

The Role of Pharmacogenetic-Based Pharmacokinetic Analysis in Precise Breast Cancer Treatment
Abstract
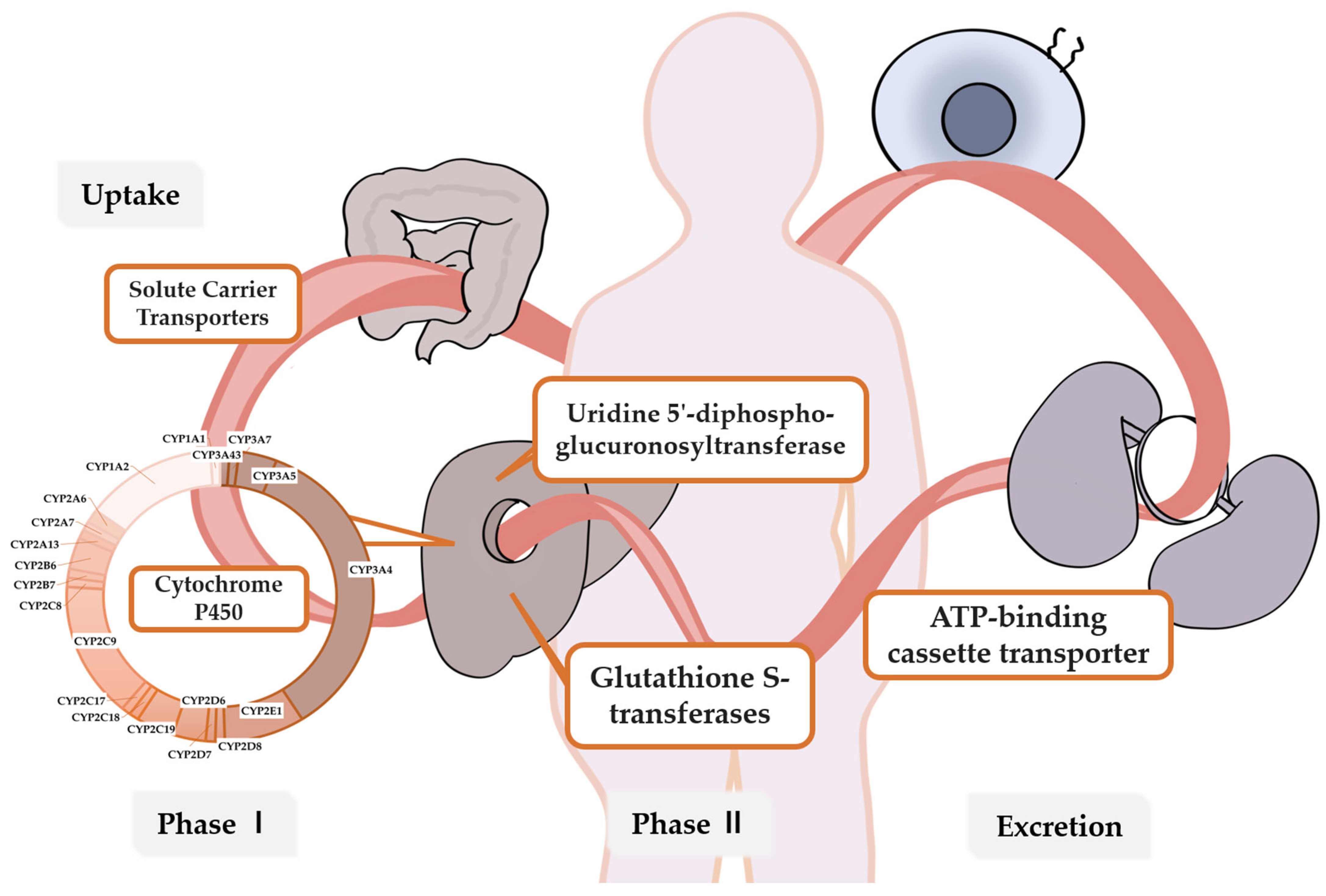
:1. Introduction
2. Metabolic Enzymes and Genes Related to Breast Cancer Drug Metabolism
3. Pharmacogenetic Variants and Breast Cancer Drugs
3.1. Endocrine Therapy
3.1.1. Tamoxifen
3.1.2. Aromatase Inhibitors (AIs)
3.1.3. Cyclin-Dependent Kinase (CDK) 4/6 Inhibitor
3.2. Chemotherapy
3.2.1. Taxanes
3.2.2. Cyclophosphamide (CTX)
3.2.3. Anthracyclines
3.3. Anti-HER2 Targeted Therapy
3.3.1. Monoclonal Antibodies
3.3.2. Tyrosine Kinase Inhibitors (TKIs)
3.3.3. Antibody-Drug Conjugate (ADC)
4. Challenges and Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Fakhri, N.; Chad, M.A.; Lahkim, M.; Houari, A.; Dehbi, H.; Belmouden, A.; El Kadmiri, N. Risk factors for breast cancer in women: An update review. Med. Oncol. 2022, 39, 197. [Google Scholar] [CrossRef] [PubMed]
- Kolak, A.; Kamińska, M.; Sygit, K.; Budny, A.; Surdyka, D.; Kukiełka-Budny, B.; Burdan, F. Primary and secondary prevention of breast cancer. Ann. Agric. Environ. Med. 2017, 24, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Bedrosian, I.; Somerfield, M.R.; Achatz, M.I.; Boughey, J.C.; Curigliano, G.; Friedman, S.; Kohlmann, W.K.; Kurian, A.W.; Laronga, C.; Lynce, F.; et al. Germline Testing in Patients with Breast Cancer: ASCO-Society of Surgical Oncology Guideline. J. Clin. Oncol. 2024, 42, 584–604. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.; Hussain, S.A.; Ghori, Q.; Naeem, N.; Fazil, A.; Giri, S.; Sathian, B.; Mainali, P.; Al Tamimi, D.M. The spectrum of genetic mutations in breast cancer. Asian Pac. J. Cancer Prev. 2015, 16, 2177–2185. [Google Scholar] [CrossRef]
- Haidar, C.E.; Crews, K.R.; Hoffman, J.M.; Relling, M.V.; Caudle, K.E. Advancing Pharmacogenomics from Single-Gene to Preemptive Testing. Annu. Rev. Genom. Hum. Genet. 2022, 23, 449–473. [Google Scholar] [CrossRef]
- Cejalvo, J.M.; Martínez de Dueñas, E.; Galván, P.; García-Recio, S.; Burgués Gasión, O.; Paré, L.; Antolín, S.; Martinello, R.; Blancas, I.; Adamo, B.; et al. Intrinsic Subtypes and Gene Expression Profiles in Primary and Metastatic Breast Cancer. Cancer Res. 2017, 77, 2213–2221. [Google Scholar] [CrossRef]
- Al Sukhun, S.; Koczwara, B.; Temin, S.; Arun, B.K. Systemic Treatment of Patients with Metastatic Breast Cancer: ASCO Resource-Stratified Guideline Q and A. JCO Glob. Oncol. 2024, 10, e2300411. [Google Scholar] [CrossRef]
- Relling, M.V.; Evans, W.E. Pharmacogenomics in the clinic. Nature 2015, 526, 343–350. [Google Scholar] [CrossRef]
- Russell, L.E.; Schwarz, U.I. Variant discovery using next-generation sequencing and its future role in pharmacogenetics. Pharmacogenomics 2020, 21, 471–486. [Google Scholar] [CrossRef]
- Kolesar, J.; Peh, S.; Thomas, L.; Baburaj, G.; Mukherjee, N.; Kantamneni, R.; Lewis, S.; Pai, A.; Udupa, K.S.; Kumar An, N.; et al. Integration of liquid biopsy and pharmacogenomics for precision therapy of EGFR mutant and resistant lung cancers. Mol. Cancer 2022, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M.; Pirmohamed, M. Precision medicine in cardiovascular therapeutics: Evaluating the role of pharmacogenetic analysis prior to drug treatment. J. Intern. Med. 2024, 295, 583–598. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. In Table of Pharmacogenetic Associations [Section 1]; US Food and Drug Administration. Available online: https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations (accessed on 16 August 2024).
