
Abiraterone and Galeterone, Powerful Tools Against Prostate Cancer: Present and Perspective



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
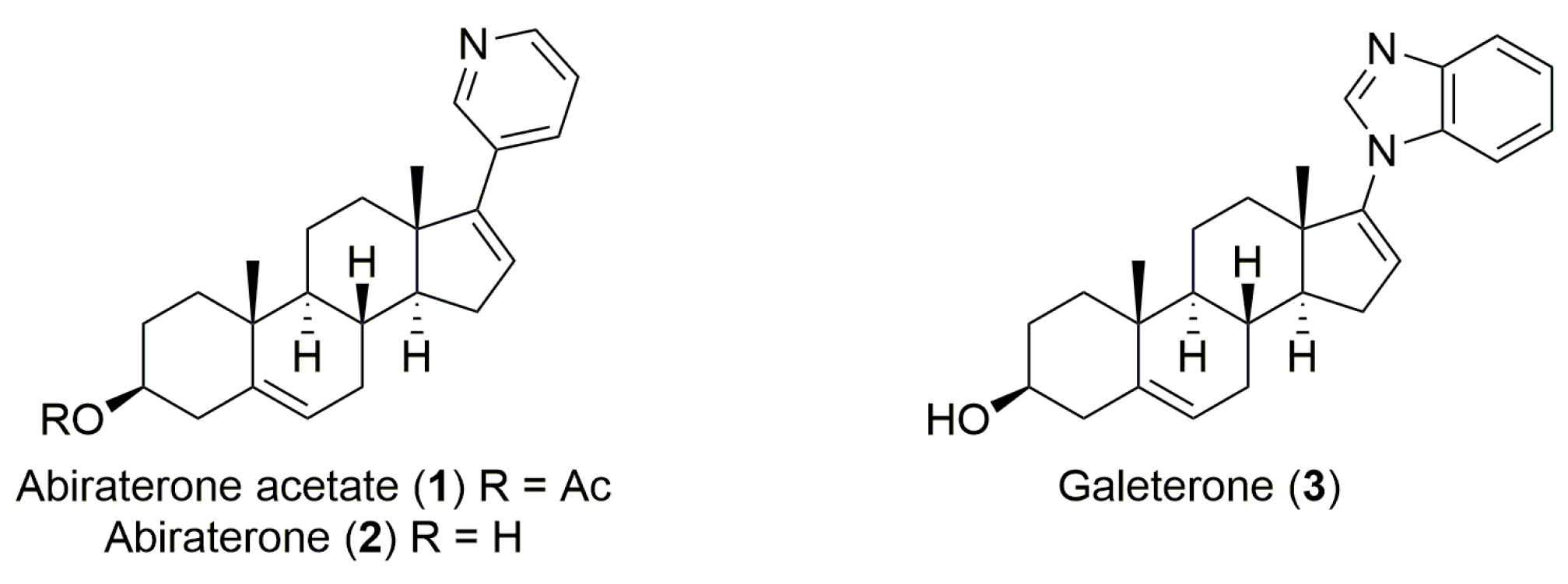
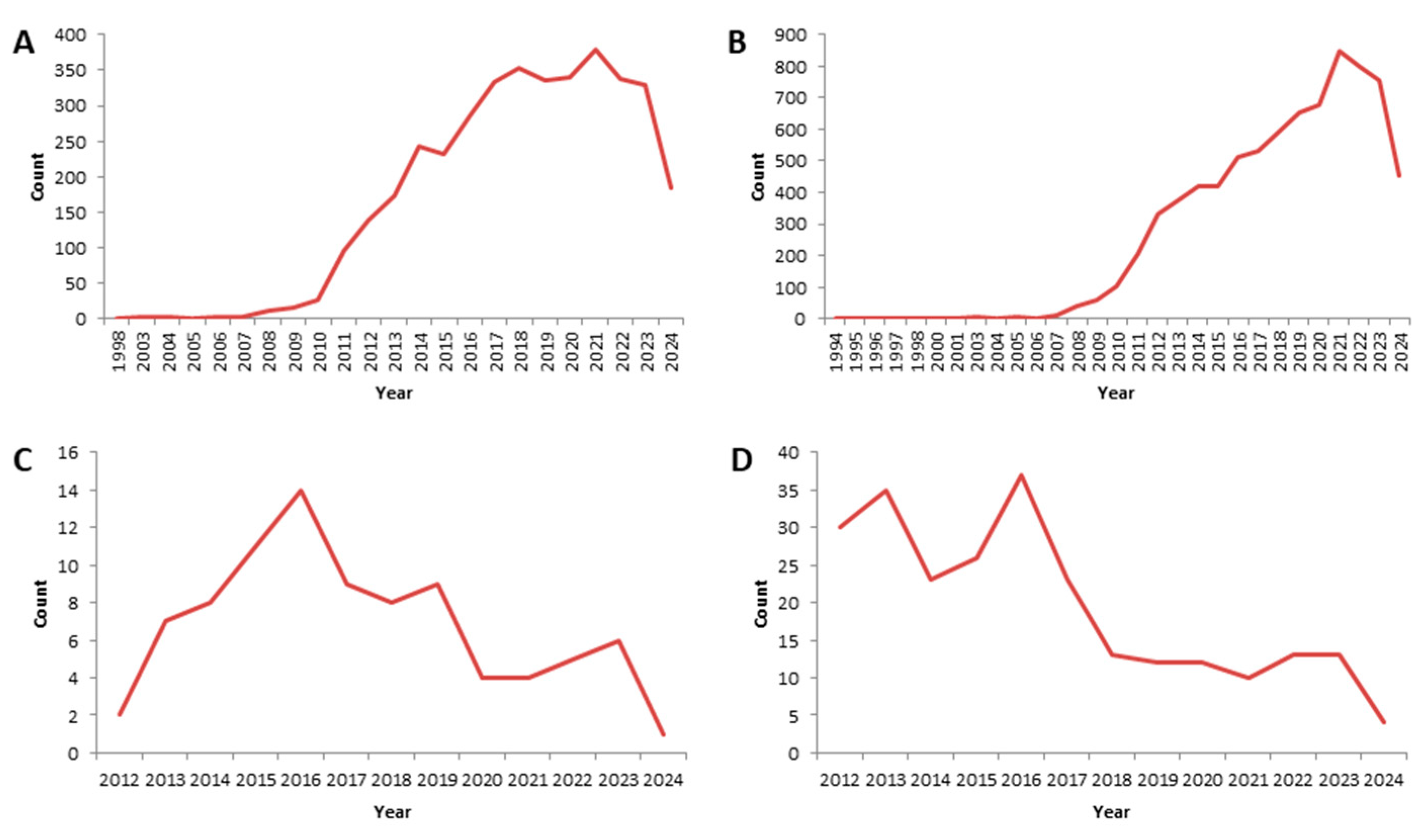
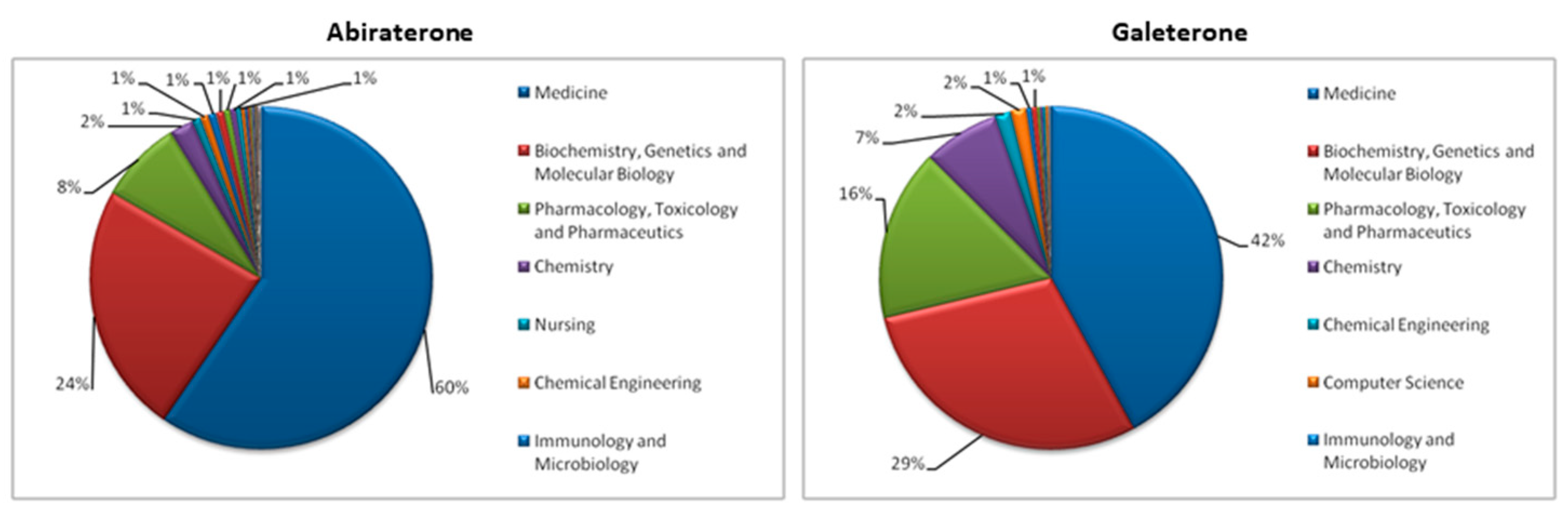
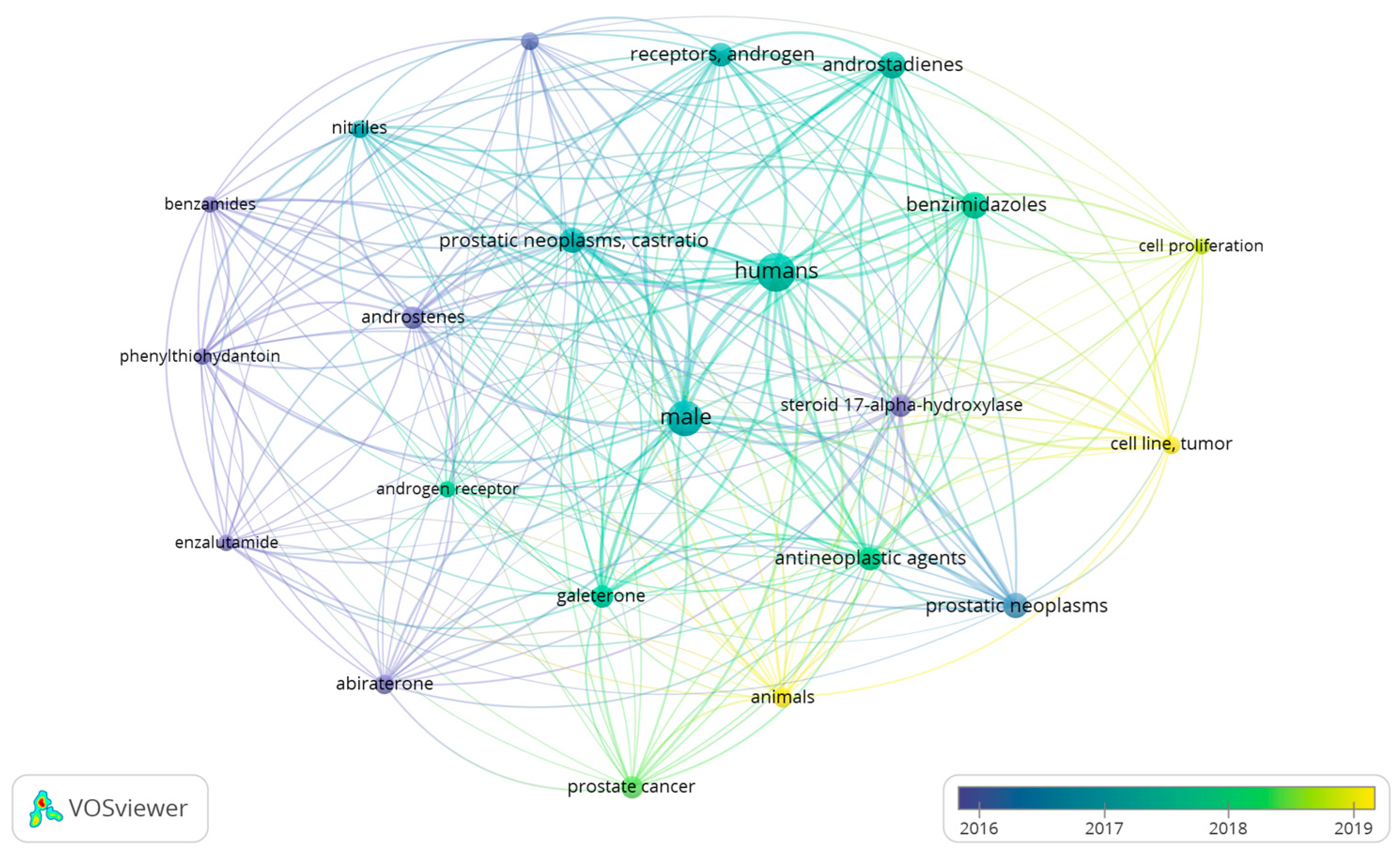
2. Literature Research Analysis
3. Synthesis
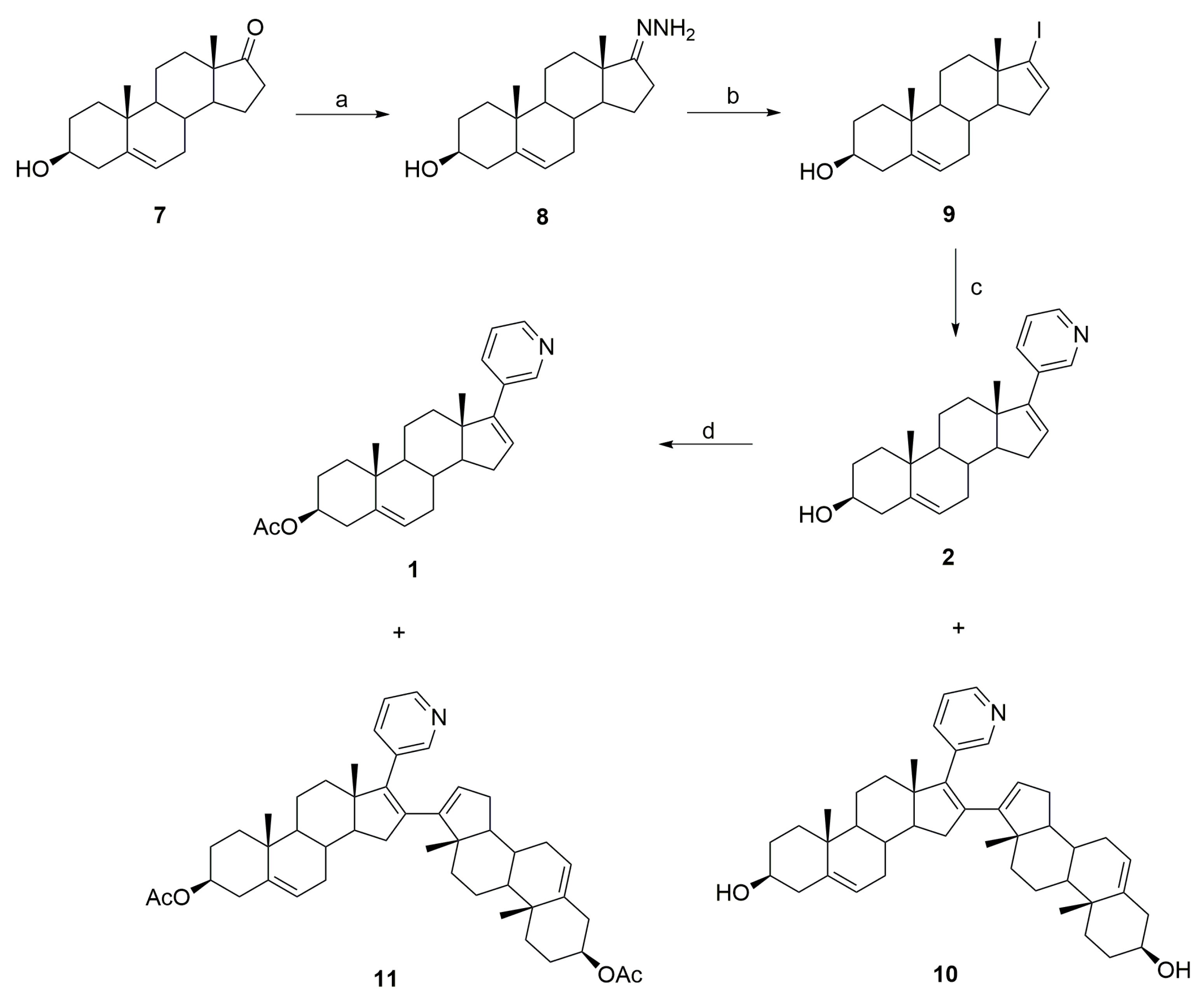
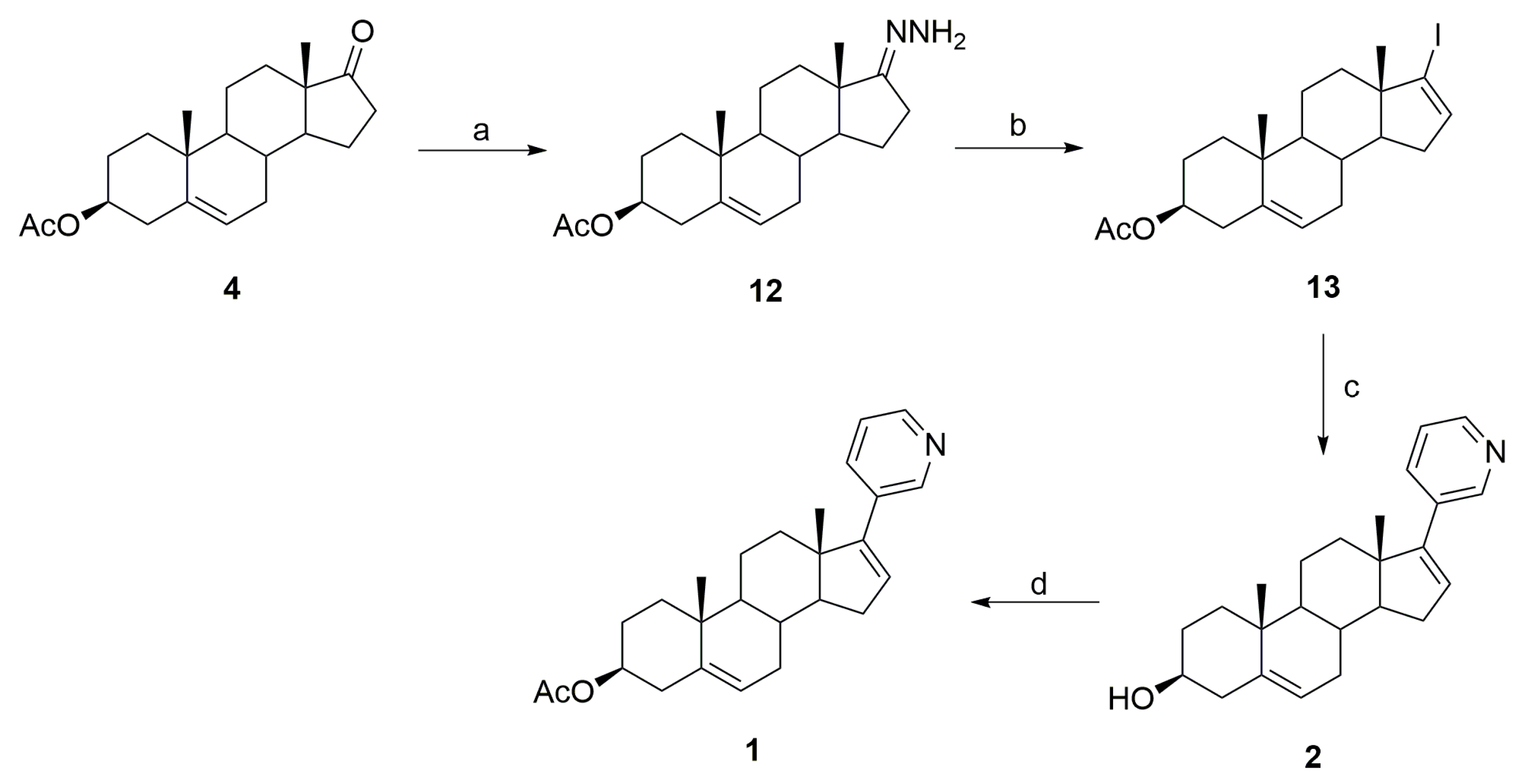
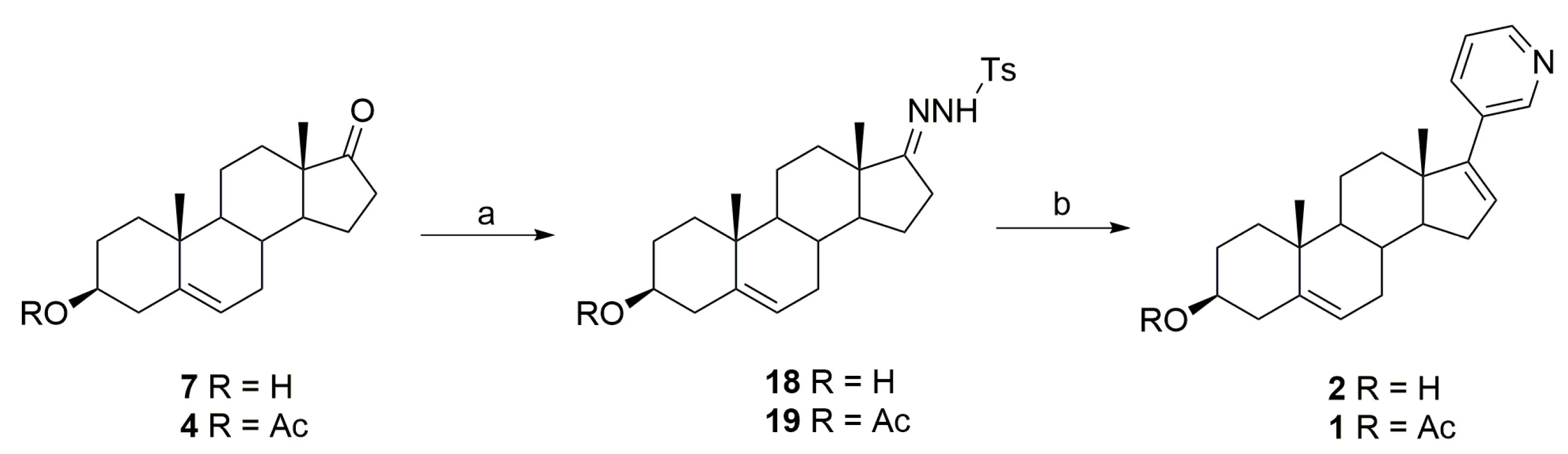
3.1. Synthesis of Abiraterone (2)
