymbery, A.J.; Prestidge, C.A. Bioavailability and Palatability of Praziquantel Incorporated into Solid-Lipid Nanoparticles Fed to Yellowtail Kingfish Seriola lalandi. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2019, 218, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Geng, Y.; Li, H.; Zhang, Y.; You, J.; Chang, Y. Enhancement the Oral Bioavailability of Praziquantel by Incorporation into Solid Lipid Nanoparticles. Pharmazie 2009, 64, 86–89. [Google Scholar] [PubMed]
- Mainardes, R.M.; Gremião, M.P.D.; Evangelista, R.C. Thermoanalytical Study of Praziquantel-Loaded PLGA Nanoparticles. Rev. Bras. Ciênc Farm. 2006, 42, 523–530. [Google Scholar] [CrossRef]
- de Souza, A.L.R.; Andreani, T.; Nunes, F.M.; Cassimiro, D.L.; de Almeida, A.E.; Ribeiro, C.A.; Sarmento, V.H.V.; Gremião, M.P.D.; Silva, A.M.; Souto, E.B. Loading of Praziquantel in the Crystal Lattice of Solid Lipid Nanoparticles. J. Therm. Anal. Calorim. 2012, 108, 353–360. [Google Scholar] [CrossRef]
- Perissutti, B.; Passerini, N.; Trastullo, R.; Keiser, J.; Zanolla, D.; Zingone, G.; Voinovich, D.; Albertini, B. An Explorative Analysis of Process and Formulation Variables Affecting Comilling in a Vibrational Mill: The Case of Praziquantel. Int. J. Pharm. 2017, 533, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Sánchez, A.; Sánchez-Espejo, R.; Albertini, B.; Passerini, N.; Cerezo, P.; Viseras, C.; Sainz-Díaz, C.I. Ground Calcium Carbonate as a Low Cost and Biosafety Excipient for Solubility and Dissolution Improvement of Praziquantel. Pharmaceutics 2019, 11, 533. [Google Scholar] [CrossRef] [PubMed]
- Zanolla, D.; Perissutti, B.; Passerini, N.; Invernizzi, S.; Voinovich, D.; Bertoni, S.; Melegari, C.; Millotti, G.; Albertini, B. Milling and Comilling Praziquantel at Cryogenic and Room Temperatures: Assessment of the Process-Induced Effects on Drug Properties. J. Pharm. Biomed. Anal. 2018, 153, 82–89. [Google Scholar] [CrossRef]
- Gaggero, A.; Jurišić Dukovski, B.; Radić, I.; Šagud, I.; Škorić, I.; Cinčić, D.; Jug, M. Co-Grinding with Surfactants as a New Approach to Enhance in Vitro Dissolution of Praziquantel. J. Pharm. Biomed. Anal. 2020, 189, 113494. [Google Scholar] [CrossRef]
- Trastullo, R.; Dolci, L.S.; Passerini, N.; Albertini, B. Development of Flexible and Dispersible Oral Formulations Containing Praziquantel for Potential Schistosomiasis Treatment of Pre-School Age Children. Int. J. Pharm. 2015, 495, 536–550. [Google Scholar] [CrossRef]
- Passerini, N.; Albertini, B.; Perissutti, B.; Rodriguez, L. Evaluation of Melt Granulation and Ultrasonic Spray Congealing as Techniques to Enhance the Dissolution of Praziquantel. Int. J. Pharm. 2006, 318, 92–102. [Google Scholar] [CrossRef]
- Silva, A.D.A.; Sarcinelli, M.A.; Patricio, B.F.d.C.; Chaves, M.H.d.C.; Lima, L.M.; Parreiras, P.M.; Pinto, P.d.F.; Prado, L.D.; Rocha, H.V.A. Pharmaceutical Development of Micro and Nanocrystals of a Poorly Water-Soluble Drug: Dissolution Rate Enhancement of Praziquantel. J. Drug Deliv. Sci. Technol. 2023, 81, 104260. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, T.; Yu, J.; Liu, Y.; Wang, Y.; He, Z. In Vitro/Vivo Assessment of Praziquantel Nanocrystals: Formulation, Characterization, and Pharmacokinetics in Beagle Dogs. Asian J. Pharm. Sci. 2019, 14, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Lara, J.C.; Guzman-Villanueva, D.; Arenas-García, J.I.; Herrera-Ruiz, D.; Rivera-Islas, J.; Román-Bravo, P.; Morales-Rojas, H.; Höpfl, H. Cocrystals of Active Pharmaceutical Ingredients-Praziquantel in Combination with Oxalic, Malonic, Succinic, Maleic, Fumaric, Glutaric, Adipic, and Pimelic Acids. Cryst. Growth Des. 2013, 13, 169–185. [Google Scholar] [CrossRef]
- Zanolla, D.; Perissutti, B.; Passerini, N.; Chierotti, M.R.; Hasa, D.; Voinovich, D.; Gigli, L.; Demitri, N.; Geremia, S.; Keiser, J.; et al. A New Soluble and Bioactive Polymorph of Praziquantel. Eur. J. Pharm. Biopharm. 2018, 127, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Zanolla, D.; Perissutti, B.; Vioglio, P.C.; Chierotti, M.R.; Gigli, L.; Demitri, N.; Passerini, N.; Albertini, B.; Franceschinis, E.; Keiser, J.; et al. Exploring Mechanochemical Parameters Using a DoE Approach: Crystal Structure Solution from Synchrotron XRPD and Characterization of a New Praziquantel Polymorph. Eur. J. Pharm. Sci. 2019, 140, 105084. [Google Scholar] [CrossRef] [PubMed]