- Chan, H.T.; Chin, Y.M.; Low, S.K. The Roles of Common Variation and Somatic Mutation in Cancer Pharmacogenomics. Oncol. Ther. 2019, 7, 1–32. [Google Scholar] [CrossRef]
- Filipski, K.K.; Mechanic, L.E.; Long, R.; Freedman, A.N. Pharmacogenomics in oncology care. Front. Genet. 2014, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, M.; Ingelman-Sundberg, M.; Evans, W.E. Pharmacogenomics and individualized drug therapy. Annu. Rev. Med. 2006, 57, 119–137. [Google Scholar] [CrossRef]
- Matthaei, J.; Brockmöller, J.; Tzvetkov, M.V.; Sehrt, D.; Sachse-Seeboth, C.; Hjelmborg, J.B.; Möller, S.; Halekoh, U.; Hofmann, U.; Schwab, M.; et al. Heritability of metoprolol and torsemide pharmacokinetics. Clin. Pharmacol. Ther. 2015, 98, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Tremmel, R.; Zhou, Y.; Schwab, M.; Lauschke, V.M. Structural variation of the coding and non-coding human pharmacogenome. NPJ Genom. Med. 2023, 8, 24. [Google Scholar] [CrossRef]
- Huddart, R.; Fohner, A.E.; Whirl-Carrillo, M.; Wojcik, G.L.; Gignoux, C.R.; Popejoy, A.B.; Bustamante, C.D.; Altman, R.B.; Klein, T.E. Standardized Biogeographic Grouping System for Annotating Populations in Pharmacogenetic Research. Clin. Pharmacol. Ther. 2019, 105, 1256–1262. [Google Scholar] [CrossRef]
- Caudle, K.E.; Dunnenberger, H.M.; Freimuth, R.R.; Peterson, J.F.; Burlison, J.D.; Whirl-Carrillo, M.; Scott, S.A.; Rehm, H.L.; Williams, M.S.; Klein, T.E.; et al. Standardizing terms for clinical pharmacogenetic test results: Consensus terms from the Clinical Pharmacogenetics Implementation Consortium (CPIC). Genet. Med. 2017, 19, 215–223. [Google Scholar] [CrossRef]
- Zhang, F.; Finkelstein, J. Inconsistency in race and ethnic classification in pharmacogenetics studies and its potential clinical implications. Pharmgenomics Pers. Med. 2019, 12, 107–123. [Google Scholar] [CrossRef]
- Rodriguez-Antona, C.; Ingelman-Sundberg, M. Cytochrome P450 pharmacogenetics and cancer. Oncogene 2006, 25, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Q.; Yang, Y.; Cai, C.-Y.; Teng, Q.-X.; Cui, Q.; Lin, J.; Assaraf, Y.G.; Chen, Z.-S. Multidrug resistance proteins (MRPs): Structure, function and the overcoming of cancer multidrug resistance. Drug Resist. Updates 2021, 54, 100743. [Google Scholar] [CrossRef]
- Pérez-Ramírez, C.; Cañadas-Garre, M.; Molina, M.; Cabeza Barrera, J.; Faus-Dáder, M.J. Impact of single nucleotide polymorphisms on the efficacy and toxicity of EGFR tyrosine kinase inhibitors in advanced non-small cell lung cancer patients. Mutat. Res. Rev. Mutat. Res. 2019, 781, 63–70. [Google Scholar] [CrossRef]
- Slavin, T.P.; Banks, K.C.; Chudova, D.; Oxnard, G.R.; Odegaard, J.I.; Nagy, R.J.; Tsang, K.W.K.; Neuhausen, S.L.; Gray, S.W.; Cristofanilli, M.; et al. Identification of Incidental Germline Mutations in Patients with Advanced Solid Tumors Who Underwent Cell-Free Circulating Tumor DNA Sequencing. J. Clin. Oncol. 2018, 36, Jco1800328. [Google Scholar] [CrossRef] [PubMed]
- Cronin-Fenton, D.P.; Damkier, P.; Lash, T.L. Metabolism and transport of tamoxifen in relation to its effectiveness: New perspectives on an ongoing controversy. Future Oncol. 2014, 10, 107–122. [Google Scholar] [CrossRef]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [CrossRef]
- Sanchez-Spitman, A.B.; Swen, J.J.; Dezentje, V.O.; Moes, D.; Gelderblom, H.; Guchelaar, H.J. Clinical pharmacokinetics and pharmacogenetics of tamoxifen and endoxifen. Expert Rev. Clin. Pharmacol. 2019, 12, 523–536. [Google Scholar] [CrossRef]
- Stearns, V.; Johnson, M.D.; Rae, J.M.; Morocho, A.; Novielli, A.; Bhargava, P.; Hayes, D.F.; Desta, Z.; Flockhart, D.A. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J. Natl. Cancer Inst. 2003, 95, 1758–1764. [Google Scholar] [CrossRef]
- Borges, S.; Desta, Z.; Li, L.; Skaar, T.C.; Ward, B.A.; Nguyen, A.; Jin, Y.; Storniolo, A.M.; Nikoloff, D.M.; Wu, L.; et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: Implication for optimization of breast cancer treatment. Clin. Pharmacol. Ther. 2006, 80, 61–74. [Google Scholar] [CrossRef]
