
The Development of an Age-Appropriate Fixed Dose Combination for Tuberculosis Using Physiologically-Based Pharmacokinetic Modeling (PBBM) and Risk Assessment

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Solution Stability and Solubility Tests
2.2. Dissolution Profiles
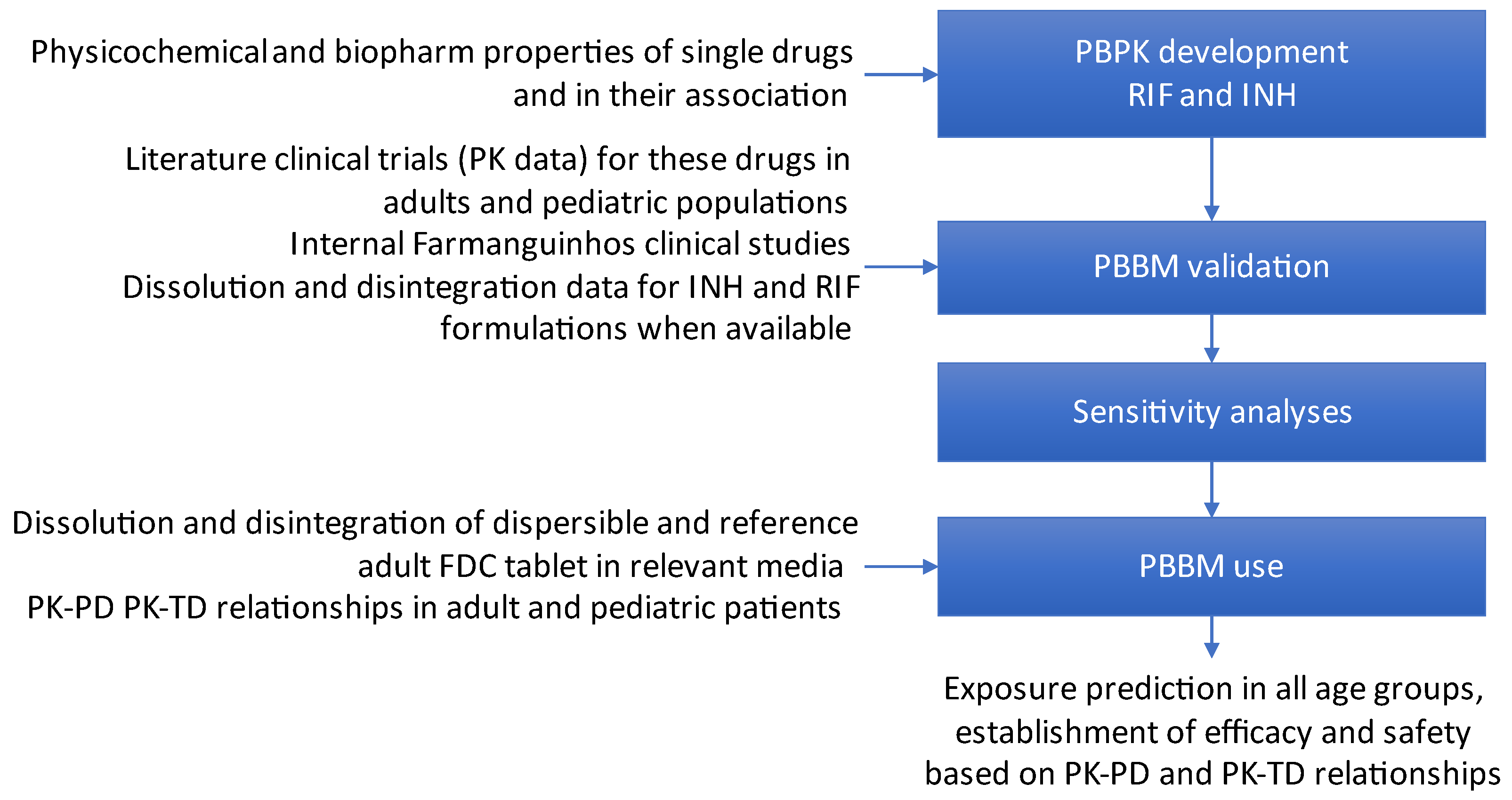
2.3. Modeling Strategy
2.4. Modeling Assumptions
2.5. Criteria for Model Validation
2.6. Integration of Disintegration in the PBBM
2.7. Integration of Dissolution in the PBBM
2.8. Dosage Forms
2.9. Clinical Studies for Model Validation
2.10. Model Application for Adult and Pediatric Simulations
2.10.1. Adult Simulations
2.10.2. Pediatric Simulations
- Group 1: 0–1 year. This group should receive 1 dispersible tablet.