- Saikia, B.; Seidel-Morgenstern, A.; Lorenz, H. Role of Mechanochemistry in Solid Form Selection and Identification of the Drug Praziquantel. Cryst. Growth Des. 2021, 21, 5854–5861. [Google Scholar] [CrossRef]
- de Moraes, M.G.F.; Barreto, A.G., Jr.; Secchi, A.R.; de Souza, M.B., Jr.; Lage, P.L.D.C.; Myerson, A.S. Polymorphism of Praziquantel: Role of Cooling Crystallization in Access to Solid Forms and Discovery of New Polymorphs. Cryst. Growth Des. 2023, 23, 1247–1258. [Google Scholar] [CrossRef]
- Salazar-Rojas, D.; Maggio, R.M.; Kaufman, T.S. Preparation and Characterization of a New Solid Form of Praziquantel, an Essential Anthelmintic Drug. Praziquantel Racemic Monohydrate. Eur. J. Pharm. Sci. 2020, 146, 105267. [Google Scholar] [CrossRef]
- Zanolla, D.; Hasa, D.; Arhangelskis, M.; Schneider-Rauber, G.; Chierotti, M.R.; Keiser, J.; Voinovich, D.; Jones, W.; Perissutti, B. Mechanochemical Formation of Racemic Praziquantel Hemihydrate with Improved Biopharmaceutical Properties. Pharmaceutics 2020, 12, 289. [Google Scholar] [CrossRef]
- MacEachern, L.; Kermanshahi-Pour, A.; Mirmehrabi, M. Transformation under Pressure: Discovery of a Novel Crystalline Form of Anthelmintic Drug Praziquantel Using High-Pressure Supercritical Carbon Dioxide. Int. J. Pharm. 2022, 619, 121723. [Google Scholar] [CrossRef]
- Zanolla, D.; Gigli, L.; Hasa, D.; Chierotti, M.R.; Arhangelskis, M.; Demitri, N.; Jones, W.; Voinovich, D.; Perissutti, B. Mechanochemical Synthesis and Physicochemical Characterization of Previously Unreported Praziquantel Solvates with 2-Pyrrolidone and Acetic Acid. Pharmaceutics 2021, 13, 1606. [Google Scholar] [CrossRef]
- Rodríguez-Ruiz, C.; Salas-Zúñiga, R.; Obdulia Sánchez-Guadarrama, M.; Delgado-Díaz, A.; Herrera-Ruiz, D.; Morales-Rojas, H.; Höpfl, H. Structural, Physicochemical, and Biopharmaceutical Properties of Cocrystals with RS- and R-Praziquantel—Generation and Prolongation of the Supersaturation State in the Presence of Cellulosic Polymers. Cryst. Growth Des. 2022, 22, 6023–6038. [Google Scholar] [CrossRef]
- Sánchez-Guadarrama, O.; Mendoza-Navarro, F.; Cedillo-Cruz, A.; Jung-Cook, H.; Arenas-García, J.I.; Delgado-Díaz, A.; Herrera-Ruiz, D.; Morales-Rojas, H.; Höpfl, H. Chiral Resolution of RS-Praziquantel via Diastereomeric Co-Crystal Pair Formation with l-Malic Acid. Cryst. Growth Des. 2015, 16, 307–314. [Google Scholar] [CrossRef]
- Yang, D.; Cao, J.; Heng, T.; Xing, C.; Yang, S.; Zhang, L.; Lu, Y.; Du, G. Theoretical Calculation and Structural Analysis of the Cocrystals of Three Flavonols with Praziquantel. Cryst. Growth Des. 2021, 21, 2292–2300. [Google Scholar] [CrossRef]
- Devogelaer, J.J.; Charpentier, M.D.; Tijink, A.; Dupray, V.; Coquerel, G.; Johnston, K.; Meekes, H.; Tinnemans, P.; Vlieg, E.; Ter Horst, J.H.; et al. Cocrystals of Praziquantel: Discovery by Network-Based Link Prediction. Cryst. Growth Des. 2021, 21, 3428–3437. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yang, D.; Chen, T.; Zhang, B.; Xing, C.; Zhang, L.; Lu, Y.; Du, G. Insights into the Solubility and Structural Features of Four Praziquantel Cocrystals. Cryst. Growth Des. 2021, 21, 6321–6331. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Q.; Ji, W.; An, Q.; Song, J.; Xing, C.; Yang, D.; Zhang, L.; Lu, Y.; Du, G. Cocrystals of Praziquantel with Phenolic Acids: Discovery, Characterization, and Evaluation. Molecules 2022, 27, 2022. [Google Scholar] [CrossRef]
- D’Abbrunzo, I.; Bianco, E.; Gigli, L.; Demitri, N.; Birolo, R.; Chierotti, M.R.; Škoríc, I.; Keiser, J.; Häberli, C.; Voinovich, D.; et al. Praziquantel Meets Niclosamide: A Dual-Drug Antiparasitic Cocrystal. Int. J. Pharm. 2023, 644, 123315. [Google Scholar] [CrossRef]