- Bousman, C.A.; Stevenson, J.M.; Ramsey, L.B.; Sangkuhl, K.; Hicks, J.K.; Strawn, J.R.; Singh, A.B.; Ruaño, G.; Mueller, D.J.; Tsermpini, E.E.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6, CYP2C19, CYP2B6, SLC6A4, and HTR2A Genotypes and Serotonin Reuptake Inhibitor Antidepressants. Clin. Pharmacol. Ther. 2023, 114, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Desta, Z.; Stearns, V.; Ward, B.; Ho, H.; Lee, K.H.; Skaar, T.; Storniolo, A.M.; Li, L.; Araba, A.; et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J. Natl. Cancer Inst. 2005, 97, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Crews, K.R.; Monte, A.A.; Huddart, R.; Caudle, K.E.; Kharasch, E.D.; Gaedigk, A.; Dunnenberger, H.M.; Leeder, J.S.; Callaghan, J.T.; Samer, C.F.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for CYP2D6, OPRM1, and COMT Genotypes and Select Opioid Therapy. Clin. Pharmacol. Ther. 2021, 110, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Eklund, M.; Borgquist, S.; Hellgren, R.; Margolin, S.; Thoren, L.; Rosendahl, A.; Lång, K.; Tapia, J.; Bäcklund, M.; et al. Low-Dose Tamoxifen for Mammographic Density Reduction: A Randomized Controlled Trial. J. Clin. Oncol. 2021, 39, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Eriksson, M.; Eliasson, E.; Grassmann, F.; Bäcklund, M.; Gabrielson, M.; Hammarström, M.; Margolin, S.; Thorén, L.; Wengström, Y.; et al. CYP2D6 genotype predicts tamoxifen discontinuation and drug response: A secondary analysis of the KARISMA trial. Ann. Oncol. 2021, 32, 1286–1293. [Google Scholar] [CrossRef]
- Vita, G.; Compri, B.; Matcham, F.; Barbui, C.; Ostuzzi, G. Antidepressants for the treatment of depression in people with cancer. Cochrane Database Syst. Rev. 2023, 3, Cd011006. [Google Scholar] [CrossRef]
- Kelly, C.M.; Juurlink, D.N.; Gomes, T.; Duong-Hua, M.; Pritchard, K.I.; Austin, P.C.; Paszat, L.F. Selective serotonin reuptake inhibitors and breast cancer mortality in women receiving tamoxifen: A population based cohort study. Bmj 2010, 340, c693. [Google Scholar] [CrossRef]
- Busby, J.; Mills, K.; Zhang, S.D.; Liberante, F.G.; Cardwell, C.R. Selective serotonin reuptake inhibitor use and breast cancer survival: A population-based cohort study. Breast Cancer Res. 2018, 20, 4. [Google Scholar] [CrossRef]
- Margolin, S.; Lindh, J.D.; Thorén, L.; Xie, H.; Koukel, L.; Dahl, M.L.; Eliasson, E. CYP2D6 and adjuvant tamoxifen: Possible differences of outcome in pre- and post-menopausal patients. Pharmacogenomics 2013, 14, 613–622. [Google Scholar] [CrossRef]
- Wang, H.; Ma, X.; Zhang, B.; Zhang, Y.; Han, N.; Wei, L.; Sun, C.; Sun, S.; Zeng, X.; Guo, H.; et al. Chinese breast cancer patients with CYP2D6*10 mutant genotypes have a better prognosis with toremifene than with tamoxifen. Asia Pac. J. Clin. Oncol. 2022, 18, e148–e153. [Google Scholar] [CrossRef]
- Napoli, N.; Rastelli, A.; Ma, C.; Yarramaneni, J.; Vattikutti, S.; Moskowitz, G.; Giri, T.; Mueller, C.; Kulkarny, V.; Qualls, C.; et al. Genetic polymorphism at Val80 (rs700518) of the CYP19A1 gene is associated with aromatase inhibitor associated bone loss in women with ER + breast cancer. Bone 2013, 55, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Straume, A.H.; Knappskog, S.; Lønning, P.E. Effect of CYP19 rs6493497 and rs7176005 haplotype status on in vivo aromatase transcription, plasma and tissue estrogen levels in postmenopausal women. J. Steroid Biochem. Mol. Biol. 2012, 128, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Ghimenti, C.; Mello-Grand, M.; Regolo, L.; Zambelli, A.; Chiorino, G. Absence of the K303R estrogen receptor α mutation in breast cancer patients exhibiting different responses to aromatase inhibitor anastrozole neoadjuvant treatment. Exp. Ther. Med. 2010, 1, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Kamdem, L.K.; Liu, Y.; Stearns, V.; Kadlubar, S.A.; Ramirez, J.; Jeter, S.; Shahverdi, K.; Ward, B.A.; Ogburn, E.; Ratain, M.J.; et al. In vitro and in vivo oxidative metabolism and glucuronidation of anastrozole. Br. J. Clin. Pharmacol. 2010, 70, 854–869. [Google Scholar] [CrossRef] [PubMed]
- Turkistani, A.; Marsh, S. Pharmacogenomics of third-generation aromatase inhibitors. Expert Opin. Pharmacother. 2012, 13, 1299–1307. [Google Scholar] [CrossRef]
- Precht, J.C.; Schroth, W.; Klein, K.; Brauch, H.; Krynetskiy, E.; Schwab, M.; Mürdter, T.E. The letrozole phase 1 metabolite carbinol as a novel probe drug for UGT2B7. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 1906–1913. [Google Scholar] [CrossRef]
- Pfister, C.U.; Martoni, A.; Zamagni, C.; Lelli, G.; De Braud, F.; Souppart, C.; Duval, M.; Hornberger, U. Effect of age and single versus multiple dose pharmacokinetics of letrozole (Femara) in breast cancer patients. Biopharm. Drug Dispos. 2001, 22, 191–197. [Google Scholar] [CrossRef]
- Lønning, P.; Pfister, C.; Martoni, A.; Zamagni, C. Pharmacokinetics of third-generation aromatase inhibitors. Semin. Oncol. 2003, 30 (Suppl. S4), 23–32. [Google Scholar] [CrossRef]
- Desta, Z.; Kreutz, Y.; Nguyen, A.T.; Li, L.; Skaar, T.; Kamdem, L.K.; Henry, N.L.; Hayes, D.F.; Storniolo, A.M.; Stearns, V.; et al. Plasma letrozole concentrations in postmenopausal women with breast cancer are associated with CYP2A6 genetic variants, body mass index, and age. Clin. Pharmacol. Ther. 2011, 90, 693–700. [Google Scholar] [CrossRef]