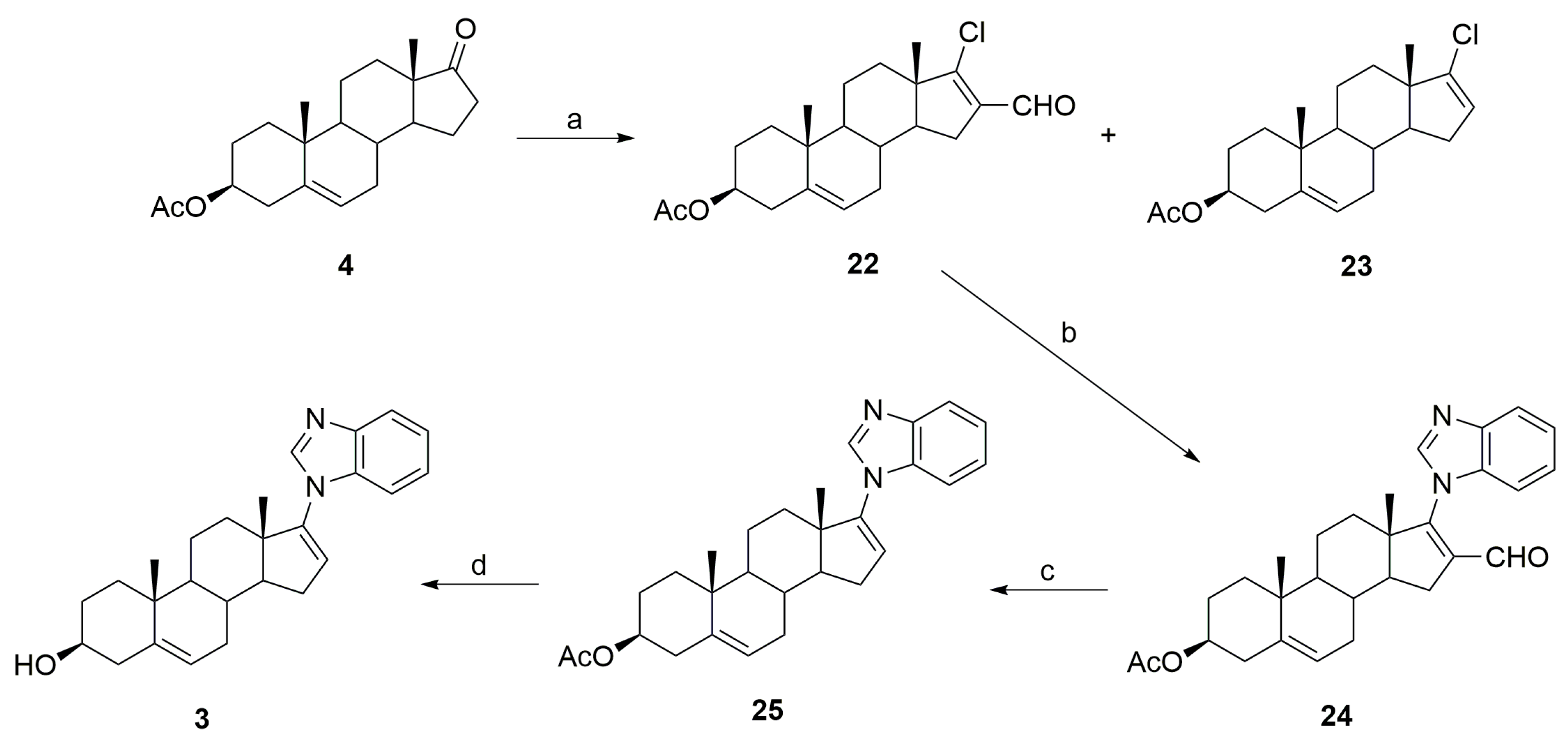
3.2. Synthesis of Galeterone (3)
4. Medicinal Uses, Benefits, and Side Effects
4.1. Abiraterone (2)
4.1.1. Medical Uses and Benefits of Abiraterone
Prostate Cancer
Breast Cancer
Ovarian Cancer
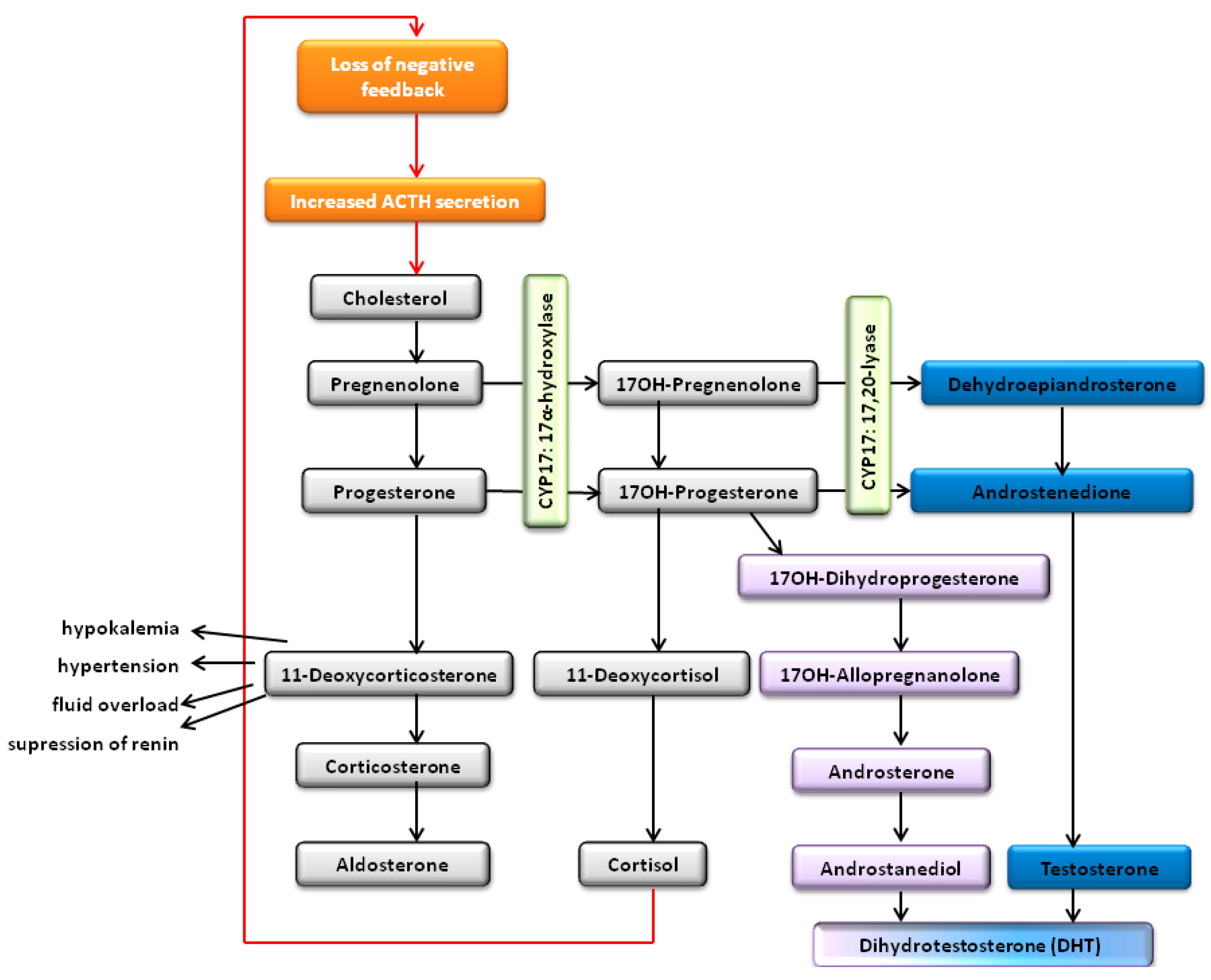
Congenital Adrenal Hyperplasia
Human Adrenocortical Cancer and Cushing’s Syndrome
Lung Cancer
Kidney Cancer
Chronic Kidney Disease
Salivary Gland Carcinoma
COVID-19
4.1.2. Metabolism of Abiraterone Acetate
4.1.3. Side Effects
4.2. Galeterone (3)
4.2.1. Enzyme Inhibition
