
Chemical Modification of Cytochrome C for Acid-Responsive Intracellular Apoptotic Protein Delivery for Cancer Eradication
 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
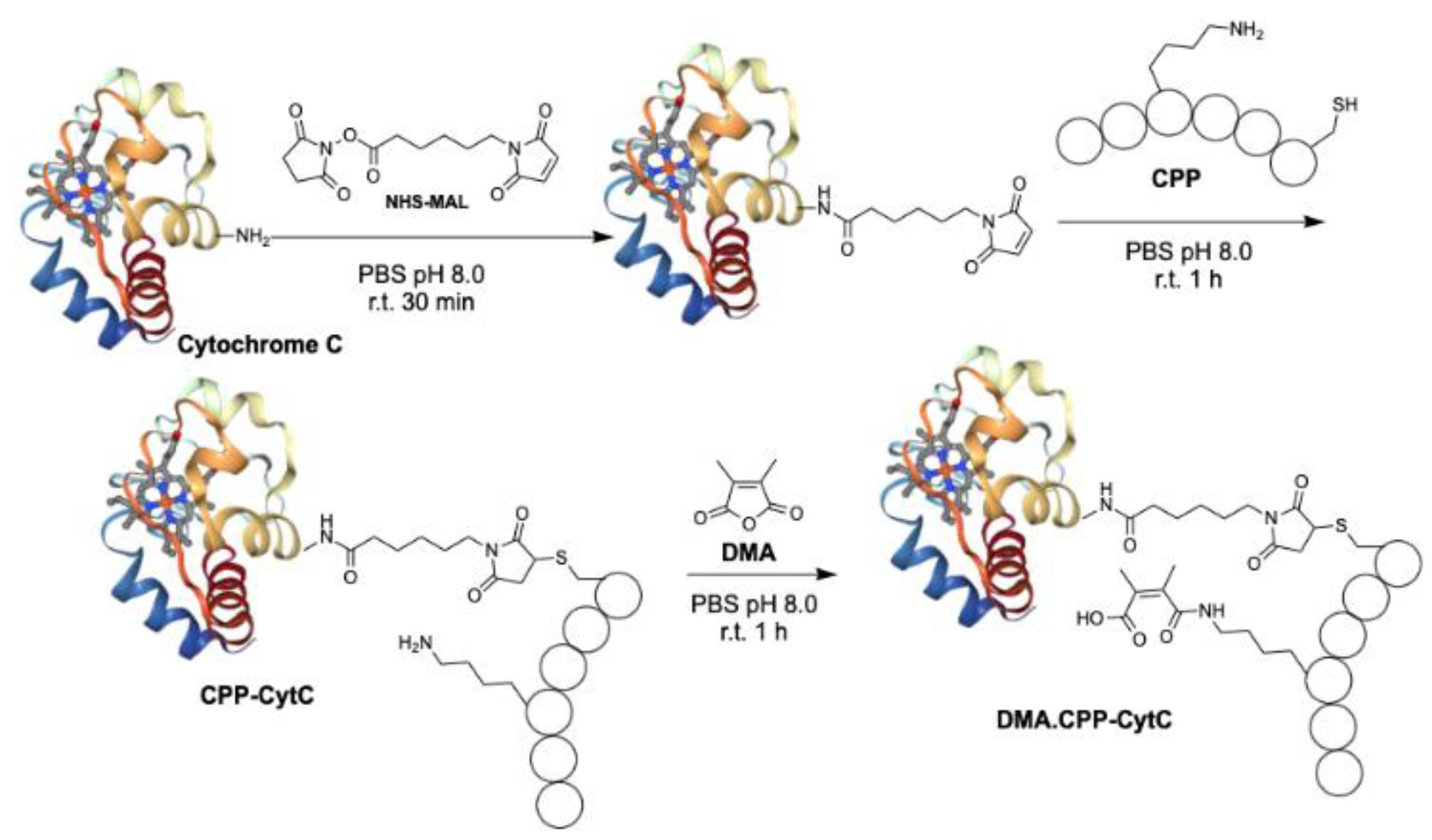
2.1. Preparation of Cell Penetrating Peptide (CPP)
2.2. Conjugation of CPP to Cytochrome C
2.3. Addition of Dimethylacetamide Anhydride (DMA)
2.4. Cell Culture
2.5. Cellular Cytotoxicity Assay
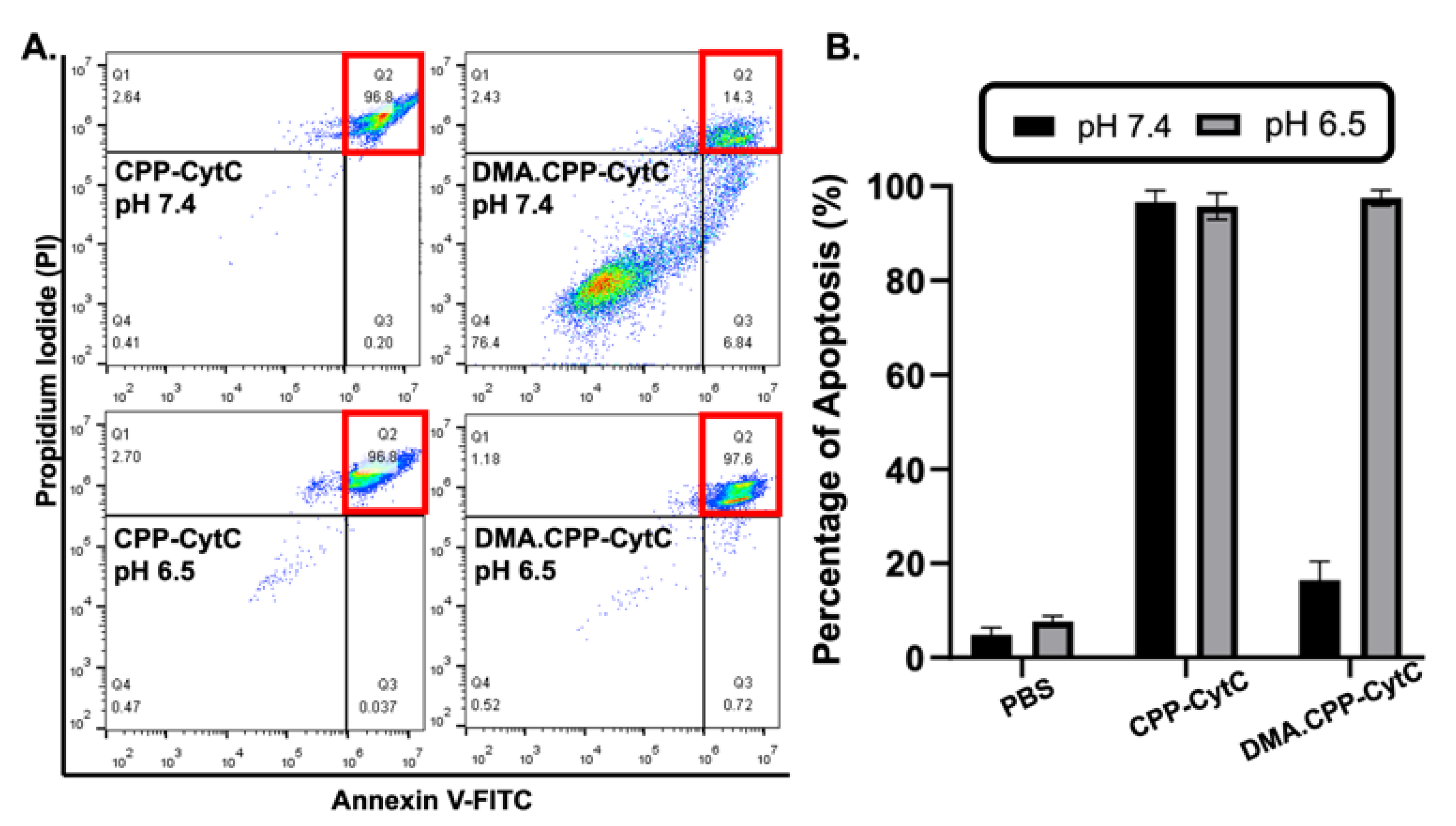
2.6. Cell Apoptosis Assay
2.7. In Vitro Imaging Experiment
2.8. HT29 Spheroid Culture and Confocal Microscopy
2.9. 3D Spheroid Viability Assay
2.10. In Vivo Tumour Regression Experiment
3. Results
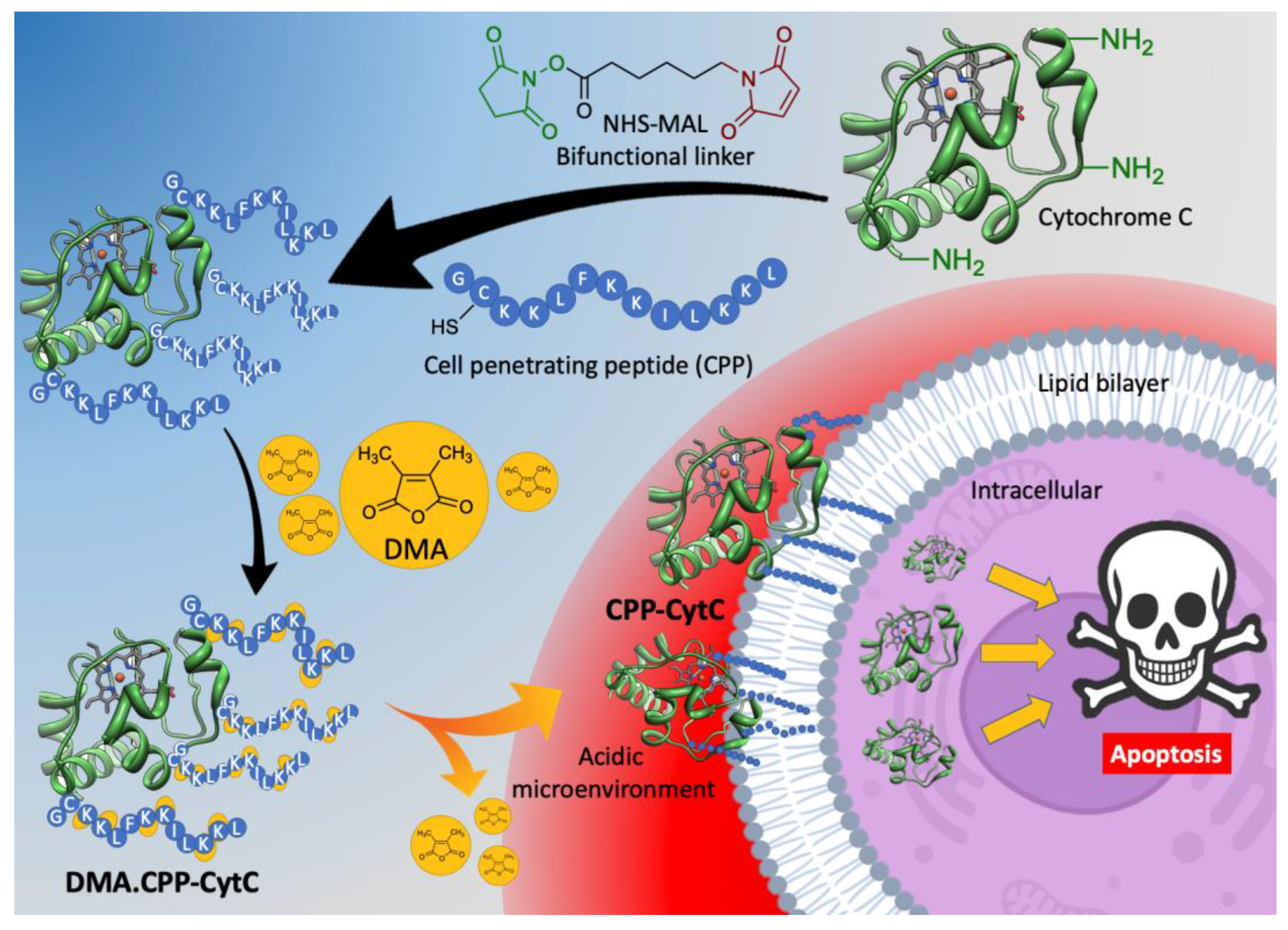
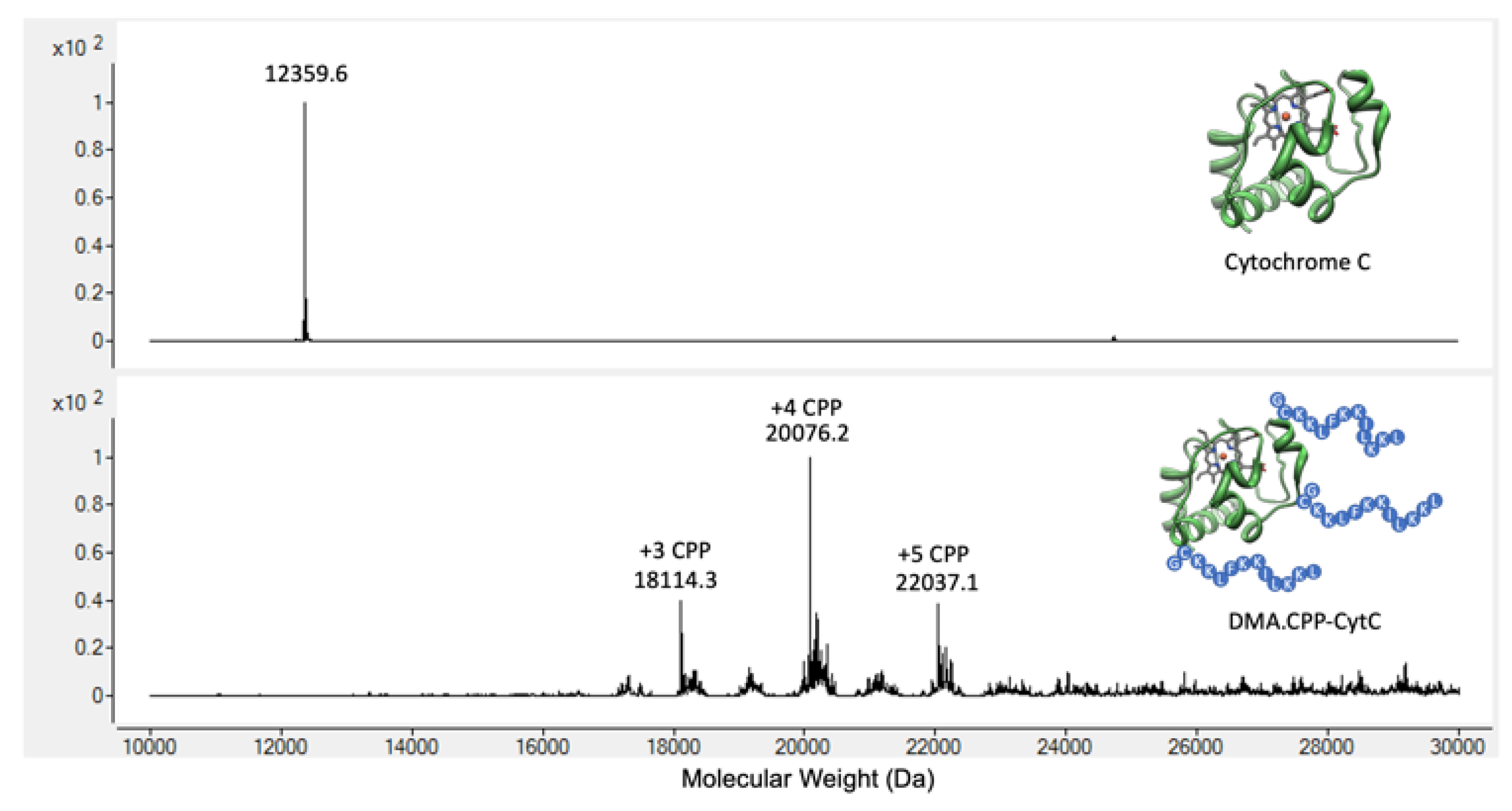
3.1. Design and Synthesis of DMA.CPP-CytC
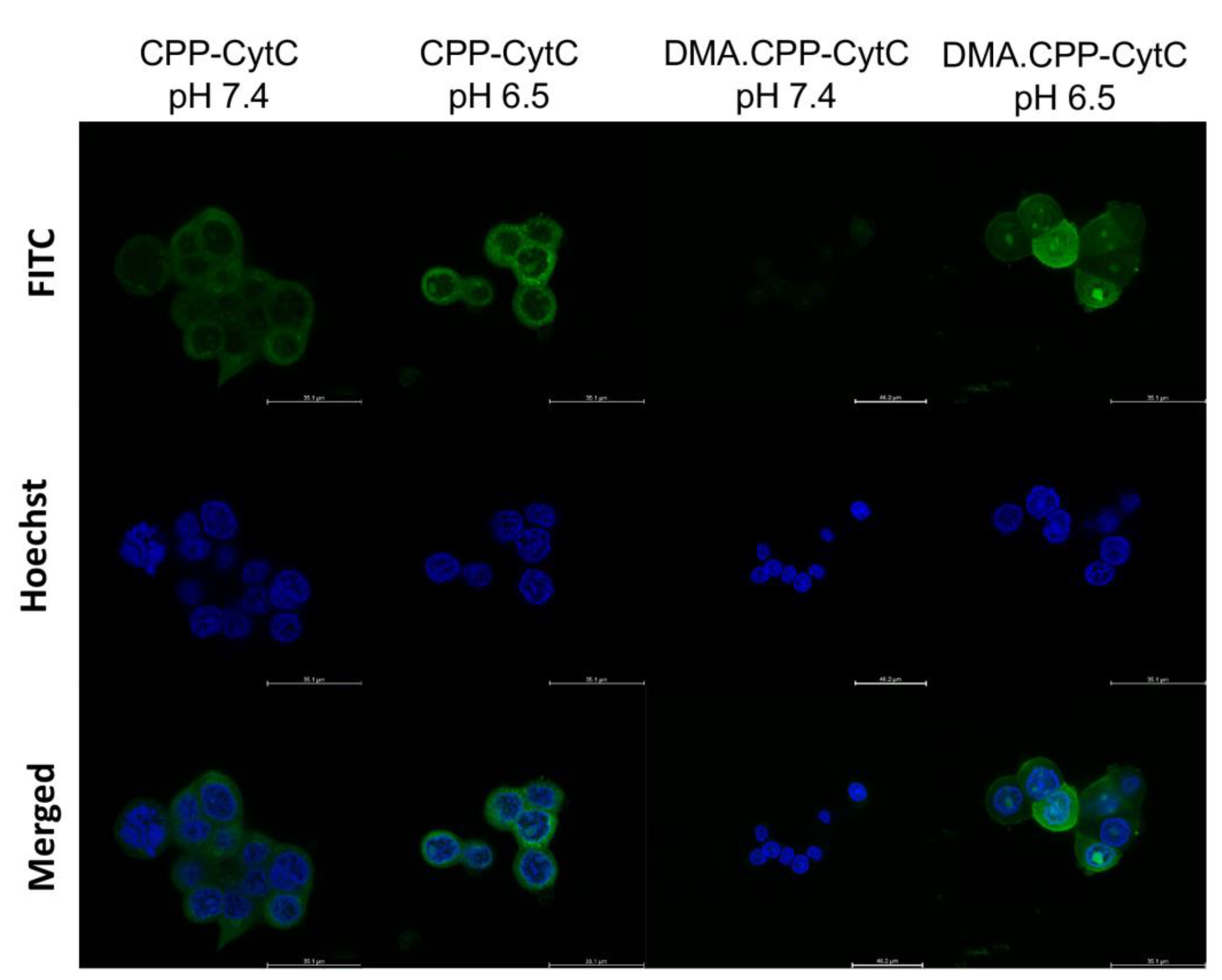
3.2. Internalisation of DMA.CPP-CytC
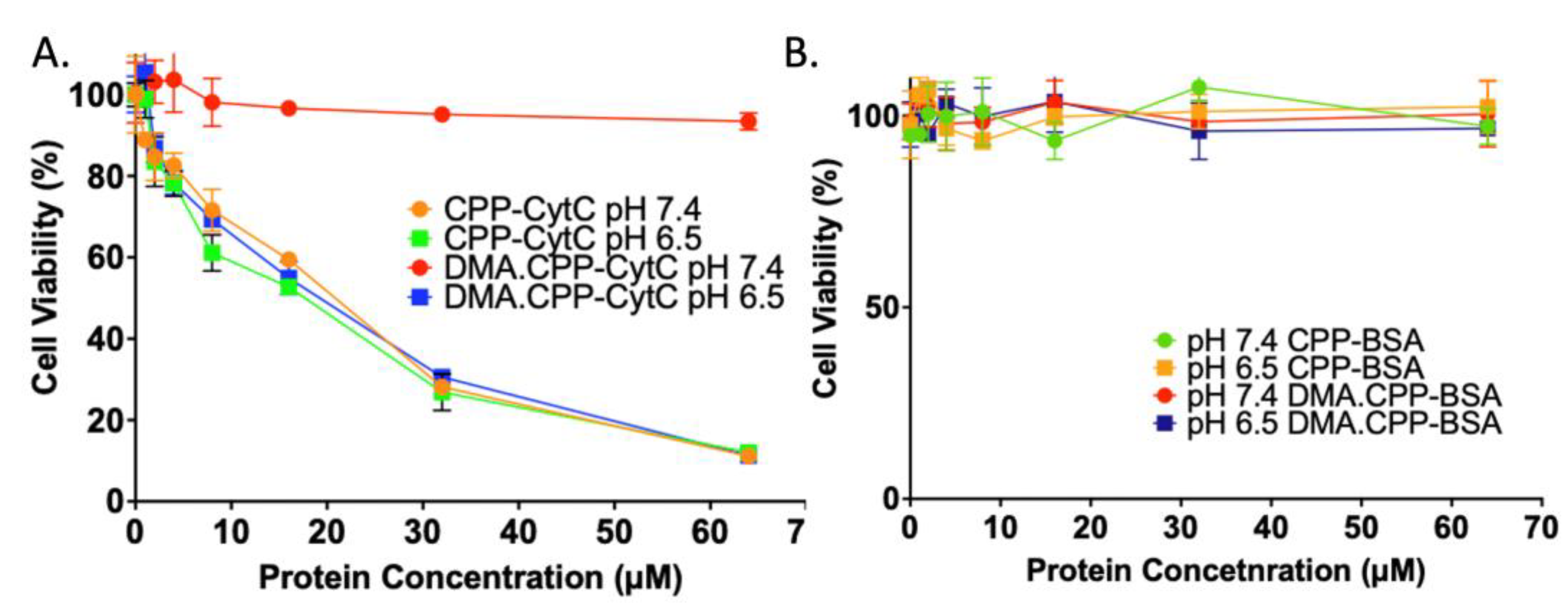
3.3. Cytotoxicity of DMA.CPP-CytC and Its Analogues at Different pHs
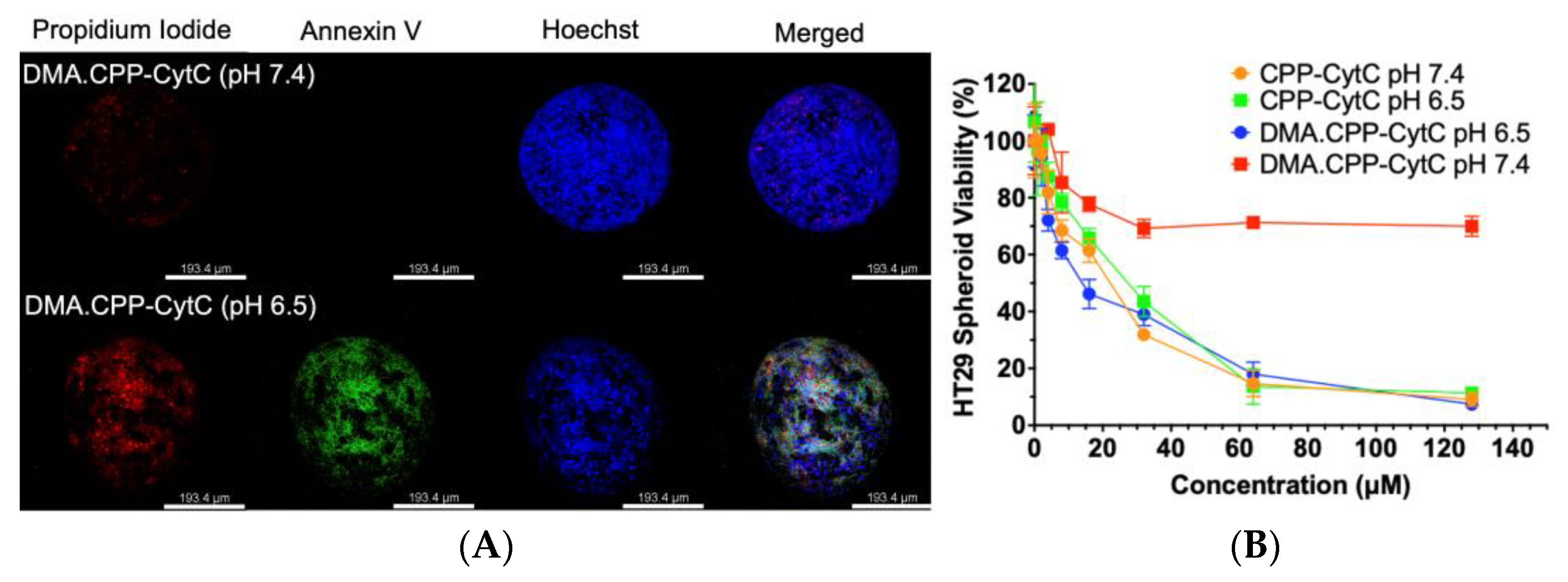
3.4. Spheroid Cytotoxicity Assay and Internalisation
3.5. In Vivo Anticancer Effect of DMA.CPP-CytC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, X.; Wu, F.; Ji, Y.; Yin, L. Recent Advances in Anti-cancer Protein/Peptide Delivery. Bioconjug. Chem. 2019, 30, 305–324. [Google Scholar] [CrossRef]
- Tian, Y.; Tirrell, M.V.; Labelle, J.L. Harnessing the Therapeutic Potential of Biomacromolecules through Intracellular Delivery of Nucleic Acids, Peptides, and Proteins. Adv. Healthc. Mater. 2022, 11, 2102600. [Google Scholar] [CrossRef]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef]
- Porello, I.; Cellesi, F. Intracellular delivery of therapeutic proteins. New advancements and future directions. Front. Bioeng. Biotechnol. 2023, 11, 1211798. [Google Scholar] [CrossRef]