- Nakajima, M.; Kuroiwa, Y.; Yokoi, T. Interindividual differences in nicotine metabolism and genetic polymorphisms of human CYP2A6. Drug Metab. Rev. 2002, 34, 865–877. [Google Scholar] [CrossRef]
- Abubakar, M.B.; Wei, K.; Gan, S.H. The influence of genetic polymorphisms on the efficacy and side effects of anastrozole in postmenopausal breast cancer patients. Pharm. Genom. 2014, 24, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Edavana, V.K.; Dhakal, I.B.; Williams, S.; Penney, R.; Boysen, G.; Yao-Borengasser, A.; Kadlubar, S. Potential role of UGT1A4 promoter SNPs in anastrozole pharmacogenomics. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Bojanic, K.; Kuna, L.; Bilic Curcic, I.; Wagner, J.; Smolic, R.; Kralik, K.; Kizivat, T.; Ivanac, G.; Vcev, A.; Wu, G.Y.; et al. Representation of CYP3A4, CYP3A5 and UGT1A4 Polymorphisms within Croatian Breast Cancer Patients’ Population. Int. J. Environ. Res. Public Health 2020, 17, 3692. [Google Scholar] [CrossRef]
- Tannenbaum, C.; Sheehan, N.L. Understanding and preventing drug-drug and drug-gene interactions. Expert Rev. Clin. Pharmacol. 2014, 7, 533–544. [Google Scholar] [CrossRef]
- Buzdar, A.U.; Robertson, J.F.; Eiermann, W.; Nabholtz, J.M. An overview of the pharmacology and pharmacokinetics of the newer generation aromatase inhibitors anastrozole, letrozole, and exemestane. Cancer 2002, 95, 2006–2016. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Chen, G.; Truica, C.I.; Baird, C.C.; Xia, Z.; Lazarus, P. Identification and Quantification of Novel Major Metabolites of the Steroidal Aromatase Inhibitor, Exemestane. Drug Metab. Dispos. Biol. Fate Chem. 2018, 46, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.L.; Kidwell, K.M.; Seewald, N.J.; Gersch, C.L.; Desta, Z.; Flockhart, D.A.; Storniolo, A.M.; Stearns, V.; Skaar, T.C.; Hayes, D.F.; et al. Polymorphisms in drug-metabolizing enzymes and steady-state exemestane concentration in postmenopausal patients with breast cancer. Pharm. J. 2017, 17, 521–527. [Google Scholar] [CrossRef]
- Teslenko, I.; Trudeau, J.; Luo, S.; Watson, C.J.W.; Chen, G.; Truica, C.I.; Lazarus, P. Influence of Glutathione-S-Transferase A1*B Allele on the Metabolism of the Aromatase Inhibitor, Exemestane, in Human Liver Cytosols and in Patients Treated with Exemestane. J. Pharmacol. Exp. Ther. 2022, 382, 327–334. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, Q.; Zhang, P.; Hu, X.; Li, W.; Tong, Z.; Sun, T.; Teng, Y.; Wu, X.; Ouyang, Q.; et al. Dalpiciclib or placebo plus fulvestrant in hormone receptor-positive and HER2-negative advanced breast cancer: A randomized, phase 3 trial. Nat. Med. 2021, 27, 1904–1909. [Google Scholar] [CrossRef]
- Lu, Y.S.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Cardoso, F.; Harbeck, N.; Hurvitz, S.; Chow, L.; Sohn, J.; et al. Updated Overall Survival of Ribociclib plus Endocrine Therapy versus Endocrine Therapy Alone in Pre- and Perimenopausal Patients with HR+/HER2- Advanced Breast Cancer in MONALEESA-7: A Phase III Randomized Clinical Trial. Clin. Cancer Res. 2022, 28, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M. GS01-12: MONARCH 3: Final Overall Survival Results with Abemaciclib-Based Therapy for Advanced Breast Cancer. In Proceedings of the 2023 San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 5–9 December 2023. [Google Scholar]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Tate, S.C.; Cai, S.; Ajamie, R.T.; Burke, T.; Beckmann, R.P.; Chan, E.M.; De Dios, A.; Wishart, G.N.; Gelbert, L.M.; Cronier, D.M. Semi-mechanistic pharmacokinetic/pharmacodynamic modeling of the antitumor activity of LY2835219, a new cyclin-dependent kinase 4/6 inhibitor, in mice bearing human tumor xenografts. Clin. Cancer Res. 2014, 20, 3763–3774. [Google Scholar] [CrossRef]
- O’Brien, N.; Conklin, D.; Beckmann, R.; Luo, T.; Chau, K.; Thomas, J.; Mc Nulty, A.; Marchal, C.; Kalous, O.; von Euw, E.; et al. Preclinical Activity of Abemaciclib Alone or in Combination with Antimitotic and Targeted Therapies in Breast Cancer. Mol. Cancer Ther. 2018, 17, 897–907. [Google Scholar] [CrossRef]
- Torres-Guzmán, R.; Calsina, B.; Hermoso, A.; Baquero, C.; Alvarez, B.; Amat, J.; McNulty, A.M.; Gong, X.; Boehnke, K.; Du, J.; et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 2017, 8, 69493–69507. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Abemaciclib for HR-Positive, HER2-Negative Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-abemaciclib-hr-positive-her2-negative-breast-cancer (accessed on 16 August 2024).
- Summary of Product Characteristics Palbociclib. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/palbociclib-ibrance (accessed on 16 August 2024).
- Summary of Product Characteristics Ribociclib (Kisqali). Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/ribociclib-kisqali (accessed on 16 August 2024).
