

Utilizing Molecular Simulations to Examine Nanosuspension Stability

Abstract
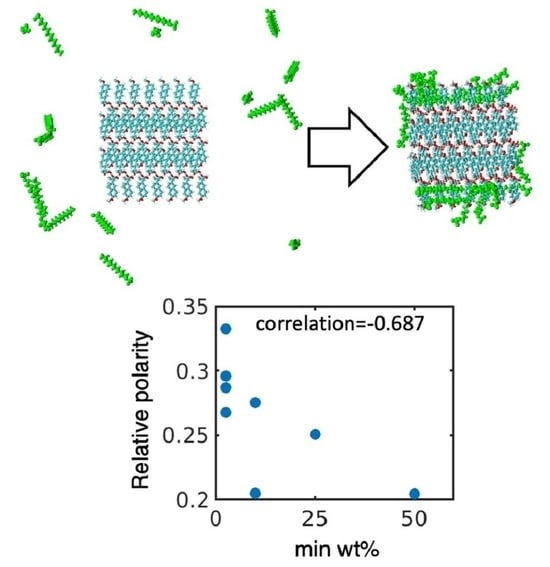
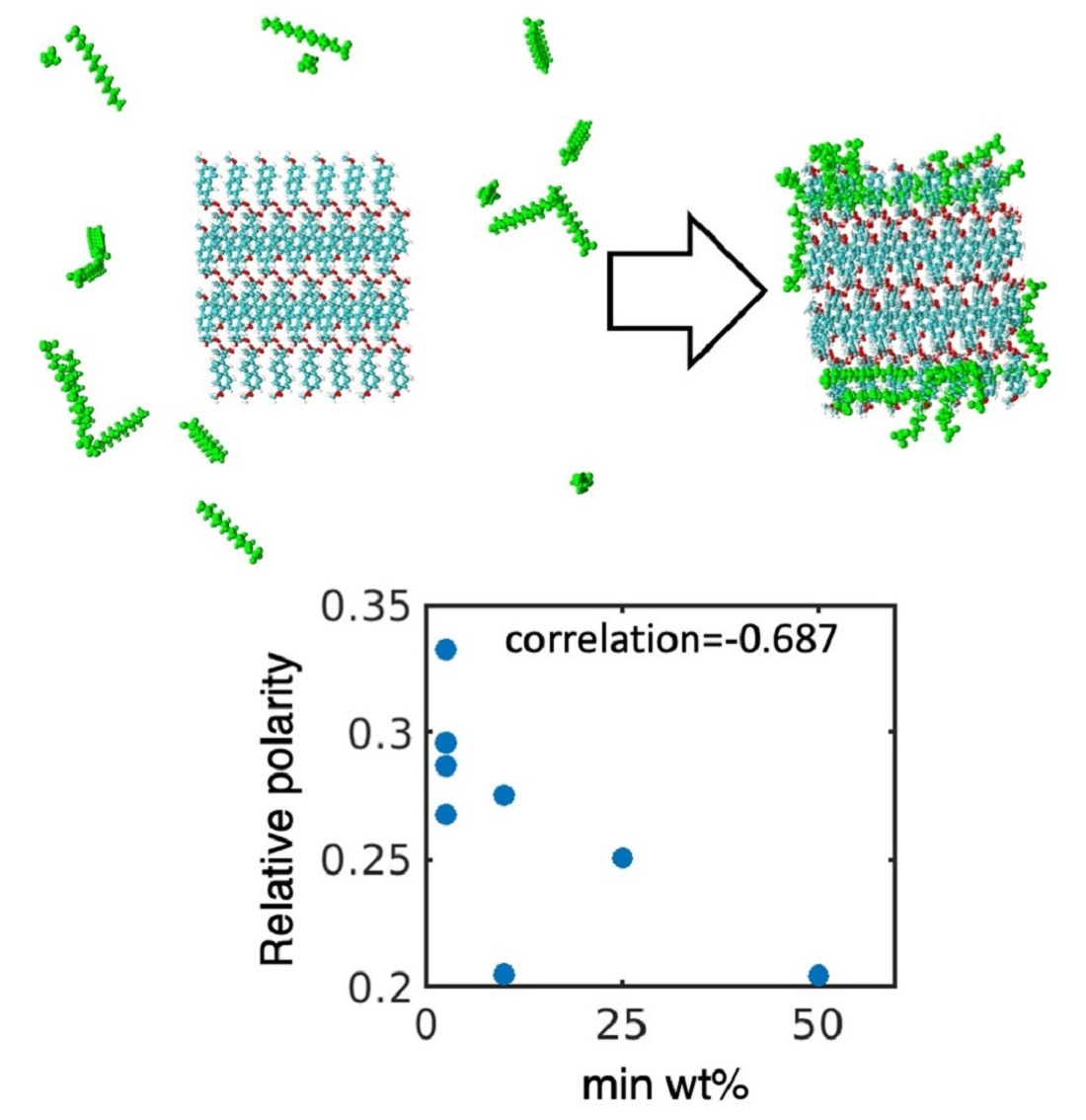
:
1. Introduction
2. Methods
2.1. Molecular Dynamics Simulation
2.2. Simulation Analysis
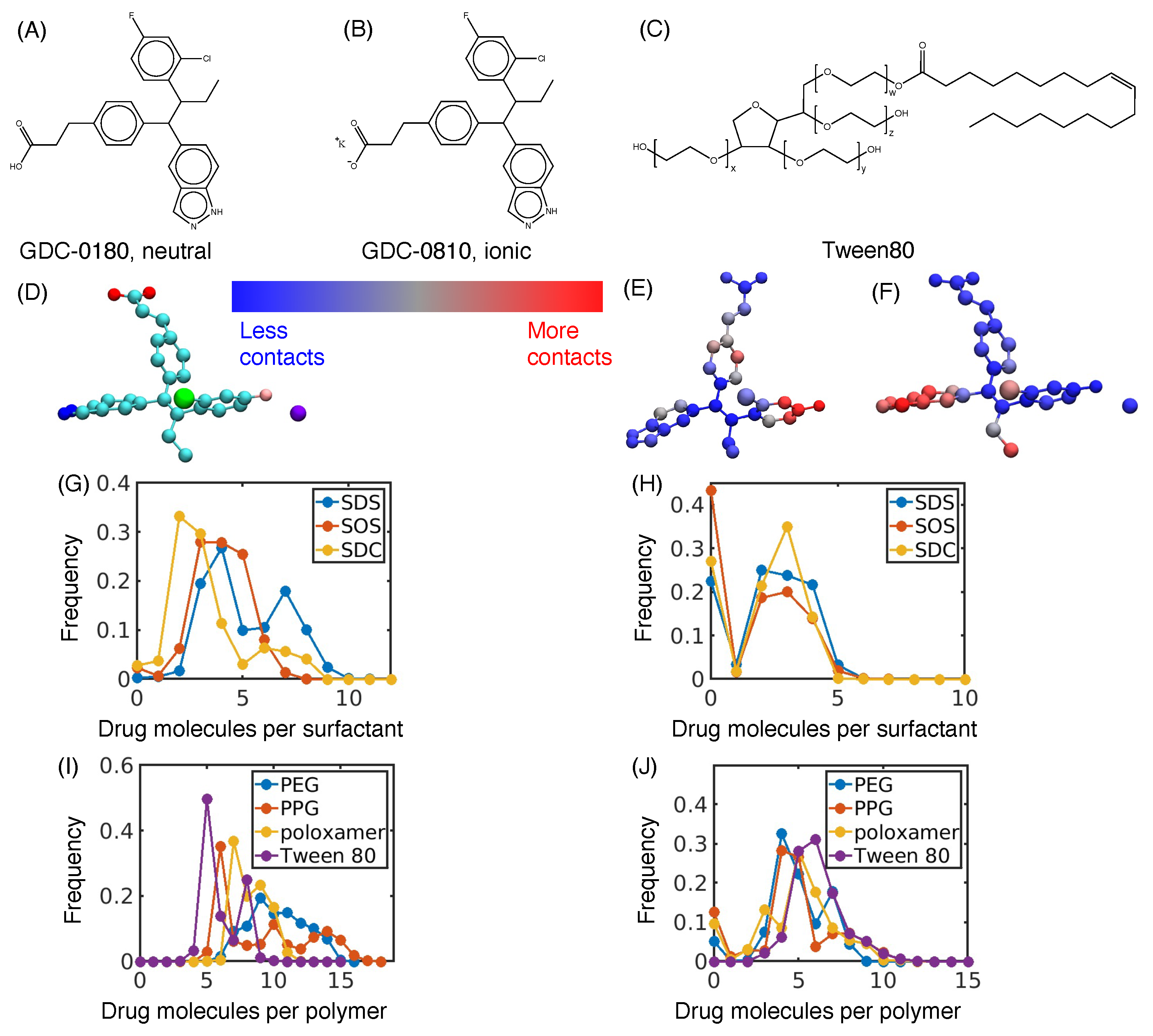
2.3. Flash Nanoprecipitation of GDC-0810
3. Results
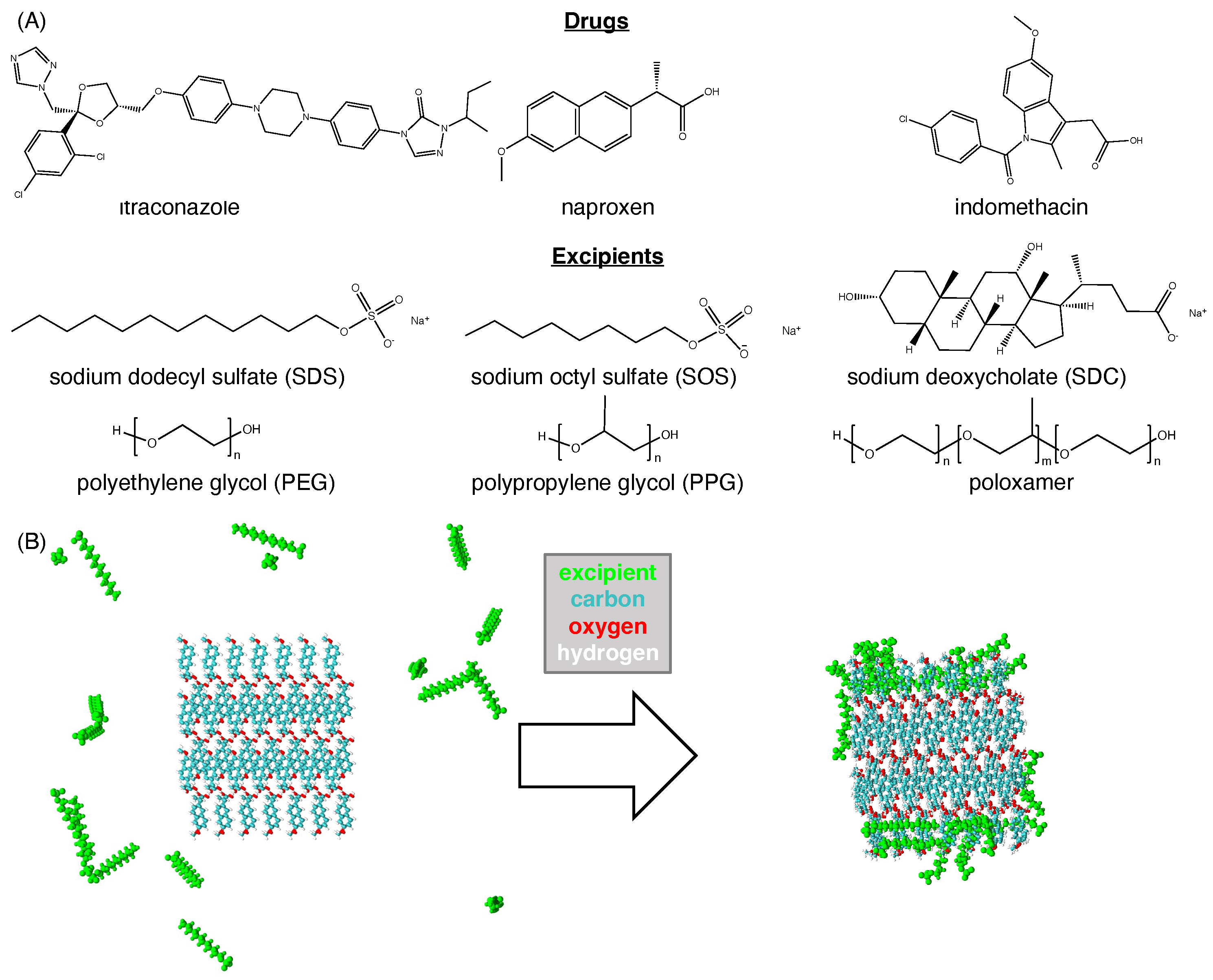
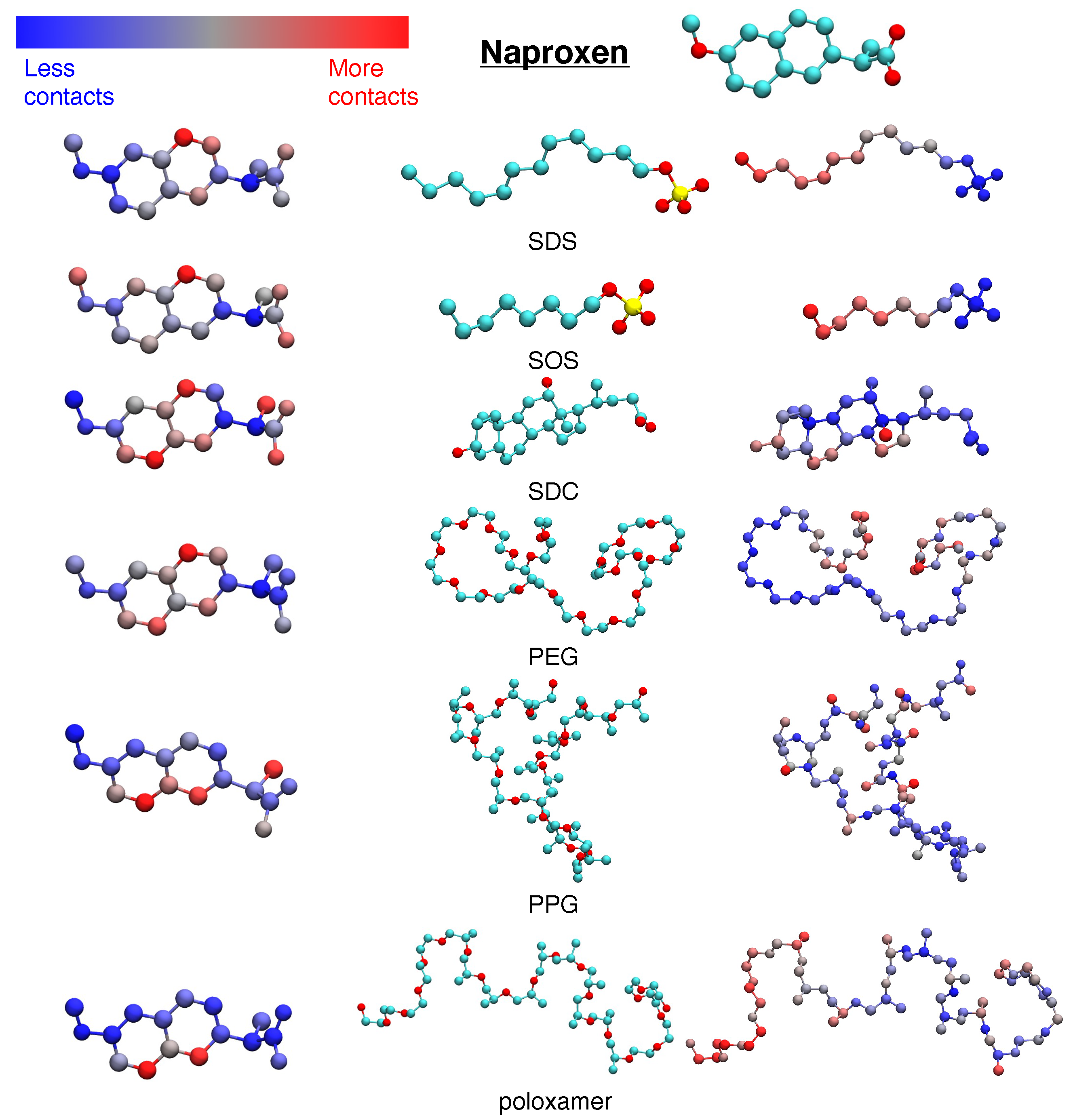
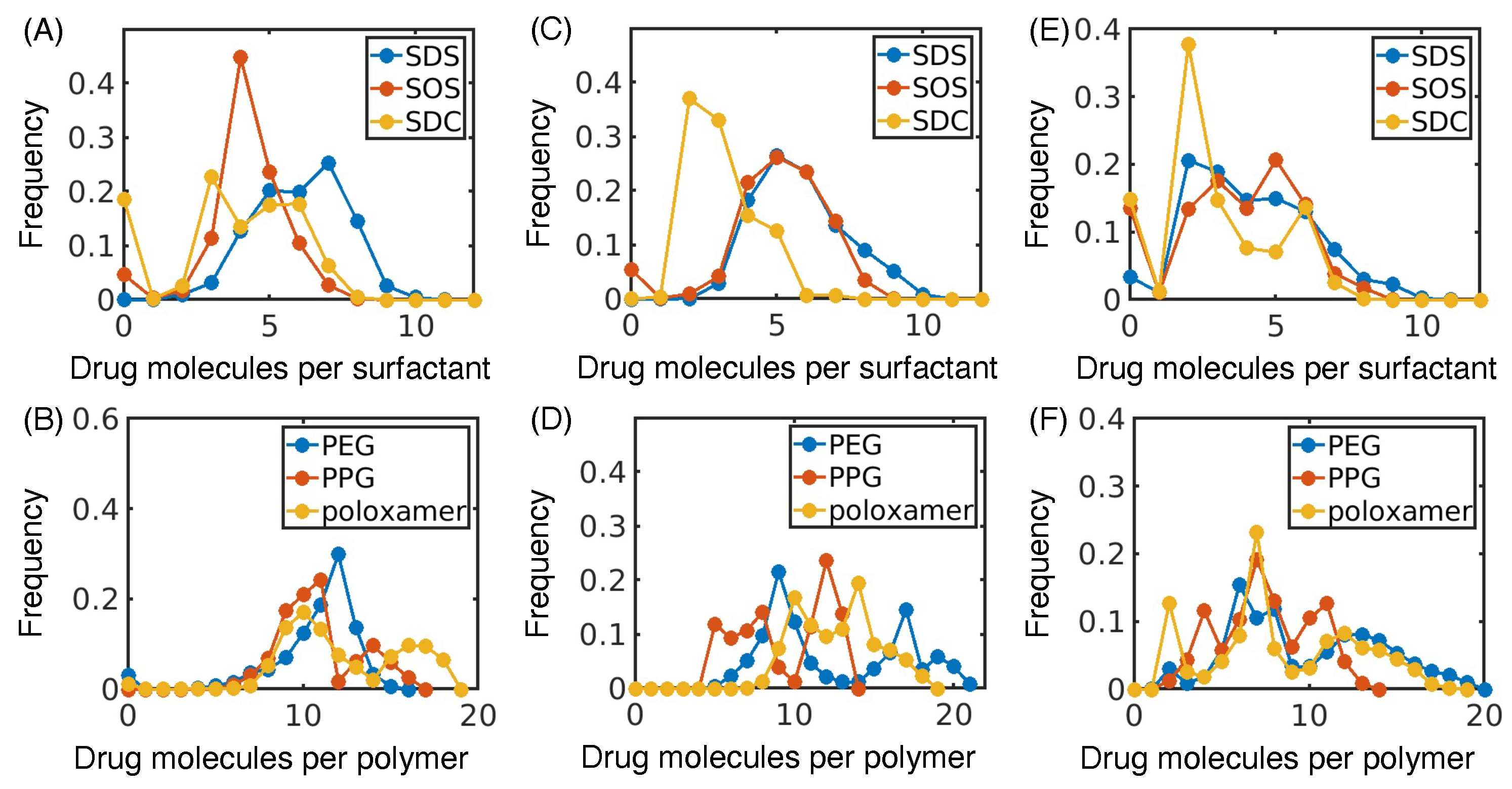
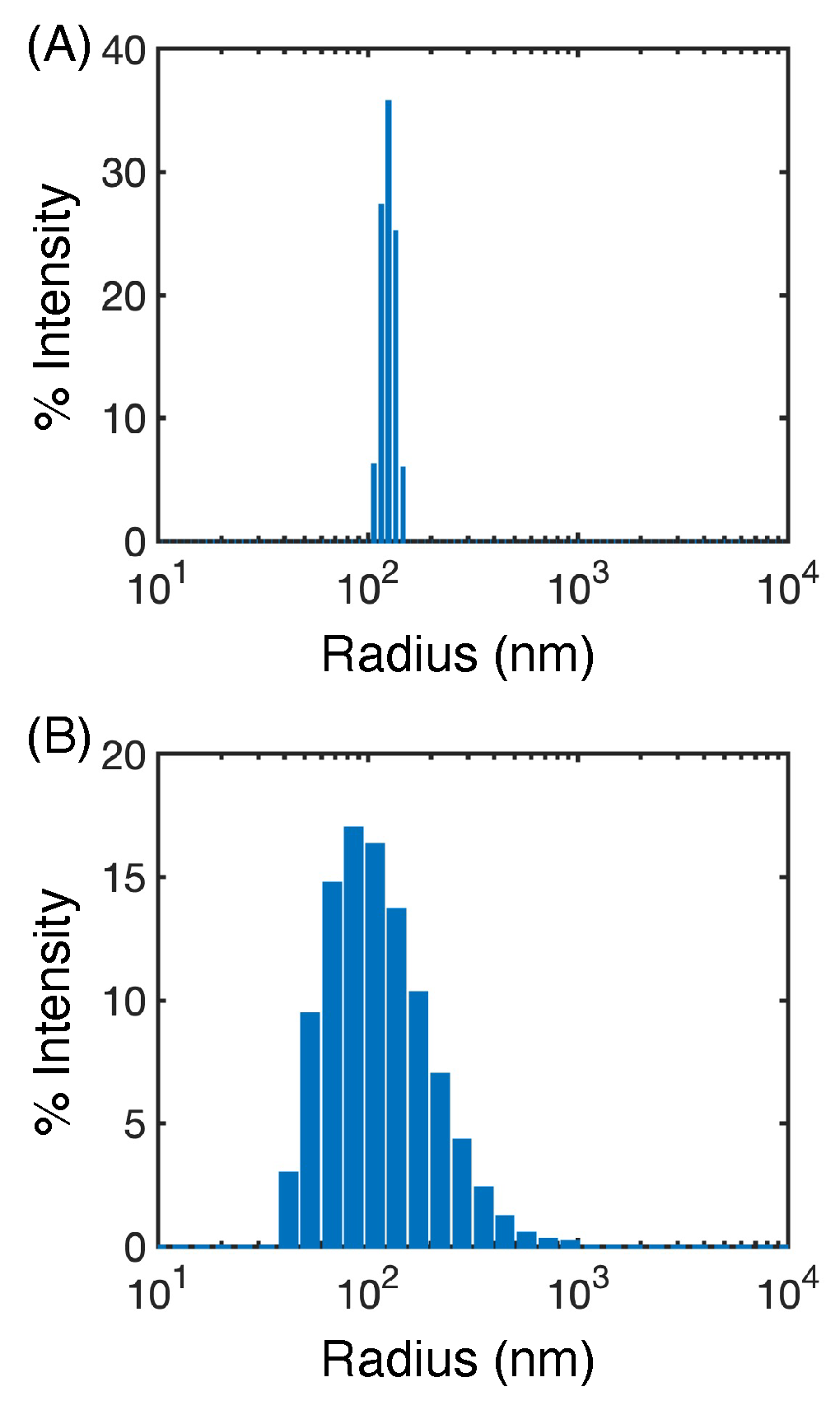
3.1. Hydrophobic Interactions Drive Excipient Adsorption to Drug Crystals
3.2. Atomistic Modeling of Nanosuspension Stability
4. Discussion
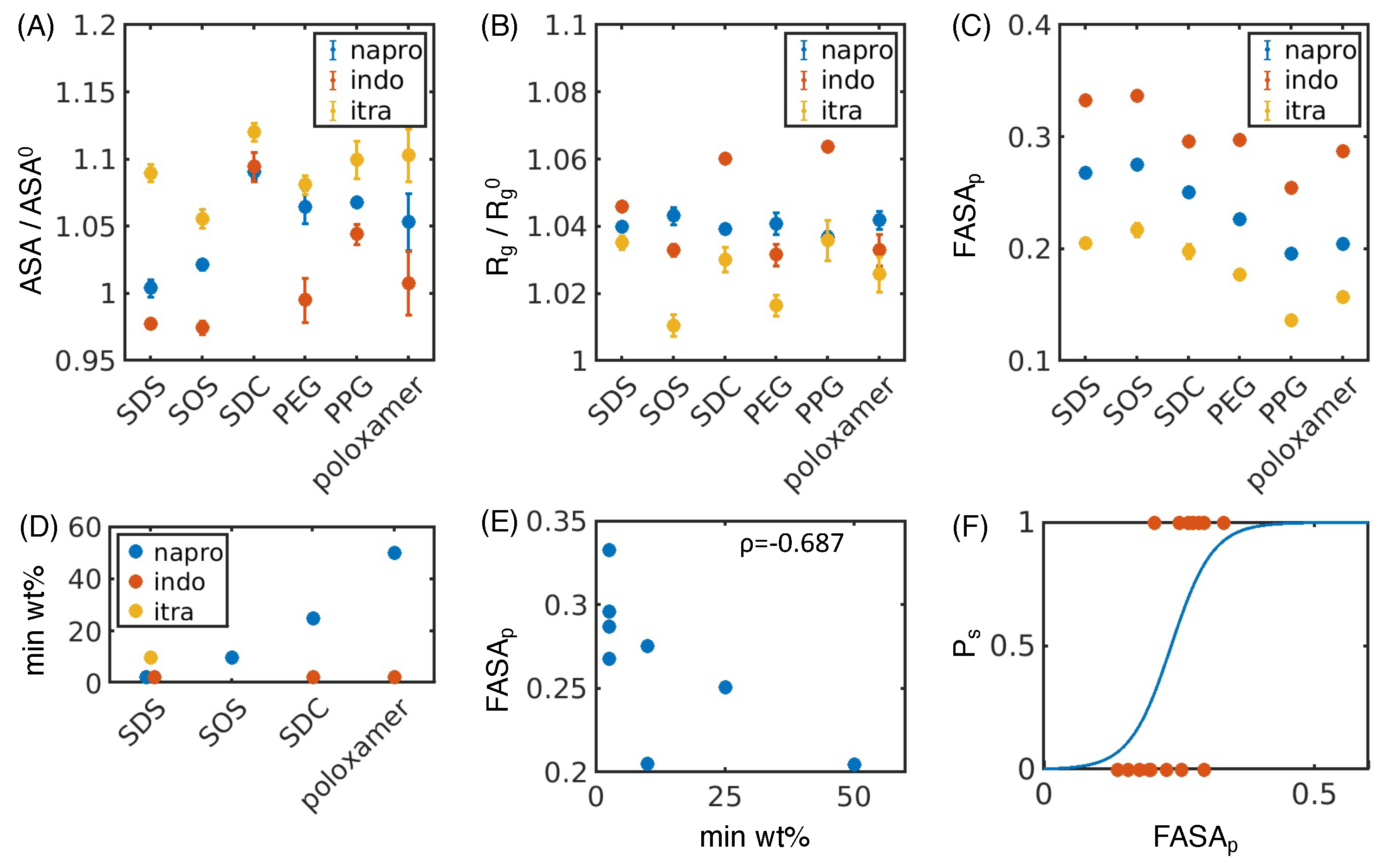
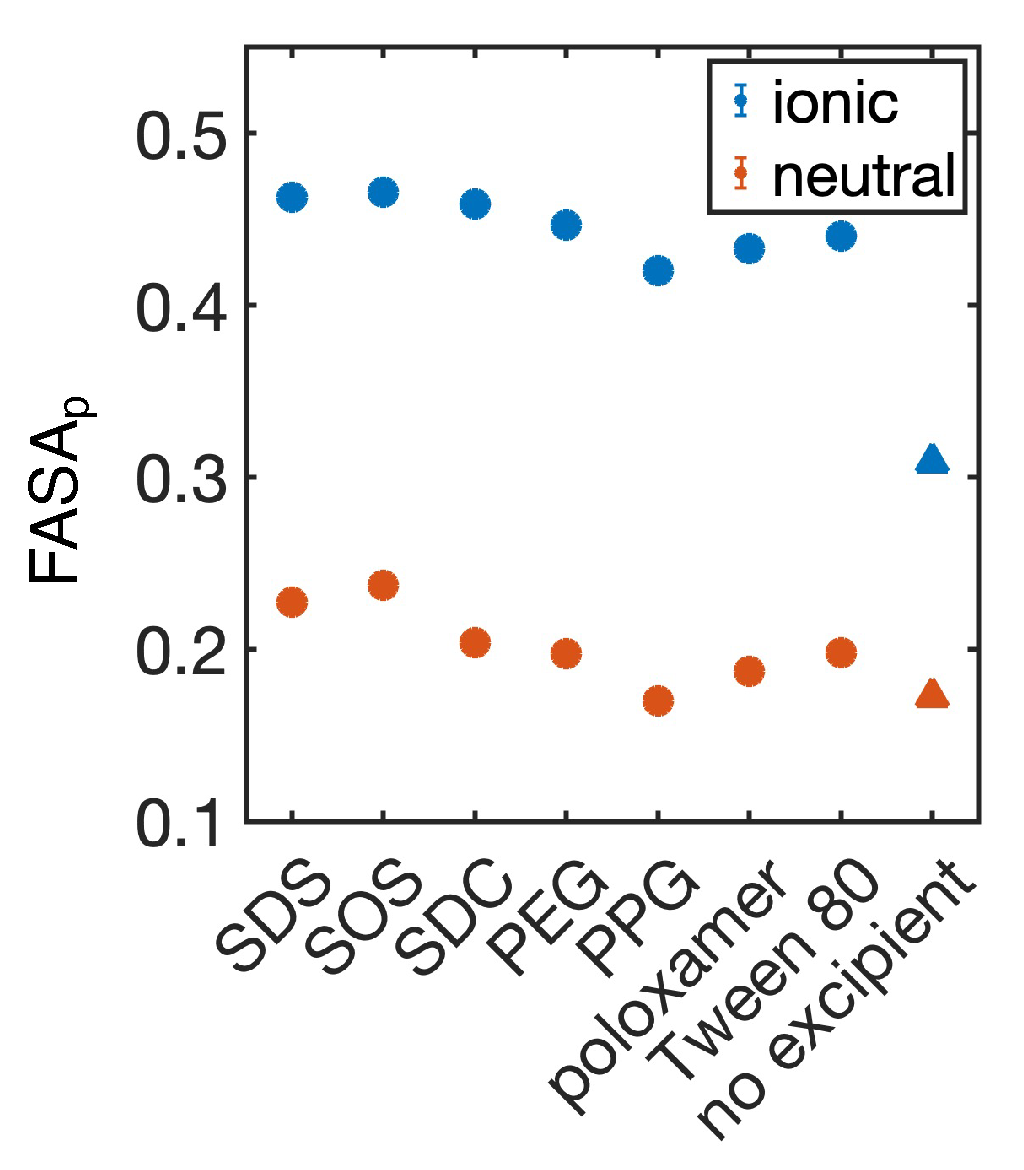
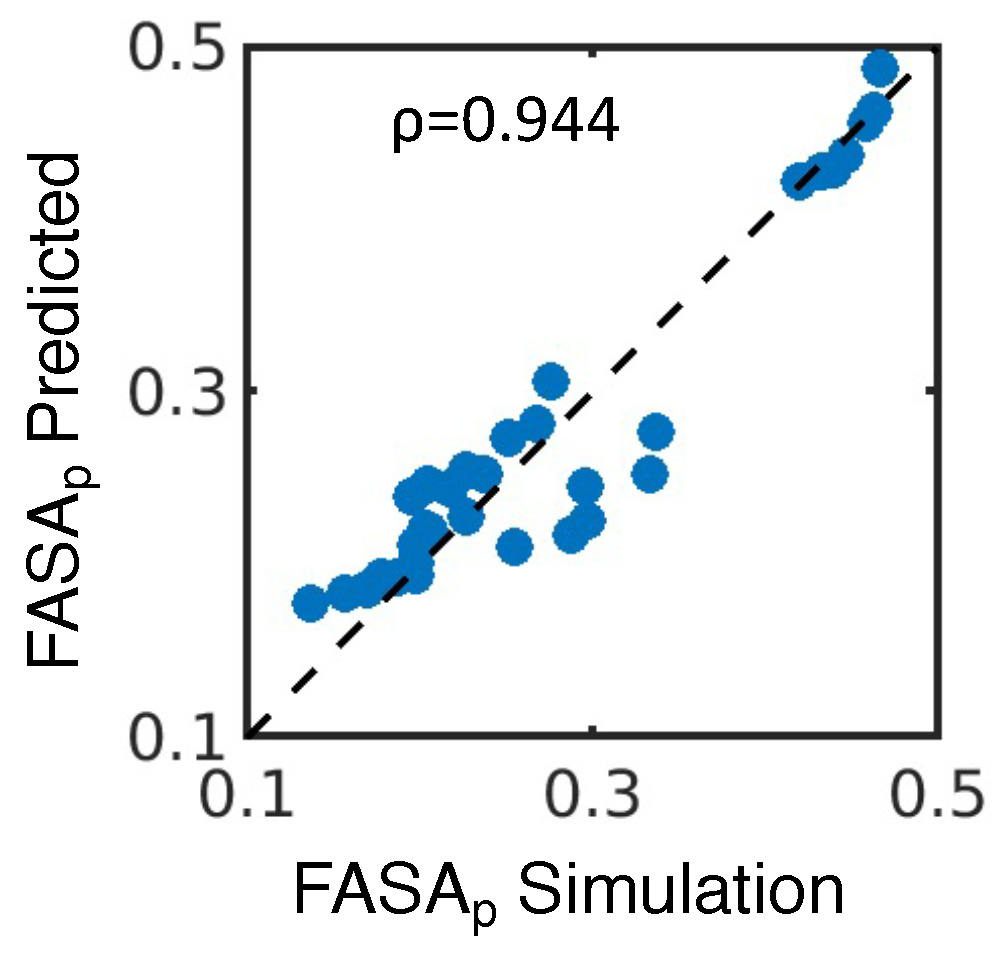
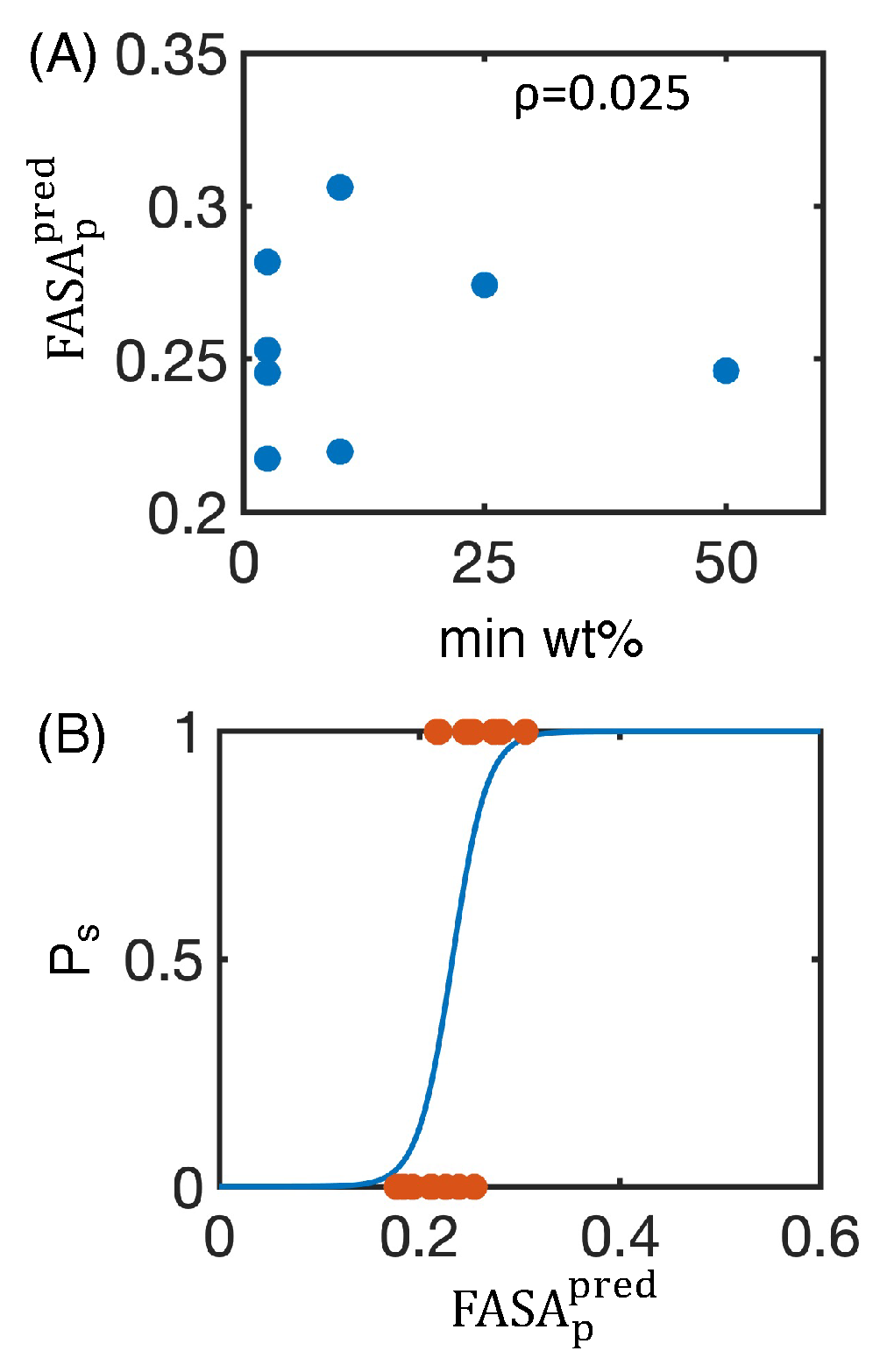
4.1. Simulated Polar Fraction of Solvent-Accessible Surface Area Successfully Predicts Experimentally Determined Nanosuspension Stability
4.2. Leveraging Two-Dimensional Properties to Predict Nanosuspension Stability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MD | Molecular dynamics |
| FNP | Flash nano precipitation |
| SDS | Sodium dodecyl sulfate |
| SOS | Sodium octyl sulfate |
| SDC | Sodium deoxycholate |
| PEG | Polyethylene glycol |
| PPG | Polypropylene glycol |
| FASAp | Fraction of polar surface area |
| ASA | Accessible surface area |
| Radius of gyration | |
| LASSO | Least absolute shrinkage and selection operator |
| MIVM | Multi-inlet vortex mixer |
| min wt.% | Minimum weight percentage of excipient needed to form a stable nanosuspension |
| % PD | Percent polydispersity reported as the polydispersity divided by the estimated |
| hydrodynamic radius of the particle size population multiplied by 100 |
References