4.2.2. Interaction with Androgen Receptors
4.2.3. Other Cancers
4.2.4. Metabolism of Galeterone
4.2.5. Galeterone Formulation
4.3. Comparative Studies of Biological Activity of Abiraterone and Galeterone
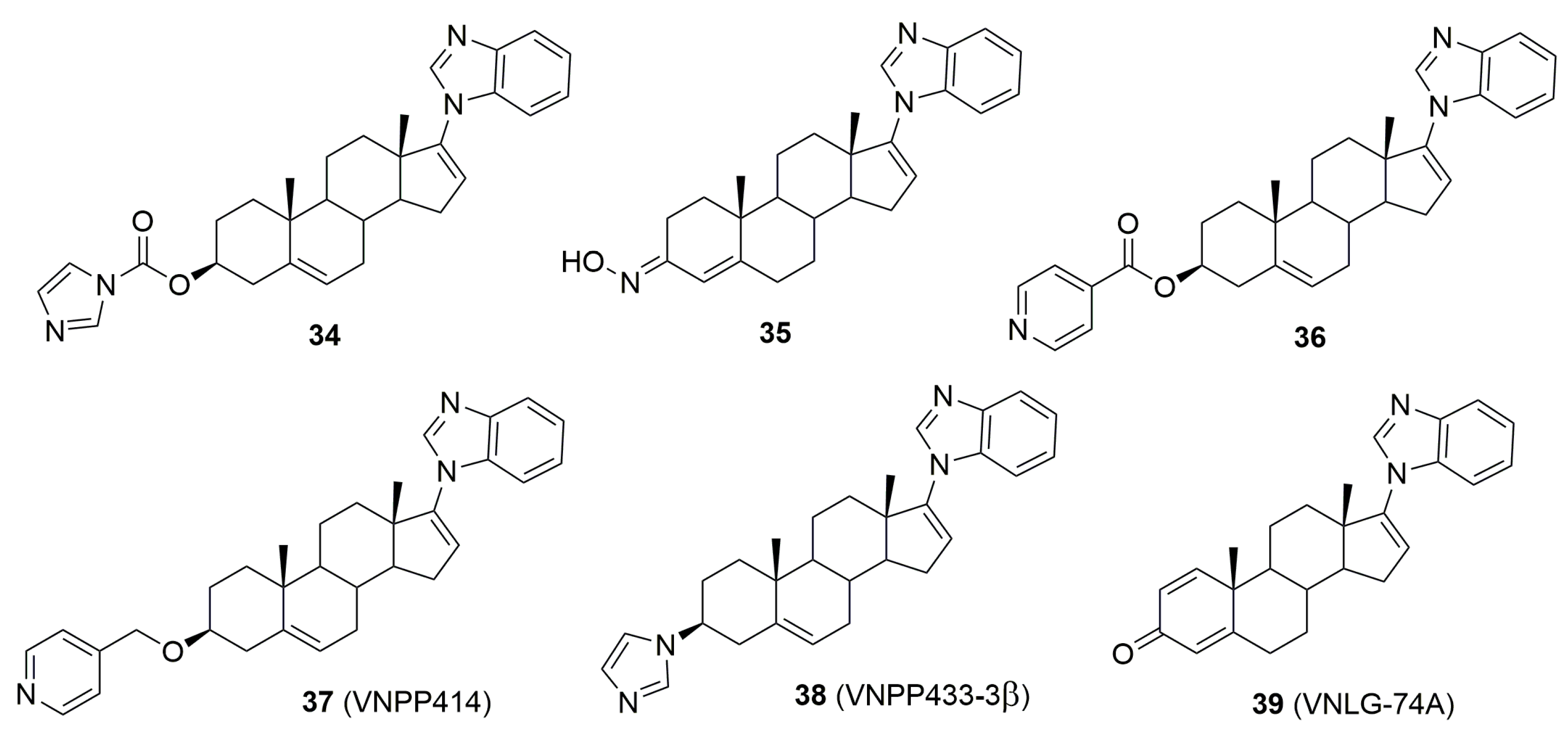
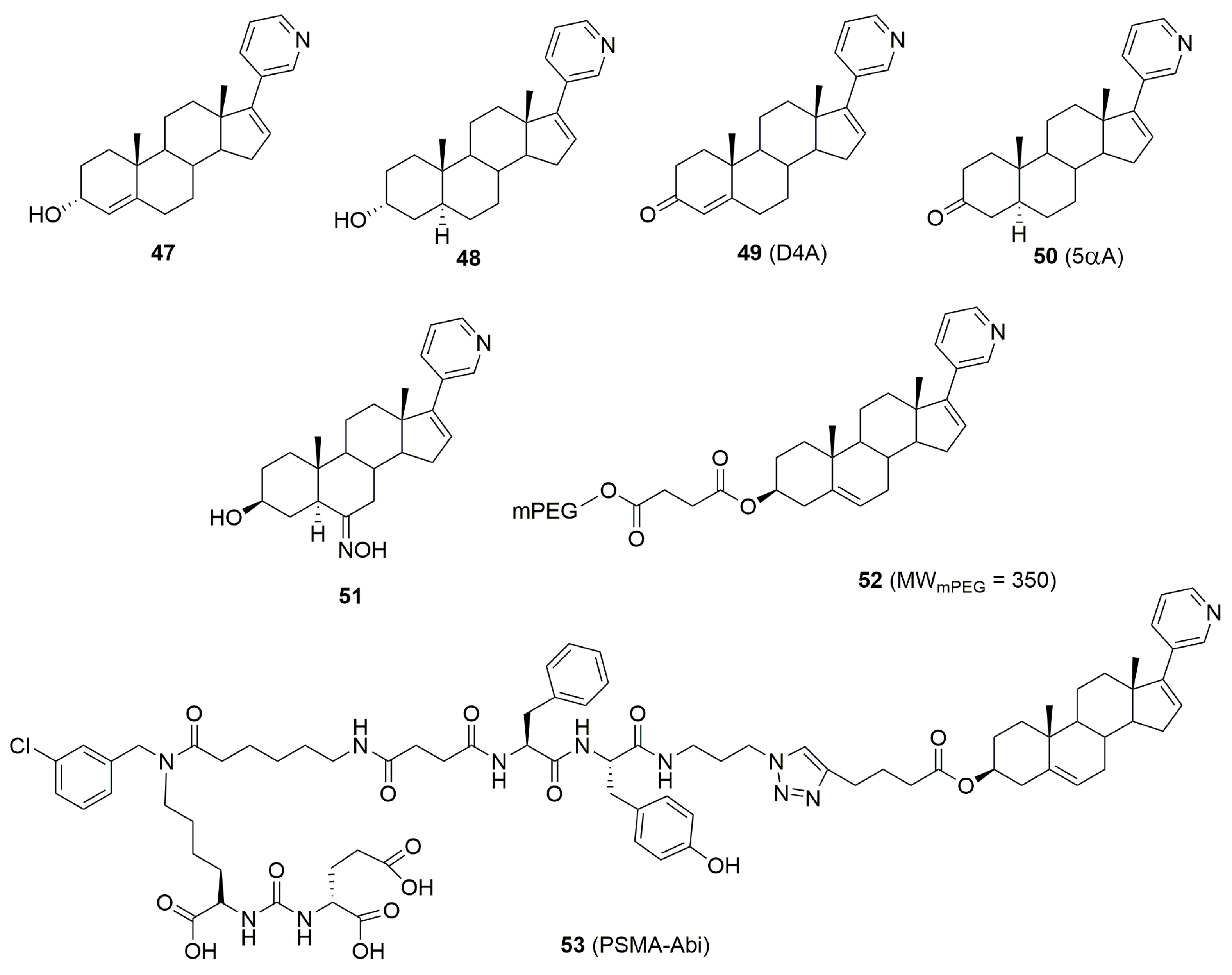
5. Biologically Active Derivatives and Analogs of Galeterone and Abiraterone
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Available online: https://www.cancer.gov/types/prostate/patient/prostate-treatment-pdq (accessed on 12 August 2024).