- Smith, N.F.; Acharya, M.R.; Desai, N.; Figg, W.D.; Sparreboom, A. Identification of OATP1B3 as a high-affinity hepatocellular transporter of paclitaxel. Cancer Biol. Ther. 2005, 4, 815–818. [Google Scholar] [CrossRef]
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in cancer treatment: Activity, chemoresistance and its overcoming. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2021, 54, 100742. [Google Scholar] [CrossRef]
- Hjorth, C.F.; Damkier, P.; Stage, T.B.; Feddersen, S.; Hamilton-Dutoit, S.; Rørth, M.; Ejlertsen, B.; Lash, T.L.; Ahern, T.P.; Sørensen, H.T.; et al. Single-nucleotide polymorphisms and the effectiveness of taxane-based chemotherapy in premenopausal breast cancer: A population-based cohort study in Denmark. Breast Cancer Res. Treat. 2022, 194, 353–363. [Google Scholar] [CrossRef]
- Bahadur, N.; Leathart, J.B.; Mutch, E.; Steimel-Crespi, D.; Dunn, S.A.; Gilissen, R.; Houdt, J.V.; Hendrickx, J.; Mannens, G.; Bohets, H.; et al. CYP2C8 polymorphisms in Caucasians and their relationship with paclitaxel 6alpha-hydroxylase activity in human liver microsomes. Biochem. Pharmacol. 2002, 64, 1579–1589. [Google Scholar] [CrossRef]
- Hertz, D.L.; Motsinger-Reif, A.A.; Drobish, A.; Winham, S.J.; McLeod, H.L.; Carey, L.A.; Dees, E.C. CYP2C8*3 predicts benefit/risk profile in breast cancer patients receiving neoadjuvant paclitaxel. Breast Cancer Res. Treat. 2012, 134, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Ando, A.; Takamura, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Prediction of response to docetaxel by CYP3A4 mRNA expression in breast cancer tissues. Int. J. Cancer 2002, 97, 129–132. [Google Scholar] [CrossRef]
- Tran, A.; Jullien, V.; Alexandre, J.; Rey, E.; Rabillon, F.; Girre, V.; Dieras, V.; Pons, G.; Goldwasser, F.; Tréluyer, J.M. Pharmacokinetics and toxicity of docetaxel: Role of CYP3A, MDR1, and GST polymorphisms. Clin. Pharmacol. Ther. 2006, 79, 570–580. [Google Scholar] [CrossRef]
- Kim, K.P.; Ahn, J.H.; Kim, S.B.; Jung, K.H.; Yoon, D.H.; Lee, J.S.; Ahn, S.H. Prospective evaluation of the drug-metabolizing enzyme polymorphisms and toxicity profile of docetaxel in Korean patients with operable lymph node-positive breast cancer receiving adjuvant chemotherapy. Cancer Chemother. Pharmacol. 2012, 69, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.L.; Roy, S.; Motsinger-Reif, A.A.; Drobish, A.; Clark, L.S.; McLeod, H.L.; Carey, L.A.; Dees, E.C. CYP2C8*3 increases risk of neuropathy in breast cancer patients treated with paclitaxel. Ann. Oncol. 2013, 24, 1472–1478. [Google Scholar] [CrossRef]
- Bosó, V.; Herrero, M.J.; Santaballa, A.; Palomar, L.; Megias, J.E.; de la Cueva, H.; Rojas, L.; Marqués, M.R.; Poveda, J.L.; Montalar, J.; et al. SNPs and taxane toxicity in breast cancer patients. Pharmacogenomics 2014, 15, 1845–1858. [Google Scholar] [CrossRef]
- Abraham, J.E.; Guo, Q.; Dorling, L.; Tyrer, J.; Ingle, S.; Hardy, R.; Vallier, A.L.; Hiller, L.; Burns, R.; Jones, L.; et al. Replication of genetic polymorphisms reported to be associated with taxane-related sensory neuropathy in patients with early breast cancer treated with Paclitaxel. Clin. Cancer Res. 2014, 20, 2466–2475. [Google Scholar] [CrossRef]
- Kim, H.J.; Im, S.A.; Keam, B.; Ham, H.S.; Lee, K.H.; Kim, T.Y.; Kim, Y.J.; Oh, D.Y.; Kim, J.H.; Han, W.; et al. ABCB1 polymorphism as prognostic factor in breast cancer patients treated with docetaxel and doxorubicin neoadjuvant chemotherapy. Cancer Sci. 2015, 106, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.R.; Kim, J.O.; Kang, D.R.; Shin, J.Y.; Zhang, X.H.; Oh, J.E.; Park, J.Y.; Kim, K.A.; Kang, J.H. Genetic Variations of Drug Transporters Can Influence on Drug Response in Patients Treated with Docetaxel Chemotherapy. Cancer Res. Treat. 2015, 47, 509–517. [Google Scholar] [CrossRef]
- Jabir, R.S.; Ho, G.F.; Annuar, M.; Stanslas, J. Association of Allelic Interaction of Single Nucleotide Polymorphisms of Influx and Efflux Transporters Genes with Nonhematologic Adverse Events of Docetaxel in Breast Cancer Patients. Clin. Breast Cancer 2018, 18, e1173–e1179. [Google Scholar] [CrossRef]
- Gréen, H.; Söderkvist, P.; Rosenberg, P.; Mirghani, R.A.; Rymark, P.; Lundqvist, E.A.; Peterson, C. Pharmacogenetic studies of Paclitaxel in the treatment of ovarian cancer. Basic. Clin. Pharmacol. Toxicol. 2009, 104, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Mysona, D.; Dorr, K.; Ward, A.; Shaver, E.; Rungruang, B.; Ghamande, S. Pharmacogenetics as a predictor chemotherapy induced peripheral neuropathy in gynecologic cancer patients treated with Taxane-based chemotherapy. Gynecol. Oncol. 2023, 168, 114–118. [Google Scholar] [CrossRef]
- Hertz, D.L.; Roy, S.; Jack, J.; Motsinger-Reif, A.A.; Drobish, A.; Clark, L.S.; Carey, L.A.; Dees, E.C.; McLeod, H.L. Genetic heterogeneity beyond CYP2C8*3 does not explain differential sensitivity to paclitaxel-induced neuropathy. Breast Cancer Res. Treat. 2014, 145, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Demurtas, S.; La Verde, N.; Rota, S.; Casazza, G.; Montrasio, C.; Cheli, S.; Cona, M.S.; Dalu, D.; Fasola, C.; Ferrario, S.; et al. Single nucleotide polymorphisms to predict taxanes toxicities and effectiveness in cancer patients. Pharm. J. 2021, 21, 491–497. [Google Scholar] [CrossRef]
- Gudur, R.A.; Bhosale, S.J.; Gudur, A.K.; Kale, S.R.; More, A.L.; Datkhile, K.D. The Effect of CYP2C19*2 (rs4244285) and CYP17 (rs743572) SNPs on Adriamycin and Paclitaxel based Chemotherapy Outcomes in Breast Cancer Patients. Asian Pac. J. Cancer Prev. 2024, 25, 1977–1986. [Google Scholar] [CrossRef]