- Cappuccino, C.; Spoletti, E.; Renni, F.; Muntoni, E.; Keiser, J.; Voinovich, D.; Perissutti, B.; Lusi, M. Co-Crystalline Solid Solution Affords a High-Soluble and Fast-Absorbing Form of Praziquantel. Mol. Pharm. 2022. [Google Scholar] [CrossRef]
- Persson, L.C.; Porter, C.J.H.; Charman, W.N.; Bergström, C.A.S. Computational Prediction of Drug Solubility in Lipid Based Formulation Excipients. Pharm. Res. 2013, 30, 3225–3237. [Google Scholar] [CrossRef]
- El-Arini, S.K.; Giron, D.; Leuenberger, H. Solubility Properties of Racemic Praziquantel and Its Enantiomers. Pharm. Dev. Technol. 1998, 3, 557–564. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Wang, M.; Ma, Y.; Tang, W. Uncovering the Effect of Solvents on Solid-Liquid Phase Equilibrium of Praziquantel. J. Mol. Liq. 2020, 297, 111917. [Google Scholar] [CrossRef]
- Li, R.; Chen, X.; He, G.; Wu, C.; Gan, Z.; He, Z.; Zhao, J.; Han, D. The Dissolution Behaviour and Thermodynamic Properties Calculation of Praziquantel in Pure and Mixed Organic Solvents. J. Chem. Thermodyn. 2020, 144, 106062. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Sánchez, A.; Viseras, C.; Aguzzi, C.; Sainz-Díaz, C.I. Molecular and Crystal Structure of Praziquantel. Spectroscopic Properties and Crystal Polymorphism. Eur. J. Pharm. Sci. 2016, 92, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Sánchez, A.; Hernández-Laguna, A.; Sainz-Díaz, C.I. Molecular Modeling and Infrared and Raman Spectroscopy of the Crystal Structure of the Chiral Antiparasitic Drug Praziquantel. J. Mol. Model. 2017, 23, 106. [Google Scholar] [CrossRef] [PubMed]
- Schepers, H.; Brasseur, R.; Goormaghtigh, E.; Duquenoy, P.; Ruysschaert, J.-M. Mode of Insertion of Praziquantel and Derivatives into Lipid Membranes. Biochem. Pharmacol. 1988, 37, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Wang, J.; Ching, C.B. Structural Characterization and Enantioseparation of the Chiral Compound Praziquantel. J. Pharm. Sci. 2004, 93, 3039–3046. [Google Scholar] [CrossRef]
- da Silva, V.B.R.; Campos, B.R.K.L.; de Oliveira, J.F.; Decout, J.-L.; do Carmo Alves de Lima, M. Medicinal Chemistry of Antischistosomal Drugs: Praziquantel and Oxamniquine. Bioorg. Med. Chem. 2017, 25, 3259–3277. [Google Scholar] [CrossRef]
- Cedillo-Cruz, A.; Aguilar, M.I.; Jung-Cook, H. (S)-(+)-Cis-4′-Benzyloxypraziquantel. Acta Crystallogr. E Struct. Sci. Cryst. Eng. Mater. 2013, 69, 1835–1836. [Google Scholar] [CrossRef]
- Valenti, G.; Tinnemans, P.; Baglai, I.; Noorduin, W.L.; Kaptein, B.; Leeman, M.; ter Horst, J.H.; Kellogg, R.M. Combining Incompatible Processes for Deracemization of a Praziquantel Derivative under Flow Conditions. Angew. Chem. Int. Ed. 2021, 60, 5279–5282. [Google Scholar] [CrossRef]
- McNaught, A.D.; Wilkinson, A. The IUPAC Compendium of Chemical Terminology, 2nd ed.; Gold, V., Ed.; International Union of Pure and Applied Chemistry (IUPAC): Research Triangle Park, NC, USA, 1997. [Google Scholar]
- Raza, K. Polymorphism: The Phenomenon Affecting the Performance of Drugs. SOJ Pharm. Pharm. Sci. 2014. [Google Scholar] [CrossRef]
- Brittain, H.G. Polymorphism and Solvatomorphism 2010. J. Pharm. Sci. 2012, 101, 464–484. [Google Scholar] [CrossRef] [PubMed]
- Singhal, D. Drug Polymorphism and Dosage Form Design: A Practical Perspective. Adv. Drug Deliv. Rev. 2004, 56, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Leopold, G.; Ungethum, W.; Groll, E.; Diekmann, H.W.; Nowak, H.; Wegner, D.H.G. Clinical Pharmacology in Normal Volunteers of Praziquantel, a New Drug against Schistosomes and Cestodes. Eur. J. Clin. Pharmacol. 1978, 14, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Bylund, G.; Bång, B.; Wikgren, K. Tests with a New Compound (Praziquantel) against Diphyllobothrium latum. J. Helminthol. 1977, 51, 115–119. [Google Scholar] [CrossRef]
- Toro, R.; Kaduk, J.; Delgado, M.; Díaz de Delgado, G. Structural Characterization of Praziquantel: A Broad Spectrum Anthelmintic, in Powder Diffraction. Annu. Spring Meet. Powder Diffr. 2014, 29, 206–207. [Google Scholar]