- Yang, M.; Li, J.; Gu, P.; Fan, X. The Application of Nanoparticles in Cancer Immunotherapy: Targeting Tumor Microenvironment. Bioactiv. Mater. 2021, 6, 1973–1987. [Google Scholar] [CrossRef]
- Shoari, A.; Tooyserkani, R.; Tahmasebi, M.; Löwik, D. Delivery of various cargos into cancer cells and tissues via cell-penetrating peptides: A review of the last decade. Pharmaceutics 2021, 13, 1391. [Google Scholar] [CrossRef]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef]
- Xu, J.; Khan, A.R.; Fu, M.; Wang, R.; Ji, J.; Zhai, G. Cell-penetrating peptide: A means of breaking through the physiological barriers of different tissues and organs. J. Control. Release 2019, 309, 106–124. [Google Scholar] [CrossRef]
- Soler, M.; González-Bártulos, M.; Figueras, E.; Massaguer, A.; Feliu, L.; Planas, M.; Ribas, X.; Costas, M. Delivering aminopyridine ligands into cancer cells through conjugation to the cell-penetrating peptide BP16. Org. Biomol. Chem. 2016, 14, 4061–4070. [Google Scholar] [CrossRef]
- Sun, X.; Zhou, C.; Xia, S.; Chen, X. Small molecule-nanobody conjugate induced proximity controls intracellular processes and modulates endogenous unligandable targets. Nat. Commun. 2023, 14, 1635. [Google Scholar] [CrossRef]
- Shadmani, N.; Makvandi, P.; Parsa, M.; Azadi, A.; Nedaei, K.; Mozafari, N.; Poursina, N.; Mattoli, V.; Tay, F.R.; Maleki, A.; et al. Enhancing Methotrexate Delivery in the Brain by Mesoporous Silica Nanoparticles Functionalized with Cell-Penetrating Peptide using in Vivo and ex Vivo Monitoring. Mol. Pharm. 2023, 20, 1531–1548. [Google Scholar] [CrossRef] [PubMed]