- Chew, S.C.; Lim, J.; Singh, O.; Chen, X.; Tan, E.H.; Lee, E.J.; Chowbay, B. Pharmacogenetic effects of regulatory nuclear receptors (PXR, CAR, RXRα and HNF4α) on docetaxel disposition in Chinese nasopharyngeal cancer patients. Eur. J. Clin. Pharmacol. 2014, 70, 155–166. [Google Scholar] [CrossRef]
- Helsby, N.A.; Burns, K.E. Molecular mechanisms of genetic variation and transcriptional regulation of CYP2C19. Front. Genet. 2012, 3, 206. [Google Scholar] [CrossRef]
- Helsby, N.A.; Yong, M.; van Kan, M.; de Zoysa, J.R.; Burns, K.E. The importance of both CYP2C19 and CYP2B6 germline variations in cyclophosphamide pharmacokinetics and clinical outcomes. Br. J. Clin. Pharmacol. 2019, 85, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Ariyoshi, N.; Miyazaki, M.; Toide, K.; Sawamura, Y.; Kamataki, T. A single nucleotide polymorphism of CYP2b6 found in Japanese enhances catalytic activity by autoactivation. Biochem. Biophys. Res. Commun. 2001, 281, 1256–1260. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Blievernicht, J.K.; Klein, K.; Saussele, T.; Schaeffeler, E.; Schwab, M.; Zanger, U.M. Aberrant splicing caused by single nucleotide polymorphism c.516G>T [Q172H], a marker of CYP2B6*6, is responsible for decreased expression and activity of CYP2B6 in liver. J. Pharmacol. Exp. Ther. 2008, 325, 284–292. [Google Scholar] [CrossRef]
- Helsby, N.A.; Tingle, M.D. Which CYP2B6 variants have functional consequences for cyclophosphamide bioactivation? Drug Metab. Dispos. Biol. Fate Chem. 2012, 40, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Haroun, F.; Al-Shaar, L.; Habib, R.H.; El-Saghir, N.; Tfayli, A.; Bazarbachi, A.; Salem, Z.; Shamseddine, A.; Taher, A.; Cascorbi, I.; et al. Effects of CYP2B6 genetic polymorphisms in patients receiving cyclophosphamide combination chemotherapy for breast cancer. Cancer Chemother. Pharmacol. 2015, 75, 207–214. [Google Scholar] [CrossRef]
- Gor, P.P.; Su, H.I.; Gray, R.J.; Gimotty, P.A.; Horn, M.; Aplenc, R.; Vaughan, W.P.; Tallman, M.S.; Rebbeck, T.R.; DeMichele, A. Cyclophosphamide-metabolizing enzyme polymorphisms and survival outcomes after adjuvant chemotherapy for node-positive breast cancer: A retrospective cohort study. Breast Cancer Res. 2010, 12, R26. [Google Scholar] [CrossRef]
- Martis, S.; Mei, H.; Vijzelaar, R.; Edelmann, L.; Desnick, R.J.; Scott, S.A. Multi-ethnic cytochrome-P450 copy number profiling: Novel pharmacogenetic alleles and mechanism of copy number variation formation. Pharm. J. 2013, 13, 558–566. [Google Scholar] [CrossRef]
- Bray, J.; Sludden, J.; Griffin, M.J.; Cole, M.; Verrill, M.; Jamieson, D.; Boddy, A.V. Influence of pharmacogenetics on response and toxicity in breast cancer patients treated with doxorubicin and cyclophosphamide. Br. J. Cancer 2010, 102, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Tulsyan, S.; Agarwal, G.; Lal, P.; Mittal, B. Significant role of CYP450 genetic variants in cyclophosphamide based breast cancer treatment outcomes: A multi-analytical strategy. Clin. Chim. Acta 2014, 434, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Kaur, R.P.; Ludhiadch, A.; Shafi, G.; Vashista, R.; Kumar, R.; Munshi, A. Association of CYP2C19*2 and ALDH1A1*1/*2 variants with disease outcome in breast cancer patients: Results of a global screening array. Eur. J. Clin. Pharmacol. 2018, 74, 1291–1298. [Google Scholar] [CrossRef]
- Mattioli, R.; Ilari, A.; Colotti, B.; Mosca, L.; Fazi, F.; Colotti, G. Doxorubicin and other anthracyclines in cancers: Activity, chemoresistance and its overcoming. Mol. Aspects Med. 2023, 93, 101205. [Google Scholar] [CrossRef]
- Mordente, A.; Meucci, E.; Silvestrini, A.; Martorana, G.E.; Giardina, B. New developments in anthracycline-induced cardiotoxicity. Curr. Med. Chem. 2009, 16, 1656–1672. [Google Scholar] [CrossRef]
- Siebel, C.; Lanvers-Kaminsky, C.; Würthwein, G.; Hempel, G.; Boos, J. Bioanalysis of doxorubicin aglycone metabolites in human plasma samples-implications for doxorubicin drug monitoring. Sci. Rep. 2020, 10, 18562. [Google Scholar] [CrossRef]
- Voon, P.J.; Yap, H.L.; Ma, C.Y.; Lu, F.; Wong, A.L.; Sapari, N.S.; Soong, R.; Soh, T.I.; Goh, B.C.; Lee, H.S.; et al. Correlation of aldo-ketoreductase (AKR) 1C3 genetic variant with doxorubicin pharmacodynamics in Asian breast cancer patients. Br. J. Clin. Pharmacol. 2013, 75, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, M.B.; Pituskin, E.; Damaraju, S.; Bies, R.R.; Vos, L.J.; Prado, C.M.; Kuzma, M.; Scarfe, A.G.; Clemons, M.; Tonkin, K.; et al. A Uridine Glucuronosyltransferase 2B7 Polymorphism Predicts Epirubicin Clearance and Outcomes in Early-Stage Breast Cancer. Clin. Breast Cancer 2016, 16, e131–e133. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.; Bardin, C.; Rigal, C.; Anthony, B.; Rousseau, R.; Dutour, A. Anti-MDR1 siRNA restores chemosensitivity in chemoresistant breast carcinoma and osteosarcoma cell lines. Anticancer. Res. 2011, 31, 2813–2820. [Google Scholar] [PubMed]
- Yin, W.; Xiang, D.; Wang, T.; Zhang, Y.; Pham, C.V.; Zhou, S.; Jiang, G.; Hou, Y.; Zhu, Y.; Han, Y.; et al. The inhibition of ABCB1/MDR1 or ABCG2/BCRP enables doxorubicin to eliminate liver cancer stem cells. Sci. Rep. 2021, 11, 10791. [Google Scholar] [CrossRef] [PubMed]
- Tulsyan, S.; Mittal, R.D.; Mittal, B. The effect of ABCB1 polymorphisms on the outcome of breast cancer treatment. Pharmgenomics Pers. Med. 2016, 9, 47–58. [Google Scholar] [CrossRef]
- Turiján-Espinoza, E.; Ruíz-Rodríguez, V.M.; Uresti-Rivera, E.E.; Martínez-Leija, E.; Zermeño-Nava, J.J.; Guel-Pañola, A.; Romano-Moreno, S.; Vargas-Morales, J.M.; Portales-Pérez, D.P. Clinical utility of ABCB1 and ABCG2 genotyping for assessing the clinical and pathological response to FAC therapy in Mexican breast cancer patients. Cancer Chemother. Pharmacol. 2021, 87, 843–853. [Google Scholar] [CrossRef]