- Lombardo, F.C.; Perissutti, B.; Keiser, J. Activity and Pharmacokinetics of a Praziquantel Crystalline Polymorph in the Schistosoma mansoni Mouse Model. Eur. J. Pharm. Biopharm. 2019, 142, 240–246. [Google Scholar] [CrossRef]
- Doehlert, D.H. Uniform Shell Designs. Appl. Stat. 1970, 19, 231. [Google Scholar] [CrossRef]
- Mathieu, D.; Nony, J.; Phan-Tan-Luu, R. New Efficient Technology for Research Using Optimal Design (NEMRODW); LPRAI: Marseille, France, 2011. [Google Scholar]
- Mayoka, G.; Keiser, J.; Häberli, C.; Chibale, K. Structure–Activity Relationship and in Vitro Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Studies of N-Aryl 3-Trifluoromethyl Pyrido[1,2-A]Benzimidazoles That Are Efficacious in a Mouse Model of Schistosomiasis. ACS Infect. Dis. 2019, 5, 418–429. [Google Scholar] [CrossRef]
- Salas-Zúñiga, R.; Mondragón-Vásquez, K.; Alcalá-Alcalá, S.; Lima, E.; Höpfl, H.; Herrera-Ruiz, D.; Morales-Rojas, H. Nanoconfinement of a Pharmaceutical Cocrystal with Praziquantel in Mesoporous Silica: The Influence of the Solid Form on Dissolution Enhancement. Mol. Pharm. 2022, 19, 414–431. [Google Scholar] [CrossRef]
- Zanolla, D.; Bertoni, S.; Passerini, N.; Albertini, B.; Zingone, G.; Perissutti, B. From Bitter to Sweet: A Preliminary Study towards a Patient-Friendly Praziquantel Dosage Form. Comptes Rendus Chim. 2022, 25, 179–188. [Google Scholar] [CrossRef]
- Alzghoul, A.; Alhalaweh, A.; Mahlin, D.; Bergström, C.A.S. Experimental and Computational Prediction of Glass Transition Temperature of Drugs. J. Chem. Inf. Model. 2014, 54, 3396–3403. [Google Scholar] [CrossRef] [PubMed]
- Khankari, R.K.; Grant, D.J.W. Pharmaceutical Hydrates. Thermochim. Acta 1995, 248, 61–79. [Google Scholar] [CrossRef]
- Tode, C.; Takeuchi, A.; Iwakawa, S.; Tatsumi, A.; Sugiura, M. Hydrogen-Deuterium (H-D) Exchange Reaction of Warfarin in D2O Solution. Chem. Pharm. Bull. 2009, 57, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Rojas, D.; Kaufman, T.S.; Maggio, R.M. A Study of the Heat-Mediated Phase Transformations of Praziquantel Hydrates. Evaluation of Their Impact on the Dissolution Rate. Heliyon 2022, 8, 11317. [Google Scholar] [CrossRef] [PubMed]
- Griesser, U.J. The Importance of Solvates. In Polymorphism; Wiley: New York, NY, USA, 2006; pp. 211–233. [Google Scholar]
- Friščić, T.; Childs, S.L.; Rizvi, S.A.A.; Jones, W. The Role of Solvent in Mechanochemical and Sonochemical Cocrystal Formation: A Solubility-Based Approach for Predicting Cocrystallisation Outcome. CrystEngComm 2009, 11, 418–426. [Google Scholar] [CrossRef]
- Von Essen, C.; Luedeker, D. In Silico Co-Crystal Design: Assessment of the Latest Advances. Drug Discov. Today 2023, 28, 103763. [Google Scholar] [CrossRef] [PubMed]
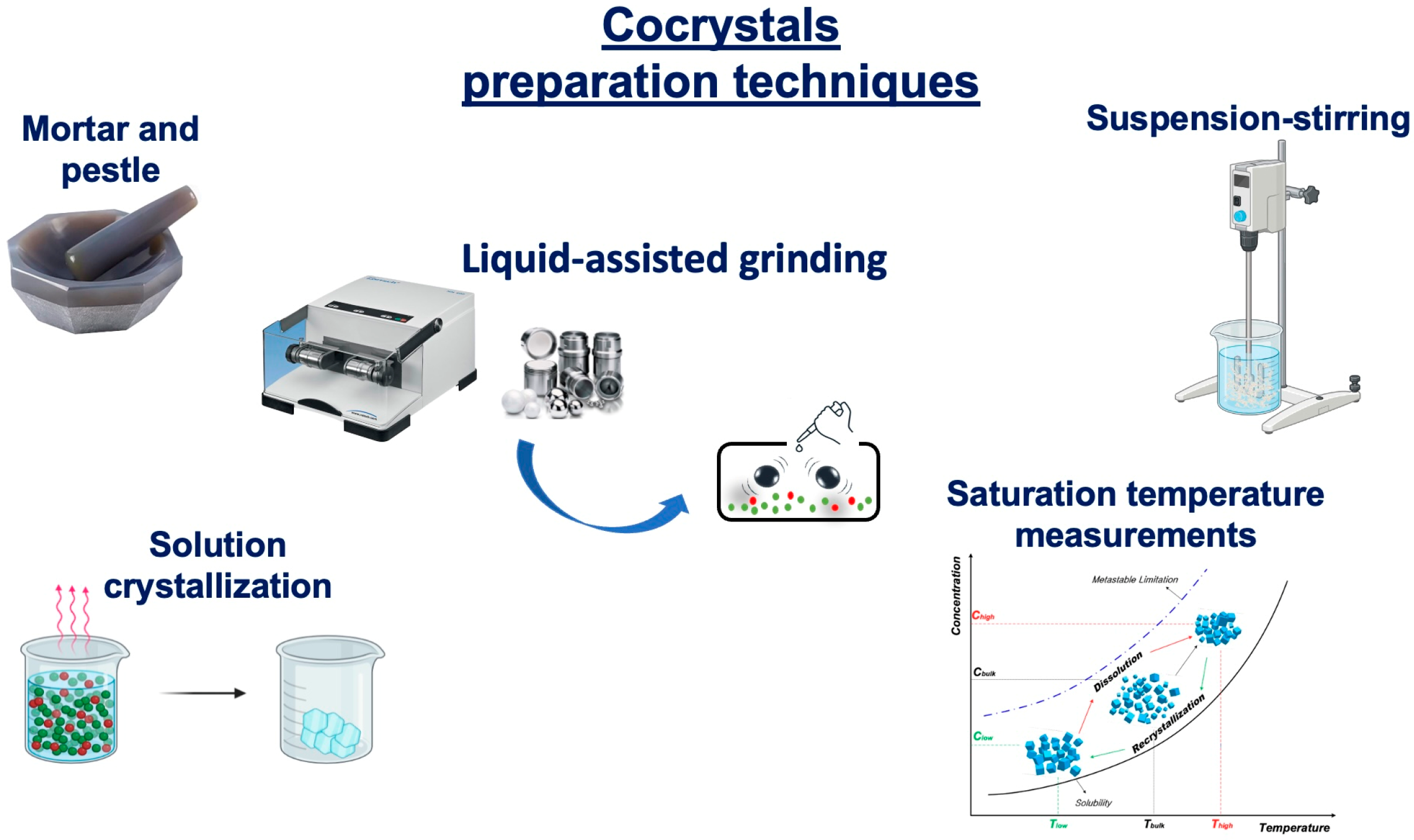
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Shaikh, R.; Singh, R.; Walker, G.M.; Croker, D.M. Pharmaceutical Cocrystal Drug Products: An Outlook on Product Development. Trends Pharmacol. Sci. 2018, 39, 1033–1048. [Google Scholar] [CrossRef]
- Douroumis, D.; Ross, S.A.; Nokhodchi, A. Advanced Methodologies for Cocrystal Synthesis. Adv. Drug Deliv. Rev. 2017, 117, 178–195. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Yang, F.; Zhang, X.; Hu, W. Cocrystal Engineering: A Collaborative Strategy toward Functional Materials. Adv. Mater. 2019, 31, e1902328. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.R.; Day, G.M. Evaluating the Energetic Driving Force for Cocrystal Formation. Cryst. Growth Des. 2018, 18, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Friščić, T.; Jones, W. Benefits of Cocrystallisation in Pharmaceutical Materials Science: An Update. J. Pharm. Pharmacol. 2010, 62, 1547–1559. [Google Scholar] [CrossRef] [PubMed]
- Urbanus, J.; Roelands, C.P.M.; Verdoes, D.; Jansens, P.J.; ter Horst, J.H. Co-Crystallization as a Separation Technology: Controlling Product Concentrations by Co-Crystals. Cryst. Growth Des. 2010, 10, 1171–1179. [Google Scholar] [CrossRef]
- Schultheiss, N.; Newman, A. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef]