- Merisko-Liversidge, E.M.; Liversidge, G.G. Drug Nanoparticles: Formulating Poorly Water-Soluble Compounds. Toxicol. Pathol. 2008, 36, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Medina, C.; Santos-Martinez, M.J.; Radomski, A.; Corrigan, O.I.; Radomski, M.W. Nanoparticles: Pharmacological and toxicological significance. Br. J. Pharmacol. 2007, 150, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Kesisoglou, F.; Mitra, A. Crystalline nanosuspensions as potential toxicology and clinical oral formulations for BCS II/IV Compounds. AAPS J. 2012, 14, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Luo, J.; Tan, S.; Otieno, B.O.; Zhang, Z. The applications of Vitamin e TPGS in drug delivery. Eur. J. Pharm. Sci. 2013, 49, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Kesisoglou, F.; Panmai, S.; Wu, Y. Nanosizing—Oral formulation development and biopharmaceutical evaluation. Adv. Drug Deliv. Rev. 2007, 59, 631–644. [Google Scholar] [CrossRef]
- Malamatari, M.; Taylor, K.M.; Malamataris, S.; Douroumis, D.; Kachrimanis, K. Pharmaceutical nanocrystals: Production by wet milling and applications. Drug Discov. Today 2018, 23, 534–547. [Google Scholar] [CrossRef]
- Merisko-Liversidge, E.; Liversidge, G.G. Nanosizing for oral and parenteral drug delivery: A perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv. Drug Deliv. Rev. 2011, 63, 427–440. [Google Scholar] [CrossRef]
- Müller, R.H.; Gohla, S.; Keck, C.M. State of the art of nanocrystals—Special features, production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm. 2011, 78, 1–9. [Google Scholar] [CrossRef]
- Peltonen, L.; Hirvonen, J. Drug nanocrystals—Versatile option for formulation of poorly soluble materials. Int. J. Pharm. 2018, 537, 73–83. [Google Scholar] [CrossRef]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785–796. [Google Scholar] [CrossRef]