- Madrid-Paredes, A.; Cañadas-Garre, M.; Sánchez-Pozo, A.; Expósito-Ruiz, M.; Calleja-Hernández, M. ABCB1 gene polymorphisms and response to chemotherapy in breast cancer patients: A meta-analysis. Surg. Oncol. 2017, 26, 473–482. [Google Scholar] [CrossRef]
- Ebaid, N.F.; Abdelkawy, K.S.; Shehata, M.A.; Salem, H.F.; Magdy, G.; Hussein, R.R.S.; Elbarbry, F. Effects of pharmacogenetics on pharmacokinetics and toxicity of doxorubicin in Egyptian breast cancer patients. Xenobiotica 2024, 54, 160–170. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, H.; Lei, T.; Liu, J.; Zhang, S.; Wu, N.; Sun, B.; Wang, M. Drug resistance gene expression and chemotherapy sensitivity detection in Chinese women with different molecular subtypes of breast cancer. Cancer Biol. Med. 2020, 17, 1014–1025. [Google Scholar] [CrossRef]
- Zeng, X.; Morgenstern, R.; Nyström, A.M. Nanoparticle-directed sub-cellular localization of doxorubicin and the sensitization breast cancer cells by circumventing GST-mediated drug resistance. Biomaterials 2014, 35, 1227–1239. [Google Scholar] [CrossRef]
- Tulsyan, S.; Chaturvedi, P.; Agarwal, G.; Lal, P.; Agrawal, S.; Mittal, R.D.; Mittal, B. Pharmacogenetic influence of GST polymorphisms on anthracycline-based chemotherapy responses and toxicity in breast cancer patients: A multi-analytical approach. Mol. Diagn. Ther. 2013, 17, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Sugishita, M.; Imai, T.; Kikumori, T.; Mitsuma, A.; Shimokata, T.; Shibata, T.; Morita, S.; Inada-Inoue, M.; Sawaki, M.; Hasegawa, Y.; et al. Pharmacogenetic association between GSTP1 genetic polymorphism and febrile neutropenia in Japanese patients with early breast cancer. Breast Cancer 2016, 23, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wu, H.; Liu, D.; Li, L.; Li, J.; Wang, Q.; Ye, M.; Huang, Q.; Yu, Z.; Zhang, J. GSTP1 c.313A > G mutation is an independent risk factor for neutropenia hematotoxicity induced by anthracycline-/paclitaxel-based chemotherapy in breast cancer patients. World J. Surg. Oncol. 2022, 20, 212. [Google Scholar] [CrossRef]
- Rocca, A.; Andreis, D.; Fedeli, A.; Maltoni, R.; Sarti, S.; Cecconetto, L.; Pietri, E.; Schirone, A.; Bravaccini, S.; Serra, P.; et al. Pharmacokinetics, pharmacodynamics and clinical efficacy of pertuzumab in breast cancer therapy. Expert. Opin. Drug Metab. Toxicol. 2015, 11, 1647–1663. [Google Scholar] [CrossRef]
- Richard, S.; Selle, F.; Lotz, J.P.; Khalil, A.; Gligorov, J.; Soares, D.G. Pertuzumab and trastuzumab: The rationale way to synergy. An. Acad. Bras. Ciências 2016, 88 (Suppl. S1), 565–577. [Google Scholar] [CrossRef]
- von Arx, C.; De Placido, P.; Caltavituro, A.; Di Rienzo, R.; Buonaiuto, R.; De Laurentiis, M.; Arpino, G.; Puglisi, F.; Giuliano, M.; Del Mastro, L. The evolving therapeutic landscape of trastuzumab-drug conjugates: Future perspectives beyond HER2-positive breast cancer. Cancer Treat. Rev. 2023, 113, 102500. [Google Scholar] [CrossRef]
- Cocca, M.; Bedognetti, D.; La Bianca, M.; Gasparini, P.; Girotto, G. Pharmacogenetics driving personalized medicine: Analysis of genetic polymorphisms related to breast cancer medications in Italian isolated populations. J. Transl. Med. 2016, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.L.; Chaiyakunapruk, N.; Tassaneeyakul, W.; Arunmanakul, P.; Nathisuwan, S.; Lee, S.W.H. Roles of pharmacogenomics in non-anthracycline antineoplastic-induced cardiovascular toxicities: A systematic review and meta-analysis of genotypes effect. Int. J. Cardiol. 2019, 280, 190–197. [Google Scholar] [CrossRef]
- Sondermann, P.; Huber, R.; Oosthuizen, V.; Jacob, U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 2000, 406, 267–273. [Google Scholar] [CrossRef]
- Varchetta, S.; Gibelli, N.; Oliviero, B.; Nardini, E.; Gennari, R.; Gatti, G.; Silva, L.S.; Villani, L.; Tagliabue, E.; Ménard, S.; et al. Elements related to heterogeneity of antibody-dependent cell cytotoxicity in patients under trastuzumab therapy for primary operable breast cancer overexpressing Her2. Cancer Res. 2007, 67, 11991–11999. [Google Scholar] [CrossRef]
- Tamura, K.; Shimizu, C.; Hojo, T.; Akashi-Tanaka, S.; Kinoshita, T.; Yonemori, K.; Kouno, T.; Katsumata, N.; Ando, M.; Aogi, K.; et al. FcγR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann. Oncol. 2011, 22, 1302–1307. [Google Scholar] [CrossRef]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Musolino, A.; Naldi, N.; Dieci, M.V.; Zanoni, D.; Rimanti, A.; Boggiani, D.; Sgargi, P.; Generali, D.G.; Piacentini, F.; Ambroggi, M.; et al. Immunoglobulin G fragment C receptor polymorphisms and efficacy of preoperative chemotherapy plus trastuzumab and lapatinib in HER2-positive breast cancer. Pharm. J. 2016, 16, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Betting, D.J.; Stern, H.M.; Quinaux, E.; Stinson, J.; Seshagiri, S.; Zhao, Y.; Buyse, M.; Mackey, J.; Driga, A.; et al. Analysis of Fcγ receptor IIIa and IIa polymorphisms: Lack of correlation with outcome in trastuzumab-treated breast cancer patients. Clin. Cancer Res. 2012, 18, 3478–3486. [Google Scholar] [CrossRef]
- Madrid-Paredes, A.; Cañadas-Garre, M.; Sánchez-Pozo, A.; Segura-Pérez, A.M.; Chamorro-Santos, C.; Vergara-Alcaide, E.; Castillo-Portellano, L.; Calleja-Hernández, M. ABCB1 C3435T gene polymorphism as a potential biomarker of clinical outcomes in HER2-positive breast cancer patients. Pharmacol. Res. 2016, 108, 111–118. [Google Scholar] [CrossRef]
- Sarah, A.; Dondi, E.; De Francia, S. Tyrosine kinase inhibitors: The role of pharmacokinetics and pharmacogenetics. Expert. Opin. Drug Metab. Toxicol. 2023, 19, 733–739. [Google Scholar] [CrossRef]