- Huggins, C. Effect of orchiectomy and irradiation on cancer of the prostate. Ann. Surg. 1942, 115, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral de novo steroid synthesis activates androgen receptor in castrationresistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef]
- Yang, L.P.H. Abiraterone Acetate. Drugs 2011, 71, 2067–2077. [Google Scholar] [CrossRef]
- Stappaerts, J.; Geboers, S.; Snoeys, J.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Rapid conversion of the ester prodrug abiraterone acetate results in intestinal supersaturation and enhanced absorption of abiraterone: In vitro, rat in situ and human in vivo studies. Eur. J. Pharm. Biopharm. 2015, 90, 1–7. [Google Scholar] [CrossRef]
- Barrie, S.E.; Jarman, M.; Potter, G.A. 17-Substituted Steroids, Used in Cancer Treatment. UK Patent Application GB 2,265,624A, 1993. [Google Scholar]
- FDA. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=202379 (accessed on 15 August 2024).
- Handratta, V.D.; Vasaitis, T.S.; Njar, V.C.O.; Gediya, L.K.; Kataria, R.; Chopra, P.; Newman, D.; Farquhar, R.; Guo, Z.; Qiu, Y.; et al. Novel C-17-Heteroaryl Steroidal CYP17 Inhibitors/Antiandrogens: Synthesis, in Vitro Biological Activity, Pharmacokinetics, and Antitumor Activity in the LAPC4 Human Prostate Cancer Xenograft Model. J. Med. Chem. 2005, 48, 2972–2984. [Google Scholar] [CrossRef] [PubMed]
- Boujonnier, F.; Lemaitre, F.; Scailteux, L.M. Pharmacokinetic Interactions Between Abiraterone, Apalutamide, Darolutamide or Enzalutamide and Antithrombotic Drugs: Prediction of Clinical Events and Review of Pharmacological Information. Cardiovasc. Drugs Ther. 2024, 38, 757–767. [Google Scholar] [CrossRef]
- Marchioni, M.; Sountoulides, P.; Bada, M.; Rapisarda, S.; De Nunzio, C.; Tamburro, F.R.; Schips, L.; Cindolo, L. Abiraterone in chemotherapy-naive patients with metastatic castration-resistant prostate cancer: A systematic review of ‘real-life’ studies. Ther. Adv. Urol. 2018, 10, 305–315. [Google Scholar] [CrossRef]
- Thakur, A.; Roy, A.; Ghosh, A.; Chhabra, M.; Banerjee, S. Abiraterone acetate in the treatment of prostate cancer. Biomed. Pharmacother. 2018, 101, 211–218. [Google Scholar] [CrossRef]
- Bastos, D.; Antonarakis, E. Galeterone for the treatment of advanced prostate cancer: The evidence to date. Drug Des. Devel. Ther. 2016, 10, 2289–2297. [Google Scholar] [CrossRef]
- Wicha, J.; Masnyk, M. Synthesis of 17β-Pyridyl- and 17β-Pyridonyl-androstane Derivatives. Heterocycles 1981, 16, 521–524. [Google Scholar] [CrossRef]
- Wicha, J.; Masnyk, M.; Schonfeld, W.; Repke, K.R.H. Synthesis and Molecular-Biological Activily of The Pyridine Analogue of Cardiotonic Steroids. Heterocycles 1983, 20, 231–234. [Google Scholar] [CrossRef]
- Barrie, S.E.; Potter, G.A.; Goddard, P.M.; Haynes, B.P.; Dowsett, M.; Jarman, M. Pharmacology of Novel Steroidal Inhibitors of Cytochrome P45017α (17α-Hydroxylase/C17-20 Lyase). J. Steroid Biochem. Mol. Biol. 1994, 50, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Potter, G.A.; Barrie, S.E.; Jarman, M.; Rowlands, M.G. Novel Steroidal Inhibitors of Human Cytochrome P45017α-Hydroxylase-C17,20-lyase): Potential Agents for the Treatment of Prostatic Cancer. J. Med. Chem. 1995, 38, 2463–2471. [Google Scholar] [CrossRef]
- Hartmann, R.W.; Hector, M.; Haidar, S.; Ehmer, P.B.; Reichert, W.; Jose, J. Synthesis and Evaluation of Novel Steroidal Oxime Inhibitors of P450 17 (17α-Hydroxylase/C17−20-Lyase) and 5α-Reductase Types 1 and 2. J. Med. Chem. 2000, 43, 4266–4277. [Google Scholar] [CrossRef] [PubMed]
- Haidar, S.; Ehmer, P.B.; Hartmann, R.W. Novel steroidal pyrimidyl inhibitors of P450 17 (17α-hydroxylase/C17-20-lyase). Arch. Pharm. 2001, 334, 373–374. [Google Scholar] [CrossRef]
- Bury, P.S. Process for the Preparation of 17-0-Vinyl-Triflates as Intermediates. International Publication Number WO2006021777A1, 2 March 2006. [Google Scholar]
- Hunt, N.J. Methanesulfonate Salts of Abiraterone-3-esters and Recovery of Salts of Abiraterone-3-esters from Solution in Methyl tert-Butyl Ether. International Publication Number WO2006021776A1, 2 March 2006. [Google Scholar]
- Dymacek, B.; Castulik, J. Process for Making the 17-Triflate Intermediate of Abiraterone-3-Acetate. International Publication Number WO2014071983A1, 15 May 2014. [Google Scholar]
- Sun, Q.; Jiang, C.; Xu, H.; Zhang, Z.; Liu, L.; Wang, C. Pd(PPh3)4/AgOAc-Catalyzed Coupling of 17-Steroidal Triflates and Alkynes: Highly Efficient Synthesis of D-ring Unsaturated 17-Alkynylsteroids. Steroids 2010, 75, 936–943. [Google Scholar] [CrossRef]
- Lenna, R.; Di Brisco, R. Process for the Preparation of Abiraterone or Abiraterone Acetate. International Publication Number WO2015014686A1, 5 February 2015. [Google Scholar]
- Bian, X.; Wang, L.; Liu, J.; Wang, C. Green Suzuki Coupling Reaction for Synthesis of Abiraterone Acetate and its Analogues. J. Chem. Res. 2016, 40, 289–292. [Google Scholar] [CrossRef]
- Potter, G.A.; Hardcastle, I.R.; Jarman, M. A convenient, large-scale synthesis of abiraterone acetate [3β-acetoxy-17-(3-pyridyl)androsta-5,16-diene] potential new drug for the treatment of prostate cancer. Org. Prep. Proced. Int. 1997, 29, 123–134. [Google Scholar] [CrossRef]
- Zhiquan, Z.; Hongbo, W. Method for Extracting Concentrated S-adenosylmethionine. China Patent CN102617681A, 1 August 2012. [Google Scholar]
- Balaev, A.N.; Gromyko, A.V.; Fedorov, V.E. Four-Step Synthesis of Abiraterone Acetate from Dehydroepiandrosterone. Pharm. Chem. J. 2016, 50, 404–406. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, L.; Shi, G.; Shi, J.; Lai, W.L. Research on the synthesis and characterization of abiraterone acetate. Bulg. Chem. Commun. 2017, 49, 199–203. [Google Scholar]
- Komati, S.K.; Madhra, M.K.; Manda, A.; Annapurna, S.C.V.; Senadi, G.C.; Maruthapillai, A.; Bandichhor, R. Efficient Process Development of Abiraterone Acetate by Employing Design of Experiments. ACS Omega 2024, 9, 29453–29470. [Google Scholar] [CrossRef] [PubMed]
- Madhra, M.K.; Sriram, H.M.; Inamdar, M.; Sharma, M.K.; Prasad, M.; Joseph, S. Improved Procedure for Preparation of Abiraterone Acetate. Org. Process Res. Dev. 2014, 18, 555–558. [Google Scholar] [CrossRef]
- Pérez Encabo, A.; Turiel Hernandez, J.A.; Gallo Nieto, F.J.; Bonde-Larsen, A.L.; Sandoval Rodríguez, C.M. Synthesis of Abiraterone and Related Compounds. International Publication Number WO2013030410A2, 7 March 2013. [Google Scholar]
- Li, J.; Ma, S.; Tang, H.; Xu, F. Synthesis of the Anti-Prostate Cancer Drug Abiraterone Acetate. Heterocycles 2018, 96, 461–469. [Google Scholar] [CrossRef]
- Marom, E.; Rubnov, S.; Mizhiritskii, M. Process and Intermediates for the Preparation of Abiraterone Acetate. International Publication Number WO2014016830A1, 30 January 2014. [Google Scholar]
- Clement, O.O.; Freeman, C.M.; Hartmann, R.W.; Handratta, V.D.; Vasaitis, T.S.; Brodie, A.M.H.; Njar, V.C.O. Three Dimensional Pharmacophore Modeling of Human CYP17 Inhibitors. Potential Agents for Prostate Cancer Therapy. J. Med. Chem. 2003, 46, 2345–2351. [Google Scholar] [CrossRef]
- Njar, V.C.O.; Klus, G.T.; Brodie, A.M.H. Nucleophilic vinylic “addition-elimination” substitution reaction of 3β-acetoxy-17-chloro-16-formylandrosta-5,16-diene: A novel and general route to 17-substituted steroids. Part 1—Synthesis of novel 17-azolyl-Δ16 steroids; inhibitors of 17α-hydroxylase/17,20-lyase (17α-lyase). Bioorg. Med. Chem. Lett. 1996, 6, 2777–2782. [Google Scholar] [CrossRef]
- Njar, V.C.O.; Kato, K.; Nnane, I.P.; Grigoryev, D.N.; Long, B.J.; Brodie, A.M.H. Novel 17-azolyl steroids, potent inhibitors of cytochrome 17α-hydroxylase/C17,20-lyase (P45017α): Potential agents for the treatment of prostate cancer. J. Med. Chem. 1998, 41, 902–912. [Google Scholar] [CrossRef]
- Barbieri, F.; Lenna, R. Process for the Preparation of Galeterone. International Publication Number WO2017208132A1, 7 December 2017. [Google Scholar]
- Purushottamachar, P.; Thomas, E.; Thankan, R.S.; Rudchenko, V.; Huang, G.; Njar, V.C.O. Large-scale synthesis of galeterone and lead next generation galeterone analog VNPP433-3β. Steroids 2022, 185, 109062. [Google Scholar] [CrossRef]
- Potter, G.A.; Barrie, S.E. Discovery of highly potent enzyme inhibitors with potential for the treatment of prostate cancer; The important dual role of transition metal chemistry in both drug design and synthesis. In Organometallic Reagents in Organic Synthesis; Bateson, J.H., Mitchell, M.B., Eds.; Academic Press: London, UK, 1994; p. 235. ISBN 978-012-499-150-7. [Google Scholar]
- O’Donnell, A.; Judson, I.; Dowsett, M.; Raynaud, F.; Dearnaley, D.; Mason, M.; Harland, S.; Robbins, A.; Halbert, G.; Nutley, B.; et al. Hormonal impact of the 17α-hydroxylase/C17,20-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br. J. Cancer 2004, 90, 2317–2325. [Google Scholar] [CrossRef]
- Solymosi, T.; Ötvös, Z.; Angi, R.; Ordasi, B.; Jordán, T.; Semsey, S.; Molnár, L.; Ránky, S.; Filipcsei, G.; Heltovics, G.; et al. Development of an abiraterone acetate formulation with improved oral bioavailability guided by absorption modeling based on in vitro dissolution and permeability measurements. Int. J. Pharmaceut. 2017, 532, 427–434. [Google Scholar] [CrossRef]
- Goldwater, R.; Hussaini, A.; Bosch, B.; Nemeth, P. Comparison of a Novel Formulation of Abiraterone Acetate vs. the Originator Formulation in Healthy Male Subjects: Two Randomized, Open-Label, Crossover Studies. Clin. Pharmacokinet. 2017, 56, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Burris-Hiday, S.D.; Scott, E.E. Steroidogenic cytochrome P450 17A1 structure and function. Mol. Cell. Endocrinol. 2021, 528, 111261. [Google Scholar] [CrossRef] [PubMed]