- Shah, D.A.; Murdande, S.B.; Dave, R.H. A Review: Pharmaceutical and Pharmacokinetic Aspect of Nanocrystalline Suspensions. J. Pharm. Sci. 2016, 105, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, I.S.; Hu, H.; Yin, L.; He, W. Drug nanocrystals: Fabrication methods and promising therapeutic applications. Int. J. Pharm. 2019, 562, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Shirui, M.; Shi, Y.; Li, C.L.; Fang, L. Nanonization of Itraconazole by High Pressure Homogenization: Stabilizer Optimization and Effect of Particle Size on Oral Absorption. J. Pharm. Sci. 2011, 100, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Mitri, K.; Shegokar, R.; Gohla, S.; Anselmi, C.; Müller, R.H. Lutein nanocrystals as antioxidant formulation for oral and dermal delivery. Int. J. Pharm. 2011, 420, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lv, H.; Jiang, K.; Gao, Y. Enhanced bioavailability after oral and pulmonary administration of baicalein nanocrystal. Int. J. Pharm. 2011, 420, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Paxton, J.W.; Baguley, B.C.; Garg, S. Formulation and pharmacokinetic evaluation of an asulacrine nanocrystalline suspension for intravenous delivery. Int. J. Pharm. 2009, 367, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Yeo, Y. Nanocrystals for the parenteral delivery of poorly water-soluble drugs. Curr. Opin. Solid State Mater. Sci. 2012, 16, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, L.; Hirvonen, J. Pharmaceutical nanocrystals by nanomilling: Critical process parameters, particle fracturing and stabilization methods. J. Pharm. Pharmacol. 2010, 62, 1569–1579. [Google Scholar] [CrossRef]
- Leung, D.H.; Lamberto, D.J.; Liu, L.; Kwong, E.; Nelson, T.; Rhodes, T.; Bak, A. A new and improved method for the preparation of drug nanosuspension formulations using acoustic mixing technology. Int. J. Pharm. 2014, 473, 10–19. [Google Scholar] [CrossRef]