- Tietz, O.; Cortezon-Tamarit, F.; Chalk, R.; Able, S.; Vallis, K.A. Tricyclic cell-penetrating peptides for efficient delivery of functional antibodies into cancer cells. Nat. Chem. 2022, 14, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, D.V.; Allevato, M.M.; Camargo, M.F.; Lesperance, J.; Quraishi, M.A.; Aguilera, J.; Franiak-Pietryga, I.; Scanderbeg, D.J.; Wang, Z.; Molinolo, A.A.; et al. Monomethyl auristatin antibody and peptide drug conjugates for trimodal cancer chemo-radio-immunotherapy. Nat. Commun. 2022, 13, 3869. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Nakazawa, H.; Miura, D.; Otake, M.; Umetsu, M. Enzymatic ligation of an antibody and arginine 9 peptide for efficient and cell-specific siRNA delivery. Sci. Rep. 2021, 11, 21882. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mei, L.; Xu, C.; Yu, Q.; Shi, K.; Zhang, L.; Wang, Y.; Zhang, Q.; Gao, H.; Zhang, Z.; et al. Dual receptor recognizing cell penetrating peptide for selective targeting, efficient intratumoral diffusion and synthesized anti-glioma therapy. Theranostics 2016, 6, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Lemech, C.R.; Kichenadasse, G.; Marschner, J.-P.; Alevizopoulos, K.; Otterlei, M.; Millward, M. ATX-101, a cell-penetrating protein targeting PCNA, can be safely administered as intravenous infusion in patients and shows clinical activity in a Phase 1 study. Oncogene 2023, 42, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-penetrating peptides in diagnosis and treatment of human diseases: From preclinical research to clinical application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef] [PubMed]
- Staecker, H.; Jokovic, G.; Karpishchenko, S.; Kienle-Gogolok, A.; Krzyzaniak, A.; Lin, C.-D.; Navratil, P.; Tzvetkov, V.; Wright, N.; Meyer, T. Efficacy and safety of AM-111 in the treatment of acute unilateral sudden deafness—A double-blind, randomized, placebo-controlled phase 3 study. Otol. Neurotol. 2019, 40, 584–594. [Google Scholar] [CrossRef]