- Thakuria, R.; Delori, A.; Jones, W.; Lipert, M.P.; Roy, L.; Rodríguez-Hornedo, N. Pharmaceutical Cocrystals and Poorly Soluble Drugs. Int. J. Pharm. 2013, 453, 101–125. [Google Scholar] [CrossRef]
- Wasim, M.; Abdul Mannan, A.M.; Azim, T.; Ali Khan, R.; Alotaibi, G.; Amer, M.; Shafique, M.; Asghar Khan, M.; Gul, S.; Hussain, I. Physicochemical and Pharmacokinetic Evaluation of Praziquantel Co-Crystals by Varying the Spacer Group of Co-Crystal Formers. Sains Malays. 2022, 51, 3271–3284. [Google Scholar] [CrossRef]
- Hamilton, B.D.; Ha, J.-M.; Hillmyer, M.A.; Ward, M.D. Manipulating Crystal Growth and Polymorphism by Confinement in Nanoscale Crystallization Chambers. Acc. Chem. Res. 2012, 45, 414–423. [Google Scholar] [CrossRef]
- Nartowski, K.P.; Malhotra, D.; Hawarden, L.E.; Fábián, L.; Khimyak, Y.Z. Nanocrystallization of Rare Tolbutamide Form V in Mesoporous MCM-41 Silica. Mol. Pharm. 2018, 15, 4926–4932. [Google Scholar] [CrossRef]
- Nartowski, K.P.; Tedder, J.; Braun, D.E.; Fábián, L.; Khimyak, Y.Z. Building Solids inside Nano-Space: From Confined Amorphous through Confined Solvate to Confined ‘Metastable’ Polymorph. Phys. Chem. Chem. Phys. 2015, 17, 24761–24773. [Google Scholar] [CrossRef]
- Sun, D.D.; Wen, H.; Taylor, L.S. Non-Sink Dissolution Conditions for Predicting Product Quality and In Vivo Performance of Supersaturating Drug Delivery Systems. J. Pharm. Sci. 2016, 105, 2477–2488. [Google Scholar] [CrossRef] [PubMed]
- Reddy, L.S.; Bethune, S.J.; Kampf, J.W.; Rodríguez-Hornedo, N. Cocrystals and Salts of Gabapentin: pH Dependent Cocrystal Stability and Solubility. Cryst. Growth Des. 2009, 9, 378–385. [Google Scholar] [CrossRef]
- Adachi, M.; Hinatsu, Y.; Kusamori, K.; Katsumi, H.; Sakane, T.; Nakatani, M.; Wada, K.; Yamamoto, A. Improved Dissolution and Absorption of Ketoconazole in the Presence of Organic Acids as PH-Modifiers. Eur. J. Pharm. Sci. 2015, 76, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Hunter, C.A.; Prohens, R.; Scuderi, S.; McCabe, J.F. Virtual Cocrystal Screening. Chem. Sci. 2011, 2, 883. [Google Scholar] [CrossRef]
- Devogelaer, J.-J.; Brugman, S.J.T.; Meekes, H.; Tinnemans, P.; Vlieg, E.; de Gelder, R. Cocrystal Design by Network-Based Link Prediction. CrystEngComm 2019, 21, 6875–6885. [Google Scholar] [CrossRef]
- Devogelaer, J.-J.; Meekes, H.; Vlieg, E.; de Gelder, R. Cocrystals in the Cambridge Structural Database: A Network Approach. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2019, 75, 371–383. [Google Scholar] [CrossRef]
- Charpentier, M.D.; Devogelaer, J.-J.; Tijink, A.; Meekes, H.; Tinnemans, P.; Vlieg, E.; De Gelder, R.; Johnston, K.; Ter Horst, J.H. Comparing and Quantifying the Efficiency of Cocrystal Screening Methods for Praziquantel. Cryst. Growth Des. 2022, 22, 5525. [Google Scholar] [CrossRef]
- Yang, D.; Wang, L.; Yuan, P.; An, Q.; Su, B.; Yu, M.; Chen, T.; Hu, K.; Zhang, L.; Lu, Y.; et al. Cocrystal Virtual Screening Based on the XGBoost Machine Learning Model. Chin. Chem. Lett. 2023, 34, 107964. [Google Scholar] [CrossRef]
- Dong, J.; Cao, D.-S.; Miao, H.-Y.; Liu, S.; Deng, B.-C.; Yun, Y.-H.; Wang, N.-N.; Lu, A.-P.; Zeng, W.-B.; Chen, A.F. ChemDes: An Integrated Web-Based Platform for Molecular Descriptor and Fingerprint Computation. J. Cheminform 2015, 7, 60. [Google Scholar] [CrossRef]
- Sanphui, P.; Kumar, S.S.; Nangia, A. Pharmaceutical Cocrystals of Niclosamide. Cryst. Growth Des. 2012, 12, 4588–4599. [Google Scholar] [CrossRef]
- Bhushan, B.; Dubey, P.; Kumar, S.U.; Sachdev, A.; Matai, I.; Gopinath, P. Bionanotherapeutics: Niclosamide Encapsulated Albumin Nanoparticles as a Novel Drug Delivery System for Cancer Therapy. RSC Adv. 2015, 5, 12078–12086. [Google Scholar] [CrossRef]
- Xie, Y.; Yao, Y. Octenylsuccinate Hydroxypropyl Phytoglycogen Enhances the Solubility and In-Vitro Antitumor Efficacy of Niclosamide. Int. J. Pharm. 2018, 535, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Generally Recognized as Safe (GRAS)|FDA. Available online: https://www.fda.gov/food/food-ingredients-packaging/generally-recognized-safe-gras (accessed on 6 December 2023).