- DeVore, N.; Scott, E. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 2012, 482, 116–119. [Google Scholar] [CrossRef] [PubMed]
- De Conti, P.; Atallah, Á.N.; Arruda, H.O.; Soares, B.G.O.; El Dib, R.P.; Wilt, T.J. Intermittent versus continuous androgen suppression for prostatic cancer. Cochrane Database Syst. Rev. 2007, 4, CD005009. [Google Scholar] [CrossRef]
- Le, T.K.; Duong, Q.H.; Baylot, V.; Fargette, C.; Baboudjian, M.; Colleaux, L.; Taïeb, D.; Rocchi, P. Castration-Resistant Prostate Cancer: From Uncovered Resistance Mechanisms to Current Treatments. Cancers 2023, 15, 5047. [Google Scholar] [CrossRef]
- Jacob, A.; Raj, R.; Allison, D.B.; Myint, Z.W. Androgen Receptor Signaling in Prostate Cancer and Therapeutic Strategies. Cancers 2021, 13, 5417. [Google Scholar] [CrossRef]
- Attard, G.; Belldegrun, A.S.; De Bono, J.S. Selective blockade of androgenic steroid synthesis by novel lyase inhibitors as a therapeutic strategy for treating metastatic prostate cancer. BJU Int. 2005, 96, 1241–1246. [Google Scholar] [CrossRef]
- Martins, V.; Asad, Y.; Wilsher, N.; Raynaud, F. A validated liquid chromatographic-tandem mass spectroscopy method for the quantification of abiraterone acetate and abiraterone in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 2006, 843, 262–267. [Google Scholar] [CrossRef]
- Attard, G.; Reid, A.H.M.; Yap, T.A.; Raynaud, F.; Dowsett, M.; Settatree, S.; Barret, M.; Parker, C.; Martins, V.; Folkerd, E.; et al. Phase I Clinical Trial of a Selective Inhibitor of CYP17, Abiraterone Acetate, Confirms That Castration-Resistant Prostate Cancer Commonly Remains Hormone Driven. J. Clin. Oncol. 2008, 26, 4563–4571. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; Fong, L.; Rosenberg, J.E.; Kantoff, P.; Raynaud, F.; Martins, V.; Lee, G.; Kheoh, T.; Kim, J.; et al. Phase I Clinical Trial of the CYP17 Inhibitor Abiraterone Acetate Demonstrating Clinical Activity in Patients With Castration-Resistant Prostate Cancer Who Received Prior Ketoconazole Therapy. J. Clin. Oncol. 2010, 28, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulat, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.A.; Sternberg, C.N.; Miller, K.; Logothetis, C.L.; Shore, N.D.; Small, E.J.; et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, doubleblind, placebo-controlled phase 3 study. Lancet Oncol. 2015, 16, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Saad, F.; Rathkopf, D.E.; Mulders, P.F.A.; de Bono, J.S.; Small, E.J.; Shore, N.D.; Fizazi, K.; Kheoh, T.; Li, J.; et al. Clinical Outcomes from Androgen Signaling–directed Therapy after Treatment with Abiraterone Acetate and Prednisone in Patients with Metastatic Castration-resistant Prostate Cancer: Post Hoc Analysis of COU-AA-302. Eur. Urol. 2017, 72, 10–13. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): Final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- Kotani, N.; Wilkins, J.J.; Wade, J.R.; Dang, S.; Sutaria, D.S.; Yoshida, K.; Sundrani, S.; Ding, H.; Garcia, J.; Hinton, H.; et al. Characterization of exposure-response relationships of ipatasertib in patients with metastatic castration-resistant prostate cancer in the IPATential150 study. Cancer Chemother. Pharmacol. 2022, 90, 511–521. [Google Scholar] [CrossRef]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): A multicentre, randomised, double-blind, phase 3 trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef]
- Doi, T.; Fujiwara, Y.; Matsubara, N.; Tomomatsu, J.; Iwasa, S.; Tanaka, A.; Endo-Tsukude, C.; Nakagawa, S.; Takahashi, S. Phase I study of ipatasertib as a single agent and in combination with abiraterone plus prednisolone in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2019, 84, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Roubaud, G.; Attard, G.; Boegemann, M.; Olmos, D.; Trevisan, M.; Antoni, L.; Pascoe, K.; Capone, C.; Van Sande, S.; Hashim, M.; et al. Adjustment for imbalances in baseline characteristics in the MAGNITUDE phase 3 study confirms the clinical benefit of niraparib in combination with abiraterone acetate plus prednisone in patients with metastatic prostate cancer. Eur. J. Cancer. 2024, 209, 114183. [Google Scholar] [CrossRef]
- Chi, K.N.; Rathkopf, D.; Smith, M.R.; Efstathiou, E.; Attard, G.; Olmos, D.; Lee, J.Y.; Small, E.J.; Pereira de Santana Gomes, A.J.; Roubaud, G.; et al. Niraparib and Abiraterone Acetate for Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2023, 41, 3339–3351. [Google Scholar] [CrossRef]
- Chi, K.N.; Sandhu, S.; Smith, M.R.; Attard, G.; Saad, M.; Olmos, D.; Castro, E.; Roubaud, G.; Pereira de Santana Gomes, A.J.; Small, E.J.; et al. Niraparib plus abiraterone acetate with prednisone in patients with metastatic castration-resistant prostate cancer and homologous recombination repair gene alterations: Second interim analysis of the randomized phase III MAGNITUDE trial. Ann. Oncol. 2023, 34, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhong, D.; Yu, C.; Cai, D.; Wei, Q.; Yang, M.; Li, T.; Zhu, Q.; Ye, L.; Wei, Y.; et al. Low-dose abiraterone plus Olaparib as a late-line treatment for mCRPC patients without BRCA1/2 mutations: A multicenter retrospective pilot study. Sci. Rep. 2024, 14, 19895. [Google Scholar] [CrossRef] [PubMed]
- Brenton, J.D.; Carey, L.A.; Ahmed, A.A.; Caldas, C. Molecular classification and molecular forecasting of breast cancer: Ready for clinical application? J. Clin. Oncol. 2005, 23, 7350–7360. [Google Scholar] [CrossRef]
- Shah, P.D.; Gucalp, A.; Traina, T.A. The Role of the Androgen Receptor in Triple-Negative Breast Cancer. Women’s Health 2013, 9, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Barton, V.N.; D’Amato, N.C.; Gordon, M.A.; Christenson, J.L.; Elias, A.E.; Richer, J.K. Androgen Receptor Biology in Triple Negative Breast Cancer: A Case for Classification as AR+ or Quadruple Negative Disease. Horm. Canc. 2015, 6, 206–213. [Google Scholar] [CrossRef]
- Fioretti, F.M.; Sita-Lumsden, A.; Bevan, C.L.; Brooke, G.N. Revising the role of the androgen receptor in breast cancer. J. Mol. Endocrinol. 2014, 52, R257–R265. [Google Scholar] [CrossRef]
- Basu, B.; Ang, J.E.; Crawley, D.; Folkerd, D.; Blanco Codesido, M.S.; Moran, K.; Wan, S.; Dobbs, N.; Raynaud, F.; Johnston, S.R.D.; et al. Phase I study of abiraterone acetate (AA) in patients (pts) with estrogen receptor– (ER) or androgen receptor (AR) –positive advanced breast carcinoma resistant to standard endocrine therapies. J. Clin. Oncol. 2011, 29, 2525. [Google Scholar] [CrossRef]
- Bonnefoi, H.; Grellety, T.; Tredan, O.; Saghatchian, M.; Dalenc, F.; Mailliez, A.; L’Haridon, T.; Cottu, P.; Abadie-Lacourtoisie, S.; You, B.; et al. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12-1). Ann. Oncol. 2016, 27, 812–818. [Google Scholar] [CrossRef]
- Grellety, T.; MacGrogan, G.; Chakiba, C.; Kind, M.; Bonnefoi, H. Long-Term Complete Response of an Androgen Receptor–Positive Triple-Negative Metastatic Breast Cancer to Abiraterone Acetate. J. Clin. Oncol. Precis. Oncol. 2018, 2. [Google Scholar] [CrossRef]
- Grellety, T.; Callens, C.; Richard, E.; Briaux, A.; Vélasco, V.; Pulido, M.; Gonçalves, A.; Gestraud, P.; MacGrogan, G.; Bonnefoi, H.; et al. Enhancing Abiraterone Acetate Efficacy in Androgen Receptor-positive Triple-negative Breast Cancer: Chk1 as a Potential Target. Clin. Cancer Res. 2019, 25, 856–867. [Google Scholar] [CrossRef]
- Rhanine, Y.; Bonnefoi, H.; Goncalves, A.; Debled, M.; Le Moulec, S.; Bonichon, N.; Macgrogan, G.; Arnedos, M.; Dubroca-Dehez, B.; Grellety, T. Efficacy of antiandrogens in androgen receptor-positive triple-negative metastatic breast cancer: Real-life data. Breast 2024, 73, 103667. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.H.M.; Macpherson, I.; Rea, D.; Spicer, J.; Bowman, A.; Jones, A.; Dowsett, M.; Johnston, S.R.D.; Dobbs, J.N.; de Bono, S. 325PD - Phase I/II Trial of Abiraterone Acetate (AA) in Estrogen Receptor (ERα) or Androgen Receptor (AR) Positive Metastatic Breast Cancer (MBC). Ann. Oncol. 2012, 23, ix119. [Google Scholar] [CrossRef]
- Capper, C.P.; Larios, J.M.; Sikora, M.J.; Johnson, M.D.; Rae, J.M. The CYP17A1 inhibitor abiraterone exhibits estrogen receptor agonist activity in breast cancer. Breast Cancer Res. Treat. 2016, 157, 23–30. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Campone, M.; Brain, E.; Neven, P.; Hayes, D.; Bondarenko, I.; Griffin, T.W.; Martin, J.; De Porre, P.; Kheoh, T.; et al. Abiraterone acetate, exemestane or the combination in postmenopausal patients with estrogen receptor-positive metastatic breast cancer. Ann. Oncol. 2016, 27, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; O’Shaughnessy, J.; Hayes, D.; Campone, M.; Bondarenko, I.; Zbarskaya, I.; Brain, E.; Stenina, M.; Ivanova, O.; Graas, M.P.; et al. Biomarker Associations with Efficacy of Abiraterone Acetate and Exemestane in Postmenopausal Patients with Estrogen Receptor-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 6002–6009. [Google Scholar] [CrossRef]
- Simigdala, N.; Pancholi, S.; Ribas, R.; Folkerd, E.; Liccardi, G.; Nikitorowicz-Buniak, J.; Johnston, S.R.; Dowsett, M.; Martin, L.-A. Abiraterone shows alternate activity in models of endocrine resistant and sensitive disease. Br. J. Cancer 2018, 119, 313–322. [Google Scholar] [CrossRef]
- Kwilas, A.R.; Ardiani, A.; Gameiro, S.R.; Richards, J.; Hall, A.B.; Hodge, J.W. Androgen deprivation therapy sensitizes triple negative breast cancer cells to immune-mediated lysis through androgen receptor independent modulation of osteoprotegerin. Oncotarget 2016, 7, 23498–23511. [Google Scholar] [CrossRef]
- van Doorn, H.C.; Burger, C.W.; van der Valk, P.; Bonfrèr, H.M. Oestrogen, progesterone, and androgen receptors in ovarian neoplasia: Correlation between immunohistochemical and biochemical receptor analyses. J. Clin. Pathol. 2000, 53, 201–205. [Google Scholar] [CrossRef]