- George, M.; Ghosh, I. Identifying the correlation between drug/stabilizer properties and critical quality attributes (CQAs) of nanosuspension formulation prepared by wet media milling technology. Eur. J. Pharm. Sci. 2013, 48, 142–152. [Google Scholar] [CrossRef]
- Li, M.; Zhang, L.; Davé, R.N.; Bilgili, E. An Intensified Vibratory Milling Process for Enhancing the Breakage Kinetics during the Preparation of Drug Nanosuspensions. AAPS PharmSciTech 2016, 17, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Huang, L.; Liu, F. Understanding the structure and stability of paclitaxel nanocrystals. Int. J. Pharm. 2010, 390, 242–249. [Google Scholar] [CrossRef]
- Dolenc, A.; Kristl, J.; Baumgartner, S.; Planinvsek, O. Advantages of celecoxib nanosuspension formulation and transformation into tablets. Int. J. Pharm. 2009, 376, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Lindfors, L.; Forssén, S.; Westergren, J.; Olsson, U. Nucleation and crystal growth in supersaturated solutions of a model drug. J. Colloid Interface Sci. 2008, 325, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, J.; Watanabe, W. Physical and chemical stability of drug nanoparticles. Adv. Drug Deliv. Rev. 2011, 63, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Tuomela, A.; Hirvonen, J.; Peltonen, L. Stabilizing agents for drug nanocrystals: Effect on bioavailability. Pharmaceutics 2016, 8, 16. [Google Scholar] [CrossRef]
- Master, A.M.; Rodriguez, M.E.; Kenney, M.E.; Oleinick, N.L.; Sen Gupta, A. Delivery of the photosensitizer Pc 4 in PEG-PCL micelles for in vitro PDT studies. J. Pharm. Sci. 2010, 99, 2386–2398. [Google Scholar] [CrossRef]
- Kuroiwa, Y.; Higashi, K.; Ueda, K.; Yamamoto, K.; Moribe, K. Nano-scale and molecular-level understanding of wet-milled indomethacin/poloxamer 407 nanosuspension with TEM, suspended-state NMR, and Raman measurements. Int. J. Pharm. 2018, 537, 30–39. [Google Scholar] [CrossRef]