- van Erp, N.P.; Gelderblom, H.; Guchelaar, H.J. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat. Rev. 2009, 35, 692–706. [Google Scholar] [CrossRef]
- Zhao, Q.; Wu, Z.E.; Li, B.; Li, F. Recent advances in metabolism and toxicity of tyrosine kinase inhibitors. Pharmacol. Ther. 2022, 237, 108256. [Google Scholar] [CrossRef] [PubMed]
- Bissada, J.E.; Truong, V.; Abouda, A.A.; Wines, K.J.; Crouch, R.D.; Jackson, K.D. Interindividual Variation in CYP3A Activity Influences Lapatinib Bioactivation. Drug Metab. Dispos. Biol. Fate Chem. 2019, 47, 1257–1269. [Google Scholar] [CrossRef]
- Chen, S.; Li, X.; Li, Y.; He, X.; Bryant, M.; Qin, X.; Li, F.; Seo, J.E.; Guo, X.; Mei, N.; et al. The involvement of hepatic cytochrome P450s in the cytotoxicity of lapatinib. Toxicol. Sci. 2023, 197, 69–78. [Google Scholar] [CrossRef]
- Breslin, S.; Lowry, M.C.; O’Driscoll, L. Neratinib resistance and cross-resistance to other HER2-targeted drugs due to increased activity of metabolism enzyme cytochrome P4503A4. Br. J. Cancer 2017, 116, 620–625. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Oitate, M.; Hagihara, K.; Shiozawa, H.; Furuta, Y.; Ogitani, Y.; Kuga, H. Pharmacokinetics of trastuzumab deruxtecan (T-DXd), a novel anti-HER2 antibody-drug conjugate, in HER2-positive tumour-bearing mice. Xenobiotica 2020, 50, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, C.M.; Yamaguchi, A.; Anami, Y.; Xiong, W.; Otani, Y.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; Tsuchikama, K. Antibody-drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat. Commun. 2021, 12, 3528. [Google Scholar] [CrossRef]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef]
- Trastuzumab emtansine. An inadequately assessed combination of two cytotoxic drugs. Prescrire Int. 2014, 23, 289. [Google Scholar]
- Nakada, T.; Sugihara, K.; Jikoh, T.; Abe, Y.; Agatsuma, T. The Latest Research and Development into the Antibody-Drug Conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy. Chem. Pharm. Bull. 2019, 67, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M.; Lauschke, V.M. Individualized Pharmacotherapy Utilizing Genetic Biomarkers and Novel In Vitro Systems As Predictive Tools for Optimal Drug Development and Treatment. Drug Metab. Dispos. 2024, 52, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Lauschke, V.M.; Ingelman-Sundberg, M. Prediction of drug response and adverse drug reactions: From twin studies to Next Generation Sequencing. Eur. J. Pharm. Sci. 2019, 130, 65–77. [Google Scholar] [CrossRef]
- Kang, J.S.; Lee, M.H. Overview of therapeutic drug monitoring. Korean J. Intern. Med. 2009, 24, 1–10. [Google Scholar] [CrossRef]

{kind=link}
| Drug Class | Enzyme/Genetic Variant | Effect on Pharmacokinetics |
|---|---|---|
| Tamoxifen (SERM) | CYP2D6 | Enhanced-function allele increases symptoms and discontinuation rates; the reduced-function allele diminishes tamoxifen metabolism and efficacy. |
| Aromatase Inhibitors (AIs) | CYP19A1 | Associated with baseline aromatase activity |
| CYP2A6 | Reduced-function allele elevates letrozole plasma concentrations. | |
| UGT1A4 and UGT2B7 | Affect drug conjugation and clearance. | |
| GSTA1 *B*B | inhibited metabolism | |
| CDK 4/6 Inhibitors | CYP3A4 | Higher risk of toxicity with strong CYP3A4 inhibitors |
| Taxanes | CYP2C8*3 | Higher remission rates in neoadjuvant treatment, with possible increased toxicity. |
| CYP3A4 | Reduced mRNA plasma level is associated with the docetaxel response rates | |
| ABCB1 | Increased risk of neutropenia and diarrhea | |
| SLCO1B1 521T>C | Decreased risk of mortality | |
| Cyclophosphamide (CTX) | CYP2B6 516G>T and A785A>G | Poorer overall survival (OS) |
| CYP2C19*2 | Related to an increased risk of adverse reactions (AEs) | |
| Anthracyclines | CYP2C19*2 | Increased drug-induced AEs |
| UGT2B7 161 C>T | Higher epirubicin elimination, lower risk of leukopenia | |
| ABCB1 3435 C>T | Better OS but increased risk of diarrhea and neutropenia | |
| SLC22A16 T>C | Increased risk of diarrhea and neutropenia | |
| GSTM1 and GSTT1 deletions | Decreased recurrence and mortality rates. | |
| GSTP1 313A>G | Increased risk of hematological toxicity | |
| Monoclonal Antibodies | HER2 1173A>G | Increased risk of trastuzumab-induced cardiac toxicity |
| FCGR2A 519A>G | Associated with reduced trastuzumab efficacy | |
| FCGR3A 559T>G | Higher pCR rates with trastuzumab plus lapatinib in neoadjuvant therapy | |
| ABCB1 3435 C>T | Increased resistance to chemotherapy/trastuzumab regimens | |
| Tyrosine Kinase Inhibitors (TKIs) | CYP3A4 | Increased risk of lapatinib-induced hepatotoxic toxicity and associated with resistance in neratinib-resistant cells |
| CYP3A5, CYP3A7 | May reduce lapatinib cytotoxicity and DNA damage | |
| Antibody-drug conjugate (ADCs) | Unknown | Needs further exploration |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Xiong, H. The Role of Pharmacogenetic-Based Pharmacokinetic Analysis in Precise Breast Cancer Treatment. Pharmaceutics 2024, 16, 1407. https://doi.org/10.3390/pharmaceutics16111407
Wu X, Xiong H. The Role of Pharmacogenetic-Based Pharmacokinetic Analysis in Precise Breast Cancer Treatment. Pharmaceutics. 2024; 16(11):1407. https://doi.org/10.3390/pharmaceutics16111407
Chicago/Turabian StyleWu, Xinyu, and Huihua Xiong. 2024. "The Role of Pharmacogenetic-Based Pharmacokinetic Analysis in Precise Breast Cancer Treatment" Pharmaceutics 16, no. 11: 1407. https://doi.org/10.3390/pharmaceutics16111407
APA StyleWu, X., & Xiong, H. (2024). The Role of Pharmacogenetic-Based Pharmacokinetic Analysis in Precise Breast Cancer Treatment. Pharmaceutics, 16(11), 1407. https://doi.org/10.3390/pharmaceutics16111407