- Papadatos-Pastos, D.; Dedes, K.J.; de Bono, J.S.; Kaye, S.B. Revisiting the Role of Antiandrogen Strategies in Ovarian Cancer. Oncologist 2011, 16, 1413–1421. [Google Scholar] [CrossRef]
- De Lazzari, G.; Opattova, A.; Arena, S. Novel frontiers in urogenital cancers: From molecular bases to preclinical models to tailor personalized treatments in ovarian and prostate cancer patients. J. Exp. Clin. Cancer Res. 2024, 43, 146. [Google Scholar] [CrossRef]
- Banerjee, S.; Tovey, H.; Bowen, R.; Folkerd, E.; Kilburn, L.; McLachlan, J.; Hall, M.; Tunariu, N.; Attygalle, A.; Da Silveira Nogueira Lima, J.P.; et al. Abiraterone in patients with recurrent epithelial ovarian cancer: Principal results of the phase II Cancer of the Ovary Abiraterone (CORAL) trial (CRUK—A16037). Therap. Adv. Med. Oncol. 2020, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kinaan, M.; Hamidi, O.; Yau, H.; Courtney, K.D.; Eraslan, A.; Simon, K. Congenital Adrenal Hyperplasia Causing Poor Response to Androgen Deprivation Therapy in Prostate Cancer. J. Endocrine Soc. 2021, 5, bvaa158. [Google Scholar] [CrossRef] [PubMed]
- Turcu, A.F.; Auchus, R.J. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol. Metab. Clin. North Am. 2015, 44, 275–296. [Google Scholar] [CrossRef] [PubMed]
- Prete, A.; Auchus, R.J.; Ross, R.J. Clinical advances in the pharmacotherapy of congenital adrenal hyperplasia. Eur. J. Endocrinol. 2022, 186, R1–R14. [Google Scholar] [CrossRef]
- Nordenström, A.; Lajic, S.; Falhammar, H. Clinical outcomes in 21-hydroxylase deficiency. Curr. Opin. Endocrinol. Diabetes Obes. 2021, 28, 318–324. [Google Scholar] [CrossRef]
- Auchus, R.J.; Buschur, E.O.; Chang, A.Y.; Hammer, G.D.; Ramm, C.; Madrigal, D.; Wang, G.; Gonzalez, M.; Xu, X.S.; Smit, J.W.; et al. Abiraterone acetate to lower androgens in women with classic 21-hydroxylase deficiency. J. Clin. Endocr. Metab. 2014, 99, 2763–2770. [Google Scholar] [CrossRef]
- Melau, C.; Riis, M.L.; Nielsen, J.E.; Perlman, S.; Lundvall, L.; Langhoff Thuesen, L.; Hare, K.J.; Hammerum, M.S.; Mitchell, R.T.; Frederiksen, H.; et al. The effects of selected inhibitors on human fetal adrenal steroidogenesis differs under basal and ACTH-stimulated conditions. BMC Med. 2021, 19, 204. [Google Scholar] [CrossRef]
- Malikova, J.; Brixius-Anderko, S.; Udhane, S.S.; Parween, S.; Dick, B.; Bernhardt, R.; Pandey, A.V. CYP17A1 inhibitor abiraterone, an anti-prostate cancer drug, also inhibits the 21-hydroxylase activity of CYP21A2. J. Steroid Biochem. Mol. Bio. 2017, 174, 192–200. [Google Scholar] [CrossRef]
- Sanders, K.; Wit, W.L.; Mol, J.A.; Kurlbaum, M.; Kendl, S.; Kroiss, M.; Kooistra, H.S.; Galac, S. Abiraterone acetate for Cushing syndrome: Study in a canine primary adrenocortical cell culture model. Endocrinology 2018, 159, 3689–3698. [Google Scholar] [CrossRef]
- Rijk, J.C.W.; Peijnenburg, A.A.C.M.; Blokland, M.H.; Lommen, A.; Hoogenboom, R.L.A.P.; Bovee, T.F.H. Screening for modulatory effects on steroidogenesis using the human H295R adrenocortical cell line: A metabolomics approach. Chem. Res. Toxicol. 2012, 25, 1720–1731. [Google Scholar] [CrossRef]
- Wright, C.; O’Day, P.; Alyamani, M.; Sharifi, N.; Auchus, R.J. Abiraterone acetate treatment lowers 11-oxygenated androgens. Eur. J. Endocrinol. 2020, 182, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Tritos, N.A.; Biller, B.M.K. Current management of Cushing’s disease. J. Intern. Med. 2019, 286, 526–541. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Fragni, M.; Perego, P.; Vezzoli, S.; Bonini, S.A.; Tortoreto, M.; Galli, D.; Claps, M.; Tiberio, G.A.; Terzolo, M.; et al. Antisecretive and Antitumor Activity of Abiraterone Acetate in Human Adrenocortical Cancer: A Preclinical Study. J. Clin. Endocrinol. Metab. 2016, 101, 4594–4602. [Google Scholar] [CrossRef] [PubMed]
- Claps, M.; Lazzari, B.; Grisanti, S.; Ferrari, V.; Terzolo, M.; Sigala, S.; Vezzoli, S.; Memo, M.; Castellano, M.; Berruti, A. Management of Severe Cushing Syndrome Induced by Adrenocortical Carcinoma with Abiraterone Acetate: A Case Report. AACE Clin. Case Rep. 2016, 2, e337–e341. [Google Scholar] [CrossRef]
- Chacko, R.; Abdel-Razeq, N.H.; Rous, F.A.; Loutfi, R. Abiraterone acetate for treatment of ectopic Cushing syndrome caused by ACTH-producing neuroendocrine tumor: A case report. J. Gastrointest. Oncol. 2022, 13, 2626–2632. [Google Scholar] [CrossRef]
- Ruan, T.; Jiang, L.; Xu, J.; Zhou, J. Abiraterone suppresses irradiated lung cancer cells-induced angiogenic capacities of endothelial cells. J. Bioenerg. Biomembr. 2021, 53, 343–349. [Google Scholar] [CrossRef]
- Lee, G.T.; Han, C.S.; Kwon, Y.S.; Patel, R.; Modi, P.K.; Kwon, S.J.; Faiena, I.; Patel, N.; Singer, E.A.; Ahn, H.-J.; et al. Intracrine androgen biosynthesis in renal cell carcinoma. Br. J. Cancer 2017, 116, 937–943. [Google Scholar] [CrossRef]
- Francini, E.; Petrioli, R.; Fiaschi, A.I.; Laera, L.; Roviello, G. Effects of abiraterone acetate on chronic kidney disease in 2 patients with metastatic castration-resistant prostate cancer. Medicine 2014, 93, e163. [Google Scholar] [CrossRef]
- Yeoh, C.C.; Dabab, N.; Rigby, E.; Chhikara, R.; Akaev, I.; Gomez, R.S.; Fonseca, F.; Brennan, P.A.; Rahimi, S. Androgen receptor in salivary gland carcinoma: A review of an old marker as a possible new target. J. Oral Pathol. Med. 2018, 47, 691–695. [Google Scholar] [CrossRef]
- Locati, L.D.; Perrone, F.; Cortelazzi, B.; Imbimbo, M.; Bossi, P.; Potepan, P.; Civelli, E.; Rinaldi, G.; Quattrone, P.; Licitra, L.; et al. Activity of abiraterone in rechallenging two AR-expressing salivary gland adenocarcinomas, resistant to androgen-deprivation therapy. Cancer Biol. Ther. 2014, 15, 678–682. [Google Scholar] [CrossRef]
- Locati, L.D.; Cavalieri, S.; Bergamini, C.; Resteghini, C.; Colombo, E.; Calareso, G.; Mariani, L.; Quattrone, P.; Alfieri, S.; Bossi, P.; et al. MD1,5, Abiraterone Acetate in Patients with Castration-Resistant, Androgen Receptor–Expressing Salivary Gland Cancer: A Phase II Trial. J. Clin. Oncol. 2021, 39, 4061–4068. [Google Scholar] [CrossRef] [PubMed]
- Urban, D.; Rischin, D.; Angel, C.; D’Costa, I.; Solomon, B. Abiraterone in metastatic salivary duct carcinoma. J. Natl. Compr. Canc. Netw. 2015, 13, 288–290. [Google Scholar] [CrossRef]
- Viscuse, P.V.; Price, K.A.; Garcia, J.J.; Schembri-Wismayer, D.J.; Chintakuntlawar, A.V. First Line Androgen Deprivation Therapy vs. Chemotherapy for Patients with Androgen Receptor Positive Recurrent or Metastatic Salivary Gland Carcinoma—A Retrospective Study. Front. Oncol. 2019, 9, 701. [Google Scholar] [CrossRef]
- Montopoli, M.; Zumerle, S.; Vettor, R.; Rugge, M.; Zorzi, M.; Catapano, C.V.; Carbone, G.M.; Cavalli, A.; Pagano, F.; Ragazzi, E.; et al. Androgen-deprivation therapies for prostate cancer and risk of infection by SARS-CoV-2: A population-based study (N = 4532). Ann. Oncol. 2020, 31, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.G.; Zhong, X.; Liaw, B.; Tremblay, D.; Tsao, C.-K.; Galsky, M.D.; Oh, W.K. Does androgen deprivation therapy protect against severe complications from COVID-19? Ann. Oncol. 2020, 31, 1419–1420. [Google Scholar] [CrossRef]
- Welén, K.; Rosendal, E.; Gisslén, M.; Lenman, A.; Freyhult, E.; Fonseca-Rodríguez, O.; Bremell, D.; Stranne, J.; Östholm Balkhed, Å.; Niward, K.; et al. A Phase 2 Trial of the Effect of Antiandrogen Therapy on COVID-19 Outcome: No Evidence of Benefit, Supported by Epidemiology and In Vitro Data. Eur. Urol. 2022, 81, 285–293. [Google Scholar] [CrossRef]
- Davidsson, S.; Messing Eriksson, A.; Udumyan, R.; Swanholm, P.; Lewin Lundh, M.; Widing, C.; Lindlöf, C.; Fridfelt, J.; Andersson, S.-O.; Fall, K. Androgen deprivation therapy in men with prostate cancer is not associated with COVID-19 infection. Prostate 2023, 83, 555–562. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, J.F.W.; Chik, K.K.H.; Chan, C.C.Y.; Tsang, J.O.L.; Liang, R.; Cao, J.; Tang, K.; Chen, L.-L.; Wen, K.; et al. Discovery of the FDA-approved drugs bexarotene, cetilistat, diiodohydroxyquinoline, and abiraterone as potential COVID-19 treatments with a robust two-tier screening system. Pharm. Res. 2020, 159, 104960. [Google Scholar] [CrossRef] [PubMed]
- Shahabadi, N.; Zendehcheshm, S.; Mahdavi, M.; Khademi, F. Inhibitory activity of FDA-approved drugs cetilistat, abiraterone, diiodohydroxyquinoline, bexarotene, remdesivir, and hydroxychloroquine on COVID-19 main protease and human ACE2 receptor: A comparative in silico approach. Inform. Med. Unlocked 2021, 26, 100745. [Google Scholar] [CrossRef]
- Kim, J.; Young Hwang, S.; Kim, D.; Kim, M.; Baek, K.; Kang, M.; An, S.; Gong, J.; Park, S.; Kandeel, M.; et al. Abiraterone Acetate Attenuates SARS-CoV-2 Replication by Interfering with the Structural Nucleocapsid Protein. Biomol. Ther. 2022, 30, 427–434. [Google Scholar] [CrossRef]
- Shahabadi, N.; Zendehcheshm, S.; Mahdavi, M.; Khademi, F. Repurposing FDA-approved drugs cetilistat, abiraterone, diiodohydroxyquinoline, bexarotene, and remdesivir as potential inhibitors against RNA dependent RNA polymerase of SARS-CoV-2: A comparative in silico perspective. Inform. Med. Unlocked 2023, 36, 101147. [Google Scholar] [CrossRef] [PubMed]
- Liontos, M.; Terpos, E.; Kunadis, E.; Zagouri, F.; Briasoulis, A.; Skafida, E.; Fiste, O.; Markellos, C.; Andrikopoulou, A.; Gumeni, S.; et al. Treatment with abiraterone or enzalutamide does not impair immunological response to COVID-19 vaccination in prostate cancer patients. Prostate Cancer Prostatic Dis. 2022, 25, 117–118. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Prescribing Information: Zytiga (abiraterone acetate); US Food Drug Administration: Silver Spring, MD, USA, 2011.