- Lee, J.; Choi, J.Y.; Park, C.H. Characteristics of polymers enabling nano-comminution of water-insoluble drugs. Int. J. Pharm. 2008, 355, 328–336. [Google Scholar] [CrossRef]
- Lestari, M.L.; Müller, R.H.; Möschwitzer, J.P. Systematic screening of different surface modifiers for the production of physically stable nanosuspensions. J. Pharm. Sci. 2015, 104, 1128–1140. [Google Scholar] [CrossRef]
- Yen, C.W.; Kuhn, R.; Hu, C.; Zhang, W.; Chiang, P.C.; Chen, J.Z.; Hau, J.; Estevez, A.; Nagapudi, K.; Leung, D.H. Impact of surfactant selection and incorporation on in situ nanoparticle formation from amorphous solid dispersions. Int. J. Pharm. 2021, 607, 120980. [Google Scholar] [CrossRef] [PubMed]
- Guruge, A.G.; Warren, D.B.; Pouton, C.W.; Chalmers, D.K. Molecular Dynamics Simulation Studies of Bile, Bile Salts, Lipid-Based Drug Formulations, and mRNA-Lipid Nanoparticles: A Review. Mol. Pharm. 2023, 20, 2781–2800. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Qiao, F.; Hou, Y.; Tian, B.; Yang, J. Exploring space-energy matching via quantum-molecular mechanics modeling and breakage dynamics-energy dissipation via microhydrodynamic modeling to improve the screening efficiency of nanosuspension prepared by wet media milling. Expert Opin. Drug Deliv. 2021, 18, 1643–1657. [Google Scholar] [CrossRef] [PubMed]
- Mehta, C.H.; Narayan, R.; Nayak, U.Y. Computational modeling for formulation design. Drug Discov. Today 2019, 24, 781–788. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, A.M.; Carvalho, F.C.; Teixeira, D.A.; Azevedo, D.L.; de Barros, W.M.; Gremi ao, M.P.D. Computational and experimental approaches for development of methotrexate nanosuspensions by bottom-up nanoprecipitation. Int. J. Pharm. 2017, 524, 330–338. [Google Scholar] [CrossRef]