- Mu, Y.; Gong, L.; Peng, T.; Yao, J.; Lin, Z. Advances in pH-Responsive Drug Delivery Systems. OpenNano 2021, 5, 100031. [Google Scholar] [CrossRef]
- Ding, H.; Tan, P.; Fu, S.; Tian, X.; Zhang, H.; Ma, X.; Gu, Z.; Luo, K. Preparation and Application of pH-Responsive Drug Delivery Systems. J. Control. Release 2022, 384, 206–238. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-penetrating peptides: From basic research to clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lin, W.; Yang, L.; Zhang, A.; Zhang, Y.; Liu, J.; Liu, J. Injectable and pH-responsive self-assembled peptide hydrogel for promoted tumor cell uptake and enhanced cancer chemotherapy. Biomater. Sci. 2022, 10, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Wang, Y.; Zhao, X.; Zhang, J.; Hu, H.; Qiao, M.; Zhao, X.; Chen, D. Stimuli-responsive and highly penetrable nanoparticles as a multifunctional nanoplatform for boosting nonsmall cell lung cancer siRNA therapy. ACS Biomater. Sci. Eng. 2021, 7, 3141–3155. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, Y.; Fu, C.; Li, B.; Li, L.; Zhang, R.; Xu, T.; Xu, Z.P. Charge reversion simultaneously enhances tumor accumulation and cell uptake of layered double hydroxide nanohybrids for effective imaging and therapy. Small 2020, 16, 2002115. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Deng, Y.; Jia, F.; Jin, Q.; Ji, J. Surface charge switchable supramolecular nanocarriers for nitric oxide synergistic photodynamic eradication of biofilms. ACS Nano 2020, 14, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Sui, J.; Ma, M.; Hu, J.; Sun, Y.; Yang, L.; Fan, Y.; Zhang, X. pH-Responsive charge switchable PEGylated ε-poly-l-lysine polymeric nanoparticles-assisted combination therapy for improving breast cancer treatment. J. Control. Release 2020, 326, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Chen, J.; Shen, J.