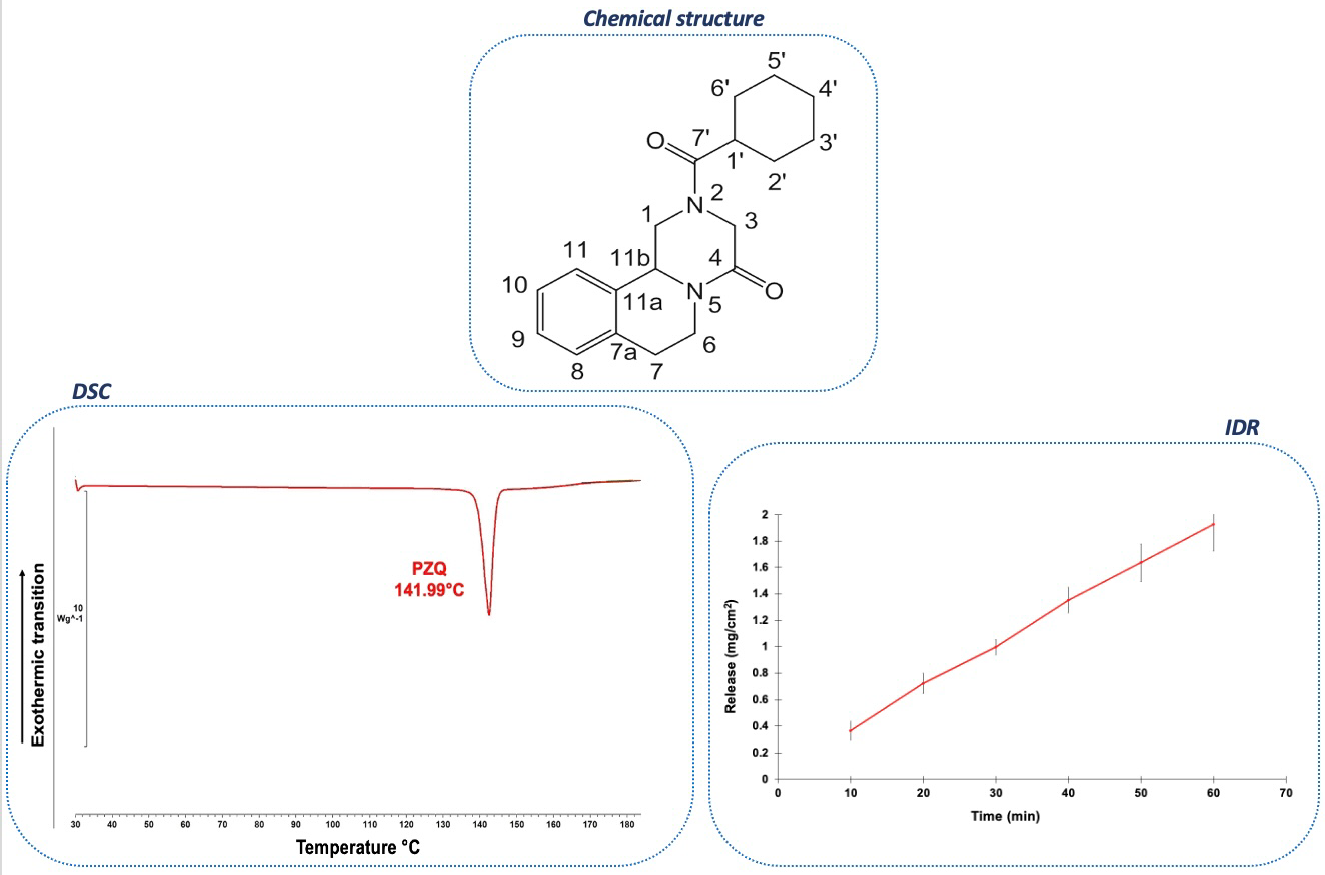








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
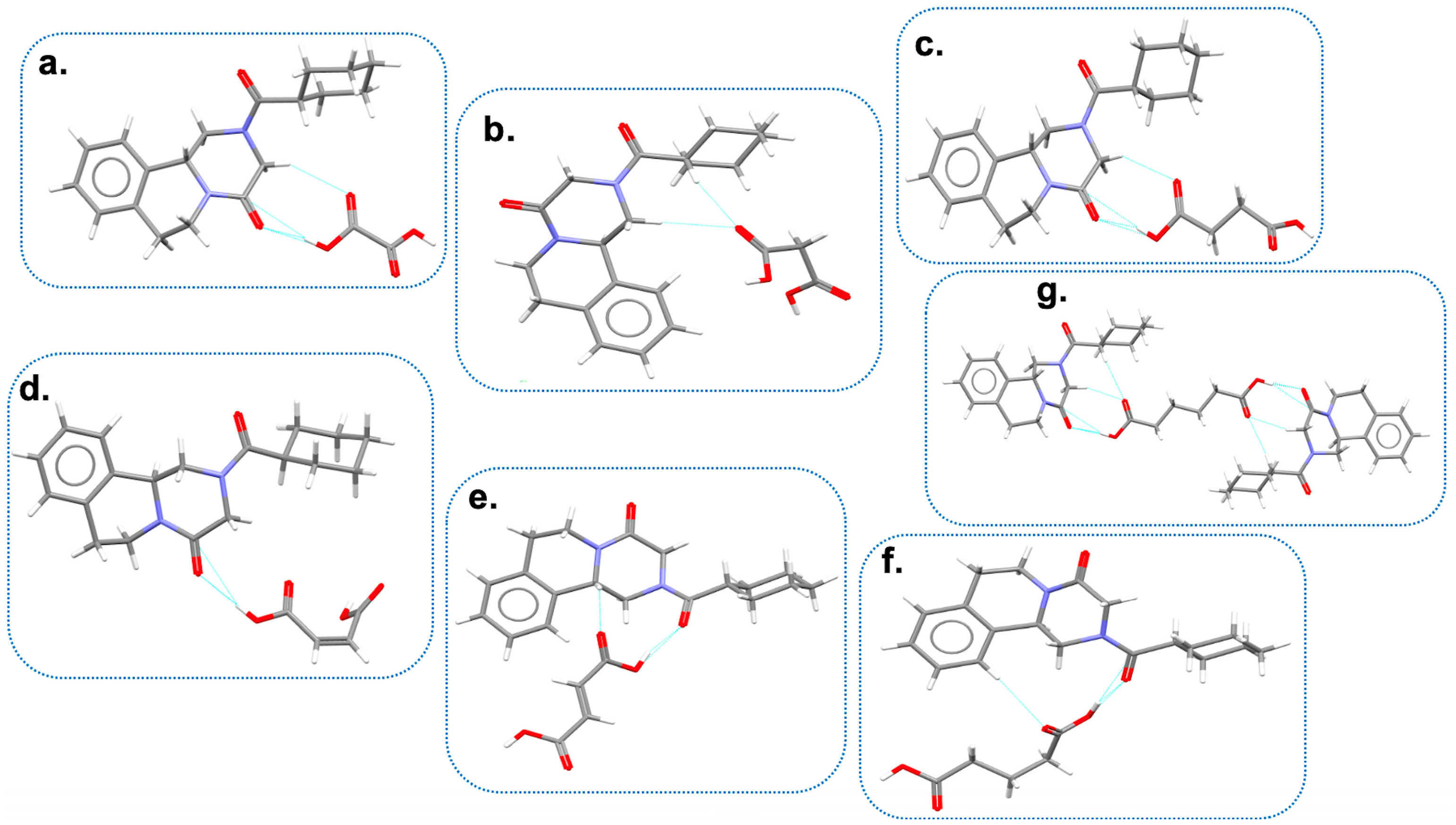
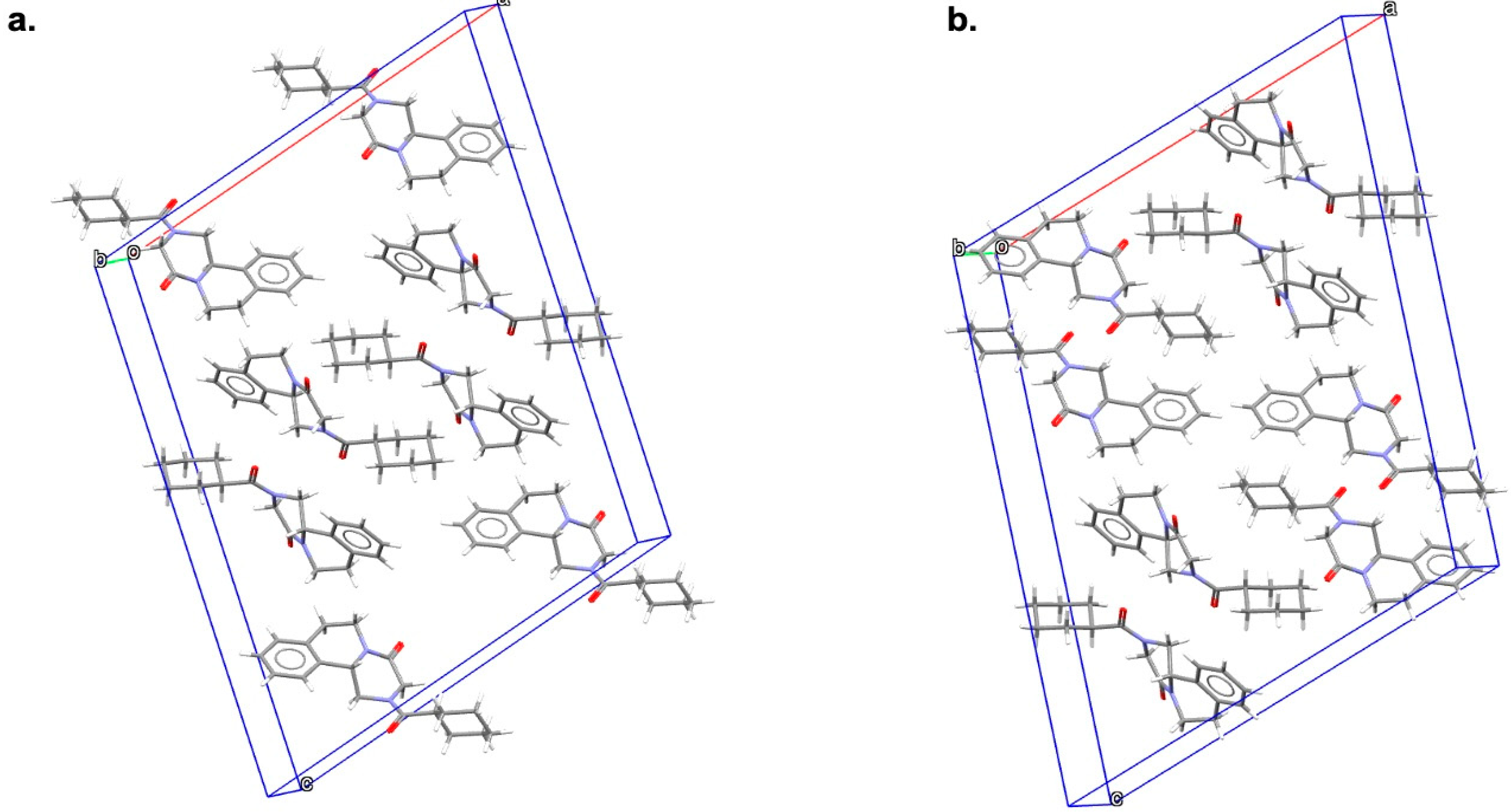{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acronym | Type of Solid Form | Stoichiometry | CSD Reference Code | References |
|---|---|---|---|---|
| Anhydrous polymorphs | ||||
| PZQ Form A | polymorph A | N/A § | TELCEU | [58] |
| PZQ Form B | polymorph B | N/A § | TELCEU01 | [59] |
| PZQ Form C | polymorph C | N/A § | GOYZOM | [60] |
| PZQ Form D | polymorph D | N/A § | not indexed | [61] |
| PZQ Form E | polymorph E | N/A § | not indexed | [61] |
| PZQ Form G | polymorph G | N/A § | not indexed | [62] |
| PZQ Form H | polymorph H | N/A § | not indexed | [62] |
| Hydrates | ||||
| (S)-PZQ-HH | (S)-praziquantel hemihydrate | 1:0.5 | SIGBUG | [25] |
| (R)-PZQ-HH | (R)-praziquantel hemihydrate | 1:0.5 | SIGBUG01 | [26] |
| (R)-PZQ-MH | (R)-praziquantel monohydrate | 1:1 | LIVFED | [19] |
| PZQ-MH | praziquantel monohydrate | 1:1 | not indexed | [63] |
| PZQ-HH | (RS)-praziquantel hemihydrate | 1:0.5 | WUHQAU | [64] |
| PZQ-HH | (RS)-praziquantel hemihydrate | 1:0.5 | not indexed | [65] |
| Solvates | ||||
| PZQ-2P | praziquantel-2-pyrrolidone | 1:1 | DAJCAW | [66] |
| PZQ-AA | praziquantel-acetic acid | 1:1 | DAJCEA | [66] |
| PZQ-DMA | praziquantel-dimethylacetamide | 1:1 | not indexed | [62] |
| Cocrystals | ||||
| PZQ-OXA | praziquantel-oxalic acid | 1:1 | TELCOE | [58] |
| PZQ-MALO | praziquantel-malonic acid | 1:1 | TELDEV | [58] |
| α-PZQ-SUC | praziquantel-succinic acid Form α | 1:1 | not indexed | [58] |
| β-PZQ-SUC | praziquantel-succinic acid Form β | 1:1 | TELDAR | [58] |
| PZQ-MALE | praziquantel-maleic acid | 1:1 | TELCIY | [58] |
| PZQ-FUM | praziquantel-fumaric acid | 1:1 | TELBUJ | [58] |
| PZQ-GLU | praziquantel-glutaric acid | 1:1 | TELDIZ | [58] |