- Ouranidis, A.; Gkampelis, N.; Vardaka, E.; Karagianni, A.; Tsiptsios, D.; Nikolakakis, I.; Kachrimanis, K. Overcoming the solubility barrier of ibuprofen by the rational process design of a nanocrystal formulation. Pharmaceutics 2020, 12, 969. [Google Scholar] [CrossRef] [PubMed]
- Parks, C.; Koswara, A.; Tung, H.H.; Nere, N.K.; Bordawekar, S.; Nagy, Z.K.; Ramkrishna, D. Nanocrystal dissolution kinetics and solubility increase prediction from molecular dynamics: The case of α-, β-, and γ-glycine. Mol. Pharm. 2017, 14, 1023–1032. [Google Scholar] [CrossRef]
- Omolo, C.A.; Kalhapure, R.S.; Agrawal, N.; Rambharose, S.; Mocktar, C.; Govender, T. Formulation and Molecular Dynamics Simulations of a Fusidic Acid Nanosuspension for Simultaneously Enhancing Solubility and Antibacterial Activity. Mol. Pharm. 2018, 15, 3512–3526. [Google Scholar] [CrossRef]
- Ferrar, J.A.; Sellers, B.D.; Chan, C.; Leung, D.H. Towards an improved understanding of drug excipient interactions to enable rapid optimization of nanosuspension formulations. Int. J. Pharm. 2020, 578, 119094. [Google Scholar] [CrossRef]
- Suo, Z.; Sun, Q.; Peng, X.; Zhang, S.; Gan, N.; Zhao, L.; Yuan, N.; Zhang, Y.; Li, H. Lentinan as a natural stabilizer with bioactivities for preparation of drug-drug nanosuspensions. Int. J. Biol. Macromol. 2021, 184, 101–108. [Google Scholar] [CrossRef]
- Shamay, Y.; Shah, J.; Işık, M.; Mizrachi, A.; Leibold, J.; Tschaharganeh, D.F.; Roxbury, D.; Budhathoki-Uprety, J.; Nawaly, K.; Sugarman, J.L.; et al. Quantitative self-assembly prediction yields targeted nanomedicines. Nat. Mater. 2018, 17, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Felberg, L.E.; Piunova, V.A.; Rice, J.E.; Head-Gordon, T.; Swope, W.C. Effect of Hydrophobic Core Topology and Composition on the Structure and Kinetics of Star Polymers: A Molecular Dynamics Study. J. Phys. Chem. B 2017, 121, 2902–2918. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Piunova, V.A.; Maarof, H.; Rice, J.E.; Swope, W.C. Influence of Solvent on the Drug-Loading Process of Amphiphilic Nanogel Star Polymers. J. Phys. Chem. B 2018, 122, 5356–5367. [Google Scholar] [CrossRef] [PubMed]
- Swope, W.C.; Rice, J.E.; Piunova, V.A.; Carr, A.C.; Miller, R.D.; Sly, J. Simulation and Experiments to Identify Factors Allowing Synthetic Control of Structural Features of Polymeric Nanoparticles. J. Phys. Chem. B 2016, 120, 7546–7568. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.; McClory, A.; Zhang, H.; Cravillion, T.; Lim, N.K.; Masui, C.; Robinson, S.J.; Han, C.; Ochs, C.; Rege, P.D.; et al. Synthesis of Selective Estrogen Receptor Degrader GDC-0810 via Stereocontrolled Assembly of a Tetrasubstituted All-Carbon Olefin. J. Org. Chem. 2018, 83, 11571–11576. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE), 2020.09. Available online: https://www.chemcomp.com/Products.htm (accessed on 21 June 2021).
- Berendsen, H.J.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Maestro, Schrodinger, LLC. Schrödiner Release 2020.4; Maestro, Schrodinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, SC’06, Tampa, FL, USA, 11–17 November 2006. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Gowers, R.; Linke, M.; Barnoud, J.; Reddy, T.; Melo, M.; Seyler, S.; Domański, J.; Dotson, D.; Buchoux, S.; Kenney, I.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016. [Google Scholar] [CrossRef]
- MATLAB. Statistics and Machine Learning Toolbox User’s Guide, 2019th ed.; Matwork Inc.: Natick, MA, USA, 2019; Available online: https://www.mathworks.com/help/stats/ (accessed on 8 September 2021).