-M.; Zhuang, R.; Zhang, S.-Q.; Zhu, Z.-Y.; Ma, J.-B. pH-Sensitive nanoparticles as smart carriers for selective intracellular drug delivery to tumor. Int. J. Pharm. 2018, 545, 274–285. [Google Scholar] [CrossRef]
- Guo, X.; Wang, L.; Duval, K.; Fan, J.; Zhou, S.; Chen, Z. Dimeric drug polymeric micelles with acid-active tumor targeting and FRET-traceable drug release. Adv. Mater. 2018, 30, 1705436. [Google Scholar] [CrossRef]
- Zhang, J.; Li, M.; Yuan, Z.; Wu, D.; Chen, J.-D.; Feng, J.J. Stepwise-activable multifunctional peptide-guided prodrug micelles for cancerous cells intracellular drug release. Nanopart. Res. 2016, 18, 299. [Google Scholar] [CrossRef]
- Li, L.; Sun, W.; Zhong, J.; Yang, Q.; Zhu, X.; Zhou, Z.; Zhang, Z.; Huang, Y. Multistage Nanovehicle Delivery System Based on Stepwise Size Reduction and Charge Reversal for Programmed Nuclear Targeting of Systemically Administered Anticancer Drugs. Adv. Funct. Mater. 2015, 25, 4101–4113. [Google Scholar] [CrossRef]
- Jin, E.; Zhang, B.; Sun, X.; Zhou, Z.; Ma, X.; Sun, Q.; Tang, J.; Shen, Y.; Van Kirk, E.; Murdoch, W.J.; et al. Acid-active cell-penetrating peptides for in vivo tumor-targeted drug delivery. J. Am. Chem. Soc. 2013, 135, 933–940. [Google Scholar] [CrossRef]
- Lamberti, M.; Zappavigna, S.; Sannolo, N.; Porto, S.; Caraglia, M. Advantages and Risks of Nanotechnologies in Cancer Patients and Occupationally Exposed Worker. Expert Opin. Drug Deliv. 2014, 11, 1087–1101. [Google Scholar] [CrossRef] [PubMed]