| PZQ-ADI | praziquantel-adipic acid | 2:1 | TELCAQ | [58] |
| PZQ-PIM | praziquantel-pimelic acid | 2:1 | not indexed | [58] |
| (R)-PZQ-GLU | (R)-praziquantel-glutaric acid | 1:1 | KEQVEL | [67] |
| (R)-PZQ-SUC | (R)-praziquantel-succinic acid | 1:1 | KEQVAH | [67] |
| (R)-PZQ/L-MAL | (R)-praziquantel-(L)-malic acid | 1:1 | CUZPIY | [68] |
| (S)-PZQ/L-MAL | (S)-praziquantel-(L)-malic acid | 1:1 | CUZPEU | [68] |
| PZQ-CA | praziquantel-citric acid | 1:1 | not indexed | [39] |
| PZQ-MAL | praziquantel-malic acid | 1:1 | not indexed | [39] |
| PZQ-SA | praziquantel-salicylic acid | 1:1 | not indexed | [39] |
| PZQ-TA | praziquantel-tartaric acid | 1:1 | not indexed | [39] |
| PZQ-KAE | praziquantel-kaempferol | 2:1 | ANAYOG | [69] |
| PZQ-QUE 1 | praziquantel-quercetin 1 | 2:1 | ANAYUM | [69] |
| PZQ-QUE 2 | praziquantel-quercetin 2 | 1:1 | not indexed | [69] |
| PZQ-MYR | praziquantel-myricetin | 1:1 | ANAZAT | [69] |
| PZQ-DITFB | praziquantel-1,2,4,5-tetrafluoro-3,6-di-iodobenzene | 1:1 | AVEKAQ | [70] |
| PZQ-4-HA | praziquantel-4-hydroxybenzoic acid | 1:1 | AVEJIX | [70] |
| PZQ-3,5-DNA | praziquantel-3,5-dinitrobenzoic acid | 1:1 | AVEJET | [70] |
| PZQ-HQ | praziquantel-hydroquinone | 1:1 | AVEKIJ | [70] |
| PZQ-VA | praziquantel-vanillic acid | 1:1 | AVEJAP | [70] |
| PZQ-VA | praziquantel-vanillic acid | 1:2 | not indexed | [70] |
| PZQ-2,5-DHA | praziquantel-2,5-dihydroxybenzoic acid | 1:1 | AVEHUH | [70] |
| PZQ-2,4-HA | praziquantel-2,4-dihydroxybenzoic acid | 1:1 | AVEJUJ | [70] |
| PZQ-ORC | praziquantel-orcinol | 1:1 | AVEHOB | [70] |
| PZQ-SA MH | praziquantel-salicylic acid monohydrate | 1:1 | AVEHER | [70] |
| PZQ-4-ASA ACN | praziquantel-4-aminosalicylic acid acetonitrile solvate | 1:1:1 | AVEJOD | [70] |
| PZQ-2,5-DHA ACN | praziquantel-2,5-dihydroxybenzoic acid acetonitrile solvate | 1:1:1 | AVEHIV | [70] |
| PZQ-3,5-DHA ACN | praziquantel-3,5-dihydroxybenzoic acid acetonitrile solvate | 1:1:1 | AVEKEU | [70] |