- Li, Z.; Bishop, A.; Alyamani, M.; Garcia, J.A.; Dreicer, R.; Bunch, D.; Liu, J.; Upadhyay, S.K.; Auchus, R.J.; Sharifi, N. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature 2015, 523, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Hu, Q. CYP17 inhibitors—Abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat. Rev. Urol. 2014, 11, 32–42. [Google Scholar] [CrossRef]
- Logothetis, C.J.; Basch, E.; Molina, A.; Fizazi, K.; North, S.A.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Mainwaring, P.N.; Sternberg, C.N.; et al. Effect of abiraterone acetate and prednisone compared with placebo and prednisone on pain control and skeletal-related events in patients with metastatic castration-resistant prostate cancer: Exploratory analysis of data from the COU-AA-301 randomised trial. Lancet Oncol. 2012, 13, 1210–1217. [Google Scholar] [CrossRef]
- Chan, J.S.K.; Lee, Y.H.A.; Leung, C.H.; Leung, D.K.W.; Dee, E.C.; Ng, K.; Tse, G.; Ng, C.F. Associations between glucocorticoid use and major adverse cardiovascular events in patients with prostate cancer receiving antiandrogen: A retrospective cohort study. Prostate Cancer Prostatic Dis. 2024. [Google Scholar] [CrossRef]
- Attard, G.; Reid, A.M.H.; A’Hern, R.; Parker, C.; Babu Oommen, N.; Folkerd, E.; Messiou, C.; Molife, R.L.; Maier, G.; Thompson, E.; et al. Selective Inhibition of CYP17 With Abiraterone Acetate Is Highly Active in the Treatment of Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2009, 27, 3742–3748. [Google Scholar] [CrossRef]
- Attard, G.; Reid, A.M.H.; Auchus, R.J.; Hughes, B.A.; Mulick Cassidy, A.; Thompson, E.; Babu Oommen, N.; Folkerd, E.; Dowsett, M.; Arlt, W.; et al. Clinical and Biochemical Consequences of CYP17A1 Inhibition with Abiraterone Given with and without Exogenous Glucocorticoids in Castrate Men with Advanced Prostate Cancer. J. Clin. Endocrinol. Metab. 2012, 97, 507–516. [Google Scholar] [CrossRef]
- Bird, I.M.; Abbott, D.H. The hunt for a selective 17,20 lyase inhibitor; learning lessons from nature. J. Steroid Biochem. Mol. Biol. 2016, 163, 136–146. [Google Scholar] [CrossRef]
- Njar, V.C.O.; Brodie, A.M.H. Discovery and Development of Galeterone (TOK-001 or VN/124-1) for the Treatment of All Stages of Prostate Cancer. J. Med. Chem. 2015, 58, 2077–2087. [Google Scholar] [CrossRef]
- Schayowitz, A.; Sabnis, G.; Njar, V.C.O.; Brodie, A.M.H. Synergistic effect of a novel antiandrogen, VN/124-1, and signal transduction inhibitors in prostate cancer progression to hormone independence in vitro. Mol. Cancer Ther. 2008, 7, 121–132. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov ARMOR1: Study of TOK-001 to Treat Castration Resistant Prostate Cancer (ARMOR1). Available online: https://clinicaltrials.gov/study/NCT00959959 (accessed on 10 September 2024).
- Taplin, M.E.; Chu, F.; Morrison, J.P.; Pili, R.; Rettig, M.B.; Stephenson, J.; Vogelzang, N.J.; Montgomery, R.B. Abstract CT-07: ARMOR1: Safety of galeterone (TOK-001) in a Phase 1 clinical trial in chemotherapy naive patients with castration resistant prostate cancer (CRPC). Cancer Res. 2012, 72, CT-07. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A 2 Part, Phase 2 Trial of Galeterone in the Treatment of Castration Resistant Prostate Cancer (ARMOR2). Available online: https://clinicaltrials.gov/study/NCT01709734?term=%20TOK-001&rank=2 (accessed on 10 September 2024).
- Montgomery, B.; Eisenberger, M.A.; Rettig, M.B.; Chu, F.; Pili, R.; Stephenson, J.J.; Vogelzang, N.J.; Koletsky, A.J.; Nordquist, L.T.; Edenfield, W.J.; et al. Androgen Receptor Modulation Optimized for Response (ARMOR) Phase I and II Studies: Galeterone for the Treatment of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.R.; Werner, L.; Fiorillo, M.; Roberts, J.; Heath, E.I.; Bubley, G.J.; Montgomery, R.B.; Taplin, M.E. Efficacy of Therapies After Galeterone in Patients with Castration-resistant Prostate Cancer. Clin. Genitourin. Cancer 2017, 15, 463–471. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study of Galeterone Compared to Enzalutamide in Men Expressing Androgen Receptor Splice Variant-7 mRNA (AR-V7) Metastatic CRPC (ARMOR3-SV). Available online: https://clinicaltrials.gov/study/NCT02438007?term=%20TOK-001&rank=3 (accessed on 10 September 2024).
- Lorente, D.; Fizazi, K.; Sweeney, C.; de Bono, J.S. Optimal Treatment Sequence for Metastatic Castration-resistant Prostate Cancer. Eur. Urol. Focus 2016, 2, 488–498. [Google Scholar] [CrossRef]
- Taplin, M.E.; Antonarakis, E.S.; Ferrante, K.J.; Horgan, K.; Blumenstein, B.; Saad, F.; Luo, J.; de Bono, J.S. Androgen Receptor Modulation Optimized for Response—Splice Variant: A Phase 3, Randomized Trial of Galeterone Versus Enzalutamide in Androgen Receptor Splice Variant-7–expressing Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2019, 76, 843–851. [Google Scholar] [CrossRef]
- Purushottamachar, P.R.; Ramalingam, S.; Njar, V.C.O. Development of benzimidazole compounds for cancer therapy. In Chemistry Applications Benzimidazole Derivatives; IntechOpen: London, UK, 2019; pp. 1–15. [Google Scholar]
- Udhane, S.S.; Dick, B.; Hu, Q.; Hartmann, R.W.; Pandey, A.V. Specificity of anti-prostate cancer CYP17A1 inhibitors on androgen biosynthesis. Biochem. Biophys. Res. Commun. 2016, 477, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Masamrekh, R.A.; Filippova, T.A.; Sherbakov, K.A.; Veselovsky, A.V.; Shumyantseva, V.V.; Kuzikov, A.V. Interactions of galeterone and its 3-keto-Δ4 metabolite (D4G) with one of the key enzymes of corticosteroid biosynthesis—Steroid 21-monooxygenase (CYP21A2). Fundam. Clin. Pharmacol. 2021, 35, 423–431. [Google Scholar] [CrossRef]
- McClurg, U.L.; Azizyan, M.; Dransfield, D.T.; Namdev, N.; Chit, N.C.T.H.; Nakjang, S.; Robson, C.N. The novel anti-androgen candidate galeterone targets deubiquitinating enzymes, USP12 and USP46, to control prostate cancer growth and survival. Oncotarget 2018, 9, 24992–25007. [Google Scholar] [CrossRef]
- Vasaitis, T.; Belosay, A.; Schayowitz, A.; Khandelwal, A.; Chopra, P.; Gediya, L.K.; Guo, Z.; Fang, H.B.; Njar, V.C.O.; Brodie, A.M.H. Androgen receptor inactivation contributes to antitumor efficacy of 17α-hydroxylase/17,20-lyase inhibitor 3β-hydroxy-17-(1H-benzimidazole-1-yl)androsta-5,16-diene in prostate cancer. Mol. Cancer Ther. 2008, 7, 2348–2357. [Google Scholar] [CrossRef]
- Norris, J.D.; Ellison, S.J.; Baker, J.G.; Stagg, D.B.; Wardell, S.E.; Park, S.; Alley, H.M.; Baldi, R.M.; Yllanes, A.; Andreano, K.J.; et al. Androgen receptor antagonism drives cytochrome P450 17A1 inhibitor efficacy in prostate cancer. J. Clin. Investig. 2017, 127, 2326–2338. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Cai, C.; Gao, S.; Simon, N.I.; Shen, H.C.; Balk, S.P. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clin. Cancer Res. 2014, 20, 4075–4085. [Google Scholar] [CrossRef]
- Kwegyir-Afful, A.K.; Ramalingam, S.; Purushottamachar, P.; Ramamurthy, V.P.; Njar, V.C. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget 2015, 6, 27440–27460. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhong, W.; Cao, R. Phosphorylation of the mRNA cap-binding protein eIF4E and cancer. Cell. Signal. 2020, 73, 109689. [Google Scholar] [CrossRef]
- Kwegyir-Afful, A.K.; Bruno, R.D.; Purushottamachar, P.; Murigi, F.N.; Njar, V.C.O. Galeterone and VNPT55 disrupt Mnk-eIF4E to inhibit prostate cancer cell migration and invasion. FEBS J. 2016, 283, 3898–3918. [Google Scholar] [CrossRef] [PubMed]
- Kwegyir-Afful, A.K.; Murigi, F.N.; Purushottamachar, P.; Ramamurthy, V.P.; Martin, M.S.; Njar, V.C.O. Galeterone and its analogs inhibit Mnk-eIF4E axis, synergize with gemcitabine, impede pancreatic cancer cell migration, invasion and proliferation and inhibit tumor growth in mice. Oncotarget 2017, 8, 52381–52402. [Google Scholar] [CrossRef]
- Xu, Y.; Liao, S.; Wang, L.; Wang, Y.; Wei, W.; Su, K.; Tu, Y.; Zhu, S. Galeterone sensitizes breast cancer to chemotherapy via targeting MNK/eIF4E and β-catenin. Cancer Chemother. Pharmacol. 2021, 87, 85–93. [Google Scholar] [CrossRef]
- Claessens, F.; Moris, L. The influence of steroid metabolism on CYP17A1 inhibitor activity. Nat. Rev. Urol. 2017, 14, 590–592. [Google Scholar] [CrossRef]
- Alyamani, M.; Li, Z.; Berk, M.; Li, J.; Tang, J.; Upadhyay, S.; Auchus, R.J.; Sharifi, N. Steroidogenic Metabolism of Galeterone Reveals a Diversity of Biochemical Activities. Cell Chem. Biol. 2017, 24, 825–832. [Google Scholar] [CrossRef]
- Taplin, M.E.; Montgomery, R.B. ARMOR2: Galeterone in progressive CRPC patients who have failed oral therapy. J. Clin. Oncol. 2014, 32, 71. [Google Scholar] [CrossRef]
- Thompson, S.A.; Gala, U.; Davis, D.A.; Kucera, S.; Miller, D.; Williams, R.O., III. Can the Oral Bioavailability of the Discontinued Prostate Cancer Drug Galeterone Be Improved by Processing Method? KinetiSol® Outperforms Spray Drying in a Head-to-head Comparison. AAPS PharmSciTech 2023, 24, 137. [Google Scholar] [CrossRef] [PubMed]
- Zipinotti dos Santos, D.; Elbaz, M.; Branchard, E.; Schormann, W.; Brown, C.E.; Meek, A.R.; Njar, V.C.O.; Hamilton, R.J.; Reed, M.A.; Andrews, D.W.; et al. Sterol-like drugs potentiate statin-triggered prostate cancer cell death by inhibiting SREBP2 nuclear translocation. Biomed. Pharmacother. 2024, 177, 116934. [Google Scholar] [CrossRef]