- Đorđević, S.; Gonzalez, M.M.; Conejos-Sánchez, I.; Carreira, B.; Pozzi, S.; Acúrcio, R.C.; Satchi-Fainaro, R.; Florindo, H.F.; Vicent, M.J. Current hurdles to the translation of nanomedicines from bench to the clinic. Drug Deliv. Transl. Res. 2022, 12, 500–525. [Google Scholar] [CrossRef] [PubMed]
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An extreme multifunctional protein with a key role in cell fate. Int. J. Biol. Macromol. 2019, 136, 1237–1246. [Google Scholar] [CrossRef]
- Goldstein, J.C.; Waterhouse, N.J.; Juin, P.; Evan, G.I.; Green, D.R. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2000, 2, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Macone, A.; Masciarelli, S.; Palombarini, F.; Quaglio, D.; Boffi, A.; Trabuco, M.C.; Baiocco, P.; Fazi, F.; Bonamore, A. Ferritin nanovehicle for targeted delivery of cytochrome C to cancer cells. Sci. Rep. 2019, 9, 11749. [Google Scholar] [CrossRef] [PubMed]
- Morales-Cruz, M.; Figueroa, C.M.; González-Robles, T.; Delgado, Y.; Molina, A.; Méndez, J.; Morales, M.; Griebenow, K. Activation of caspase-dependent apoptosis by intracellular delivery of cytochrome c-based nanoparticles. J. Nanobiotechnol. 2014, 12, 33. [Google Scholar] [CrossRef]
- Kim, S.K.; Foote, M.B.; Huang, L. The targeted intracellular delivery of cytochrome C protein to tumors using lipid-apolipoprotein nanoparticles. Biomaterials 2012, 33, 3959–3966. [Google Scholar] [CrossRef]
- Santra, S.; Kaittanis, C.; Perez, J.M. Cytochrome c Encapsulating Theranostic Nanoparticles: A Novel Bifunctional System for Targeted Delivery of Therapeutic Membrane-Impermeable Proteins to Tumors and Imaging of Cancer Therapy. Mol. Pharm. 2010, 7, 1209–1222. [Google Scholar] [CrossRef]