| PZQ-CA | praziquantel-citric acid | 1:1 | DAJYUM | [71] |
| PZQ-PA | praziquantel-phtalic acid | 1:1 | DAJZIB | [71] |
| PZQ-3-HA | praziquantel-3-hydroxybenzoic acid | 2:1 | DAJZEX | [71] |
| PZQ-4-HA | praziquantel-4-hydroxybenzoic acid | 1:1 | AVEJIX01 | [71] |
| PZQ-PA | praziquantel-protocathecuic acid | 1:1 | not indexed | [72] |
| PZQ-GA | praziquantel-gallic acid | 1:1 | not indexed | [72] |
| PZQ-FA | praziquantel-ferulic acid | 1:1 | 2133509 * | [72] |
| PZQ-PA-ACN | praziquantel-protocathecuic acid acetonitrile solvate | 1:1:1 | 2133511 * | [72] |
| PZQ-GA-ACN | praziquantel-gallic acid acetonitrile solvate | 1:1:1 | 2133510 * | [72] |
| PZQ-NCM | praziquantel-niclosamide | 1:3 | RIPFOP | [73] |
| (R)-PZQ/(S)-PZQ/D-MAL/L-MAL | (R)-praziquantel-(S)-praziquantel-(D)-malic acid-(L)-malic acid cocrystal | 1:1:1:1 | 2205816 * | [74] |
| (R)-PZQ/(S)-PZQ/D-TA/L-TA | (R)-praziquantel-(S)-praziquantel-(D)-tartaric acid-(L)-tartaric acid cocrystal | 1:1:1:1 | 2205817 * | [74] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Abbrunzo, I.; Procida, G.; Perissutti, B. Praziquantel Fifty Years on: A Comprehensive Overview of Its Solid State. Pharmaceutics 2024, 16, 27. https://doi.org/10.3390/pharmaceutics16010027
D’Abbrunzo I, Procida G, Perissutti B. Praziquantel Fifty Years on: A Comprehensive Overview of Its Solid State. Pharmaceutics. 2024; 16(1):27. https://doi.org/10.3390/pharmaceutics16010027
Chicago/Turabian StyleD’Abbrunzo, Ilenia, Giuseppe Procida, and Beatrice Perissutti. 2024. "Praziquantel Fifty Years on: A Comprehensive Overview of Its Solid State" Pharmaceutics 16, no. 1: 27. https://doi.org/10.3390/pharmaceutics16010027
APA StyleD’Abbrunzo, I., Procida, G., & Perissutti, B. (2024). Praziquantel Fifty Years on: A Comprehensive Overview of Its Solid State. Pharmaceutics, 16(1), 27. https://doi.org/10.3390/pharmaceutics16010027