- Lee, M.L.; Aliagas, I.; Feng, J.A.; Gabriel, T.; O’Donnell, T.J.; Sellers, B.D.; Wiswedel, B.; Gobbi, A. Chemalot and chemalot-knime: Command line programs as workflow tools for drug discovery. J. Cheminform. 2017, 9, 38. [Google Scholar] [CrossRef]
- Markwalter, C.E.; Prud’homme, R.K. Design of a Small-Scale Multi-Inlet Vortex Mixer for Scalable Nanoparticle Production and Application to the Encapsulation of Biologics by Inverse Flash NanoPrecipitation. J. Pharm. Sci. 2018, 107, 2465–2471. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Excipient | Neutral | Ionic |
|---|---|---|
| SDS | 0.447 | 0.997 |
| SOS | 0.510 | 0.998 |
| SDC | 0.303 | 0.997 |
| PEG | 0.271 | 0.996 |
| PPG | 0.153 | 0.992 |
| Poloxamer | 0.221 | 0.994 |
| Tween 80 | 0.271 | 0.995 |
| no excipient | 0.161 | 0.871 |
| Excipient | Neutral | Ionic |
|---|---|---|
| SDS | 0.438 | 1.000 |
| SOS | 0.761 | 1.000 |
| SDC | 0.336 | 1.000 |
| PEG | 0.141 | 1.000 |
| PPG | 0.064 | 1.000 |
| Poloxamer | 0.091 | 1.000 |
| Tween 80 | 0.097 | 1.000 |
| no excipient | 0.004 | 1.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latham, A.P.; Levy, E.S.; Sellers, B.D.; Leung, D.H. Utilizing Molecular Simulations to Examine Nanosuspension Stability. Pharmaceutics 2024, 16, 50. https://doi.org/10.3390/pharmaceutics16010050
Latham AP, Levy ES, Sellers BD, Leung DH. Utilizing Molecular Simulations to Examine Nanosuspension Stability. Pharmaceutics. 2024; 16(1):50. https://doi.org/10.3390/pharmaceutics16010050
Chicago/Turabian StyleLatham, Andrew P., Elizabeth S. Levy, Benjamin D. Sellers, and Dennis H. Leung. 2024. "Utilizing Molecular Simulations to Examine Nanosuspension Stability" Pharmaceutics 16, no. 1: 50. https://doi.org/10.3390/pharmaceutics16010050
APA StyleLatham, A. P., Levy, E. S., Sellers, B. D., & Leung, D. H. (2024). Utilizing Molecular Simulations to Examine Nanosuspension Stability. Pharmaceutics, 16(1), 50. https://doi.org/10.3390/pharmaceutics16010050