- Sun, X.S.; Jang, M.-S.; Fu, Y.; Lee, J.H.; Lee, D.S.; Li, Y.; Yang, H.Y. Intracellular delivery of cytochrome C using hypoxia-responsive polypeptide micelles for efficient cancer therapy. Mater. Sci. Eng. C 2020, 114, 111069. [Google Scholar] [CrossRef]
- Akrami, M.; Balalaie, S.; Hosseinkhani, S.; Alipour, M.; Salehi, F.; Bahador, A.; Haririan, I. Tuning the anticancer activity of a novel pro-apoptotic peptide using gold nanoparticle platforms. Sci. Rep. 2016, 6, 31030. [Google Scholar] [CrossRef] [PubMed]
- Delinois, L.J.; De León-Vélez, O.; Vázquez-Medina, A.; Vélez-Cabrera, A.; Marrero-Sánchez, A.; Nieves-Escobar, C.; Alfonso-Cano, D.; Caraballo-Rodríguez, D.; Rodriguez-Ortiz, J.; Acosta-Mercado, J.; et al. Cytochrome c: Using Biological Insight toward Engineering an Optimized Anticancer Biodrug. Inorganics 2021, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.C.H.; Wong, C.T.T.; Ng, D.K.P. Toward Precise Antitumoral Photodynamic Therapy Using a Dual Receptor-Mediated Bioorthogonal Activation Approach. Angew. Chem. 2023, 135, e202214473. [Google Scholar] [CrossRef]
- Xiong, J.; Chu, J.C.H.; Fong, W.P.; Wong, C.T.T.; Ng, D.K.P. Specific activation of photosensitizer with extrinsic enzyme for precisive photodynamic therapy. J. Am. Chem. Soc. 2022, 144, 10647–10658. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.C.H.; Fong, W.P.; Wong, C.T.T.; Ng, D.K.P. Facile Synthesis of Cyclic Peptide-Phthalocyanine Conjugates for Epidermal Growth Factor Receptor-Targeted Photodynamic Therapy. J. Med. Chem. 2021, 64, 2064–2076. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.C.H.; Yang, C.; Fong, W.P.; Wong, C.T.T.; Ng, D.K.P. Facile one-pot synthesis of cyclic peptide-conjugated photosensi-tisers for targeted photodynamic therapy. Chem. Commun. 2020, 56, 11941–11944. [Google Scholar] [CrossRef]