- Bruno, R.D.; Vasaitis, T.S.; Gediya, L.K.; Purushottamachar, P.; Godbole, A.M.; Ates-Alagoz, Z.; Brodie, A.M.H.; Njar, V.C.O. Synthesis and biological evaluations of putative metabolically stable analogs of VN/124-1 (TOK-001): Head to head anti-tumor efficacy evaluation of VN/124-1 (TOK-001) and abiraterone in LAPC-4 human prostate cancer xenograft model. Steroids 2011, 76, 1268–1279. [Google Scholar] [CrossRef]
- Soifer, H.S.; Souleimanian, N.; Wu, S.; Voskresenskiy, A.M.; Kisaayak Collak, F.; Cinar, B.; Stein, C.A. Direct Regulation of Androgen Receptor Activity by Potent CYP17 Inhibitors in Prostate Cancer Cells. J. Biol. Chem. 2012, 287, 3777–3787. [Google Scholar] [CrossRef]
- Petrunak, E.M.; Rogers, S.A.; Aubé, J.; Scott, E.E. Structure and Function of Cytochrome P450 17A1 Inhibitors. Drug Metab. Dispos. 2017, 45, 635–645. [Google Scholar] [CrossRef]
- Yan Yip, C.K.; Bansal, S.; Ying Wong, S.; Jiang Lau, A. Inhibition of DHEA Sulfonation by Galeterone and Abiraterone. Drug Metab. Dispos. 2018, 46, 470–482. [Google Scholar] [CrossRef]
- Masamrekh, R.; Kuzikov, A.; Veselovsky, A.; Toropygin, I.; Shkel, T.; Strushkevich, N.; Gilep, A.; Usanov, S.; Archakov, A.; Shumyantseva, V. Interaction of 17α-hydroxylase, 17(20)-lyase (CYP17A1) inhibitors—Abiraterone and galeterone—With human sterol 14α-demethylase (CYP51A1). J. Inorg. Biochem. 2018, 186, 24–33. [Google Scholar] [CrossRef]
- Moreira, V.M.; Vasaitis, T.S.; Njar, V.C.; Salvador, J.A.R. Synthesis and evaluation of novel 17-indazole androstene derivatives designed as CYP17 inhibitors. Steroids 2007, 72, 939–948. [Google Scholar] [CrossRef]
- Salvador, J.A.R.; Pinto, R.M.A.; Silvestre, S.M. Steroidal 5α-reductase and 17α-hydroxylase/17,20-lyase (CYP17) inhibitors useful in the treatment of prostatic diseases. J. Steroid Biochem. Mol. Biol. 2013, 137, 199–222. [Google Scholar] [CrossRef]
- Purushottamachar, P.; Godbole, A.M.; Gediya, L.K.; Martin, M.S.; Vasaitis, T.S.; Kwegyir-Afful, A.K.; Ramalingam, S.; Ates-Alagoz, Z.; Njar, V.C.O. Systematic Structure Modifications of Multitarget Prostate Cancer Drug Candidate Galeterone To Produce Novel Androgen Receptor Down-Regulating Agents as an Approach to Treatment of Advanced Prostate Cancer. J. Med. Chem. 2013, 56, 4880–4898. [Google Scholar] [CrossRef]
- Purushottamachar, P.; Kwegyir-Afful, A.K.; Martin, M.S.; Ramamurthy, V.P.; Ramalingam, S.; Njar, V.C.O. Identification of Novel Steroidal Androgen Receptor Degrading Agents Inspired by Galeterone 3β-Imidazole Carbamate. ACS Med. Chem. Lett. 2016, 7, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Kwegyir-Afful, A.K.; Ramalingam, S.; Ramamurthy, V.P.; Purushottamachar, P.; Murigi, F.N.; Vasaitis, T.S.; Huang, W.; Kane, M.A.; Zhang, Y.; Ambulos, N.; et al. Galeterone and The Next Generation Galeterone Analogs, VNPP414 and VNPP433-3β Exert Potent Therapeutic Effects in Castration-/Drug-Resistant Prostate Cancer Preclinical Models In Vitro and In Vivo. Cancers 2019, 11, 1637. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Thankan, R.S.; Purushottamachar, P.; Huang, W.; Kane, M.A.; Zhang, Y.; Ambulos, N.; Weber, D.J.; Njar, V.C.O. Transcriptome profiling reveals that VNPP433-3β, the lead next-generation galeterone analog inhibits prostate cancer stem cells by downregulating epithelial-mesenchymal transition and stem cell markers. Mol. Carcinog. 2022, 61, 643–654. [Google Scholar] [CrossRef]
- Thomas, E.; Thankan, R.S.; Purushottamachar, P.; Huang, W.; Kane, M.A.; Zhang, Y.; Ambulos, N.P.; Weber, D.J.; Njar, V.C.O. Novel AR/AR-V7 and Mnk1/2 Degrader, VNPP433-3β: Molecular Mechanisms of Action and Efficacy in AR-Overexpressing Castration Resistant Prostate Cancer In Vitro and In Vivo Models. Cells 2022, 11, 2699. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Thankan, R.S.; Purushottamachar, P.; Guo, J.; Parise, R.A.; Beumer, J.H.; Njar, V.C.O. Murine toxicology and pharmacokinetics of lead next generation galeterone analog, VNPP433-3β. Steroids 2023, 192, 109184. [Google Scholar] [CrossRef]
- Thomas, E.; Thankan, R.S.; Purushottamachar, P.; Weber, D.J.; Njar, V.C.O. Targeted Degradation of Androgen Receptor by VNPP433-3β in Castration-Resistant Prostate Cancer Cells Implicates Interaction with E3 Ligase MDM2 Resulting in Ubiquitin-Proteasomal Degradation. Cancers 2023, 15, 1198. [Google Scholar] [CrossRef]
- McCarty, D.J.; Huang, W.; Kane, M.A.; Purushottamachar, P.; Gediya, L.K.; Njar, V.C.O. Novel galeterone analogs act independently of AR and AR-V7 for the activation of the unfolded protein response and induction of apoptosis in the CWR22Rv1 prostate cancer cell model. Oncotarget 2017, 8, 88501–88516. [Google Scholar] [CrossRef]
- Jorda, R.; Řezníčková, E.; Kiełczewska, U.; Maj, J.; Morzycki, J.W.; Siergiejczyk, L.; Bazgier, V.; Berka, K.; Rárová, L.; Wojtkielewicz, A. Synthesis of novel galeterone derivatives and evaluation of their in vitro activity against prostate cancer cell lines. Eur. J. Med. Chem. 2019, 179, 483–492. [Google Scholar] [CrossRef]
- Latysheva, A.S.; Zolottsev, V.A.; Veselovsky, A.V.; Scherbakov, K.A.; Morozevich, G.E.; Pokrovsky, V.S.; Novikov, R.A.; Timofeev, V.P.; Tkachev, Y.V.; Misharin, A.Y. New steroidal oxazolines, benzoxazoles and benzimidazoles related to abiraterone and galeterone. Steroids 2020, 153, 108534. [Google Scholar] [CrossRef]
- Wang, A.; Luo, X.; Wang, Y.; Meng, X.; Lu, Z.; Yang, Y. Design, Synthesis, and Biological Evaluation of Androgen Receptor Degrading and Antagonizing Bifunctional Steroidal Analogs for the Treatment of Advanced Prostate Cancer. J. Med. Chem. 2022, 65, 12460–12481. [Google Scholar] [CrossRef]
- Wang, A.; Luo, X.; Meng, X.; Lu, Z.; Chen, K.; Yang, Y. Discovery of a Novel Bifunctional Steroid Analog, YXG-158, as an Androgen Receptor Degrader and CYP17A1 Inhibitor for the Treatment of Enzalutamide-Resistant Prostate Cancer. J. Med. Chem. 2023, 66, 9972–9991. [Google Scholar] [CrossRef] [PubMed]
- Thankan, R.S.; Thomas, E.; Purushottamachar, P.; Weber, D.J.; Njar, V.C.O. Salinization Dramatically Enhance the Anti-Prostate Cancer Efficacies of AR/AR-V7 and Mnk1/2 Molecular Glue Degraders, Galeterone and VNPP433-3β Which Outperform Docetaxel and Enzalutamide in CRPC CWR22Rv1 Xenograft Mouse Model. Bioorg. Chem. 2023, 139, 106700. [Google Scholar] [CrossRef] [PubMed]
- Garrido, M.; Peng, H.-M.; Yoshimoto, F.K.; Upadhyay, S.K.; Bratoeff, E.; Auchus, R.J. A-ring modified steroidal azoles retaining similar potent and slowly reversible CYP17A1 inhibition as abiraterone. J. Steroid Biochem. Mol. Biol. 2014, 143, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Masamrekh, R.; Filippova, T.; Haurychenka, Y.; Shcherbakov, K.; Veselovsky, A.; Strushkevich, N.; Shkel, T.; Gilep, A.; Usanov, S.; Shumyantseva, V.; et al. Estimation of the inhibiting impact of abiraterone D4A metabolite on human steroid 21-monooxygenase (CYP21A2). Steroids 2020, 154, 108528. [Google Scholar] [CrossRef] [PubMed]
- Fehl, C.; Vogt, C.D.; Yadav, R.; Li, K.; Scott, E.E.; Aubé, J. Structure-Based Design of Inhibitors with Improved Selectivity for Steroidogenic Cytochrome P450 17A1 over Cytochrome P450 21A2. J. Med. Chem. 2018, 61, 4946–4960. [Google Scholar] [CrossRef]
- Meng, S.; Zhu, N.; Han, D.; Li, B.; Zhang, X.; Zhang, J.; Liu, T. Synthesis and Biological Evaluation of Methoxypolyethylene-Glycol-Substituted Abiraterone Derivatives as Potential Antiprostate Cancer Agents. Mol. Pharmaceutics 2024, 21, 3186–3203. [Google Scholar] [CrossRef]
- Machulkin, A.E.; Nimenko, E.A.; Zyk, N.U.; Uspenskaia, A.A.; Smirnova, G.B.; Khan, I.I.; Pokrovsky, V.S.; Vaneev, A.N.; Timoshenko, R.V.; Mamed-Nabizade, V.V.; et al. Synthesis and Preclinical Evaluation of Small-Molecule Prostate-Specific Membrane Antigen-Targeted Abiraterone Conjugate. Molecules 2022, 27, 8795. [Google Scholar] [CrossRef]
- Szabó, N.; Ajduković, J.J.; Djurendić, E.A.; Sakač, M.N.; Ignáth, I.; Gardi, J.; Mahmoud, G.; Klisurić, O.R.; Jovanović-Šanta, S.; Penov Gaši, K.M.; et al. Determination of 17α-hydroxylase-C17,20-lyase (P45017α) enzyme activities and their inhibition by selected steroidal picolyl and picolinylidene compounds. Acta Biol. Hung. 2015, 66, 41–51. [Google Scholar] [CrossRef]
- Stevanović, M.Z.; Bekić, S.S.; Petri, E.T.; Ćelić, A.S.; Jakimov, D.S.; Marija, N.; Sakač, M.N.; Kuzminac, I.Z. Synthesis, in vitro and in silico anticancer evaluation of novel pyridin-2-yl estra-1,3,5(10)-triene derivatives. Future Med. Chem. 2024. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuzminac, I.Z.; Nikolić, A.R.; Savić, M.P.; Ajduković, J.J. Abiraterone and Galeterone, Powerful Tools Against Prostate Cancer: Present and Perspective. Pharmaceutics 2024, 16, 1401. https://doi.org/10.3390/pharmaceutics16111401
Kuzminac IZ, Nikolić AR, Savić MP, Ajduković JJ. Abiraterone and Galeterone, Powerful Tools Against Prostate Cancer: Present and Perspective. Pharmaceutics. 2024; 16(11):1401. https://doi.org/10.3390/pharmaceutics16111401
Chicago/Turabian StyleKuzminac, Ivana Z., Andrea R. Nikolić, Marina P. Savić, and Jovana J. Ajduković. 2024. "Abiraterone and Galeterone, Powerful Tools Against Prostate Cancer: Present and Perspective" Pharmaceutics 16, no. 11: 1401. https://doi.org/10.3390/pharmaceutics16111401
APA StyleKuzminac, I. Z., Nikolić, A. R., Savić, M. P., & Ajduković, J. J. (2024). Abiraterone and Galeterone, Powerful Tools Against Prostate Cancer: Present and Perspective. Pharmaceutics, 16(11), 1401. https://doi.org/10.3390/pharmaceutics16111401