- Chu, J.C.H.; Chin, M.L.; Wong, C.T.T.; Hui, M.; Lo, P.C.; Ng, D.K.P. One-Pot Synthesis of a Cyclic Antimicrobial Peptide-Con-jugated Phthalocyanine for Synergistic Chemo-Photodynamic Killing of Multidrug-Resistant Bacteria. Adv. Ther. 2020, 4, 2000204. [Google Scholar] [CrossRef]
- Hraběta, J.; Belhajová, M.; Šubrtová, H.; Rodrigo, M.A.M.; Heger, Z.; Eckschlager, T. Drug Sequestration in Lysosomes as One of the Mechanisms of Chemoresistance of Cancer Cells and the Possibilities of Its Inhibition. Int. J. Mol. Sci. 2020, 21, 4392. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, B.; Lau, K.M.; Zhu, Y.; Shao, C.; Wong, W.-T.; Chow, L.M.C.; Wong, C.T.T. Chemical Modification of Cytochrome C for Acid-Responsive Intracellular Apoptotic Protein Delivery for Cancer Eradication. Pharmaceutics 2024, 16, 71. https://doi.org/10.3390/pharmaceutics16010071
Tang B, Lau KM, Zhu Y, Shao C, Wong W-T, Chow LMC, Wong CTT. Chemical Modification of Cytochrome C for Acid-Responsive Intracellular Apoptotic Protein Delivery for Cancer Eradication. Pharmaceutics. 2024; 16(1):71. https://doi.org/10.3390/pharmaceutics16010071
Chicago/Turabian StyleTang, Bo, Kwai Man Lau, Yunxin Zhu, Chihao Shao, Wai-Ting Wong, Larry M. C. Chow, and Clarence T. T. Wong. 2024. "Chemical Modification of Cytochrome C for Acid-Responsive Intracellular Apoptotic Protein Delivery for Cancer Eradication" Pharmaceutics 16, no. 1: 71. https://doi.org/10.3390/pharmaceutics16010071
APA StyleTang, B., Lau, K. M., Zhu, Y., Shao, C., Wong, W.-T., Chow, L. M. C., & Wong, C. T. T. (2024). Chemical Modification of Cytochrome C for Acid-Responsive Intracellular Apoptotic Protein Delivery for Cancer Eradication. Pharmaceutics, 16(1), 71. https://doi.org/10.3390/pharmaceutics16010071