- Group 2: 1–3 years. This group should receive 2 dispersible tablets.
- Group 3: 3–5 years. This group should receive 3 dispersible tablets.
- Group 4: 5–8 years. This group should receive 4 dispersible tablets.
- Group 5: 8–11 years. This group should receive 6 dispersible tablets.
3. Results
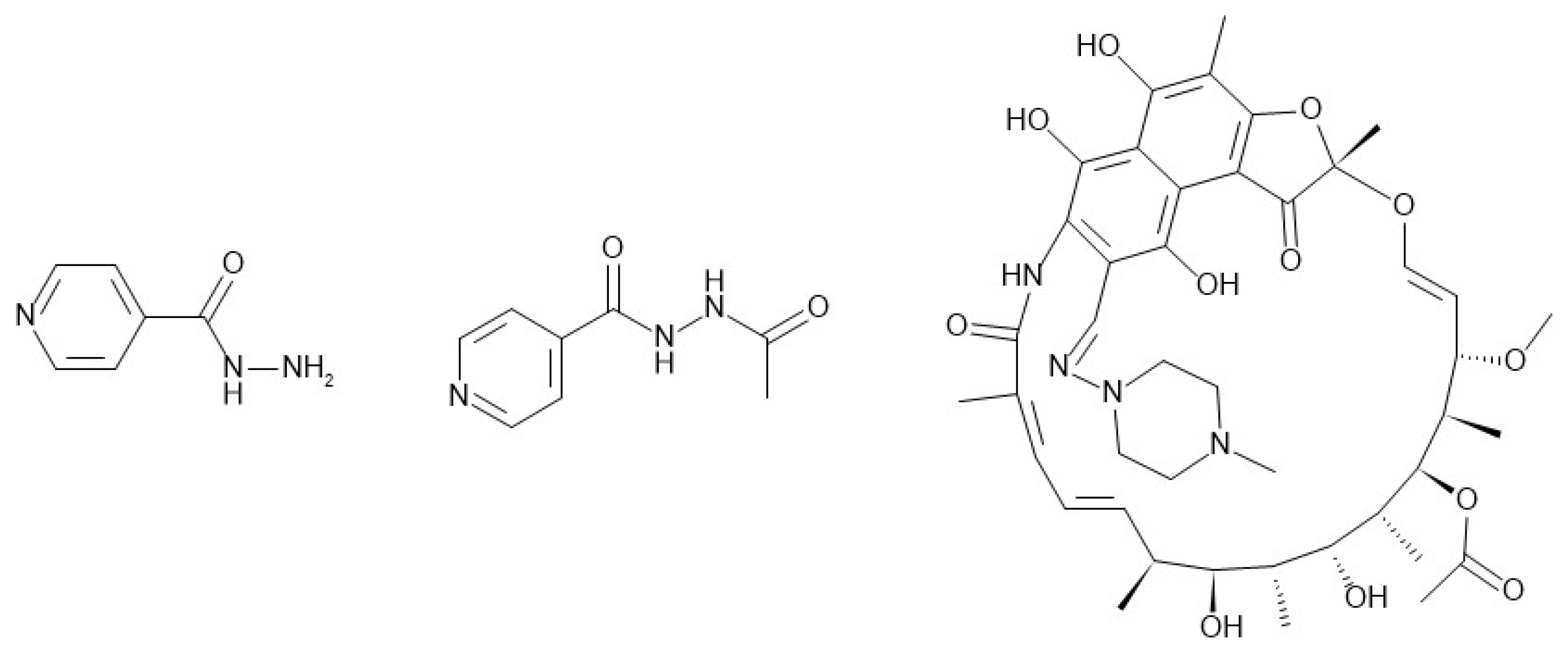
3.1. Physicochemical and Biopharmaceutical Properties
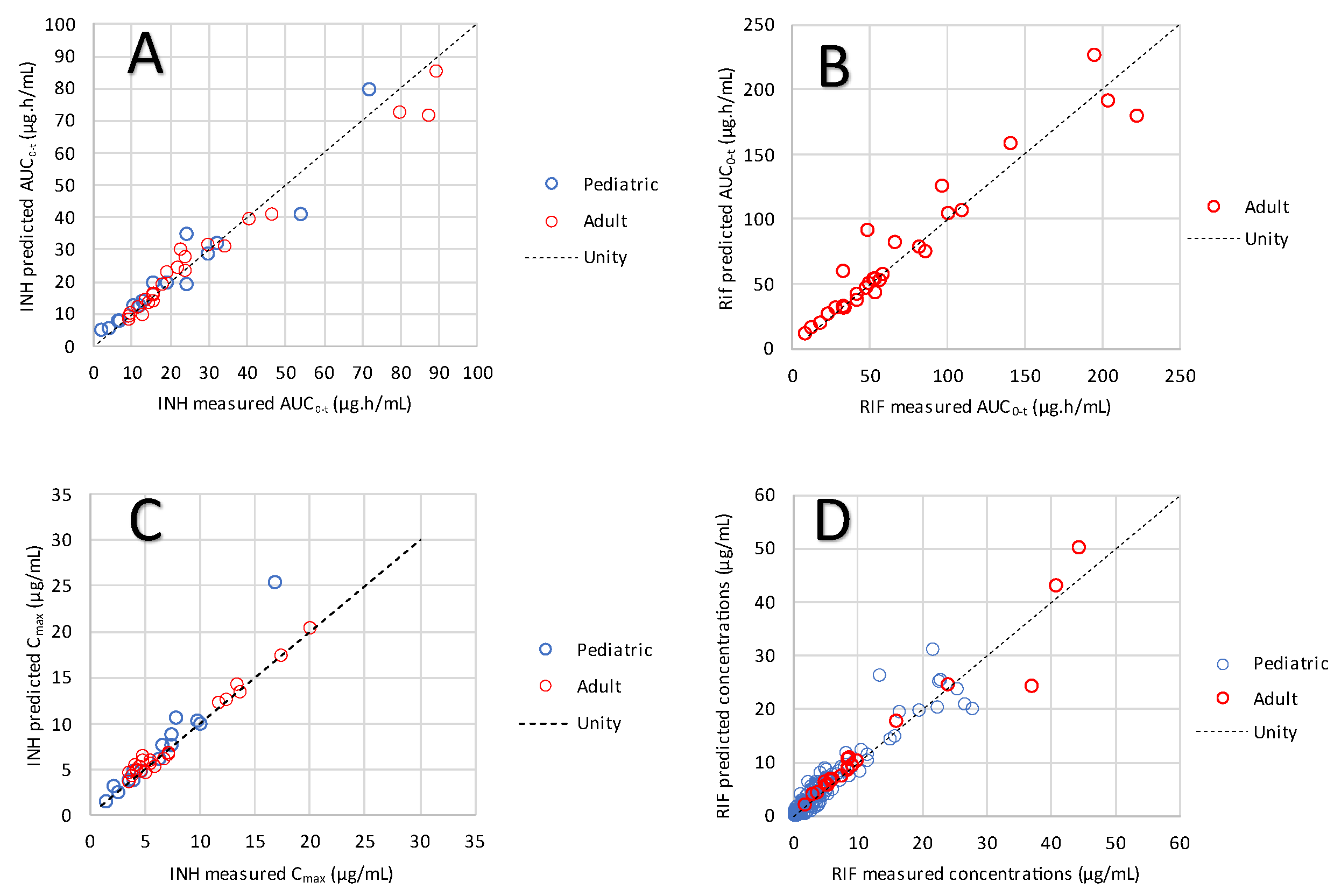
3.2. Model Validation
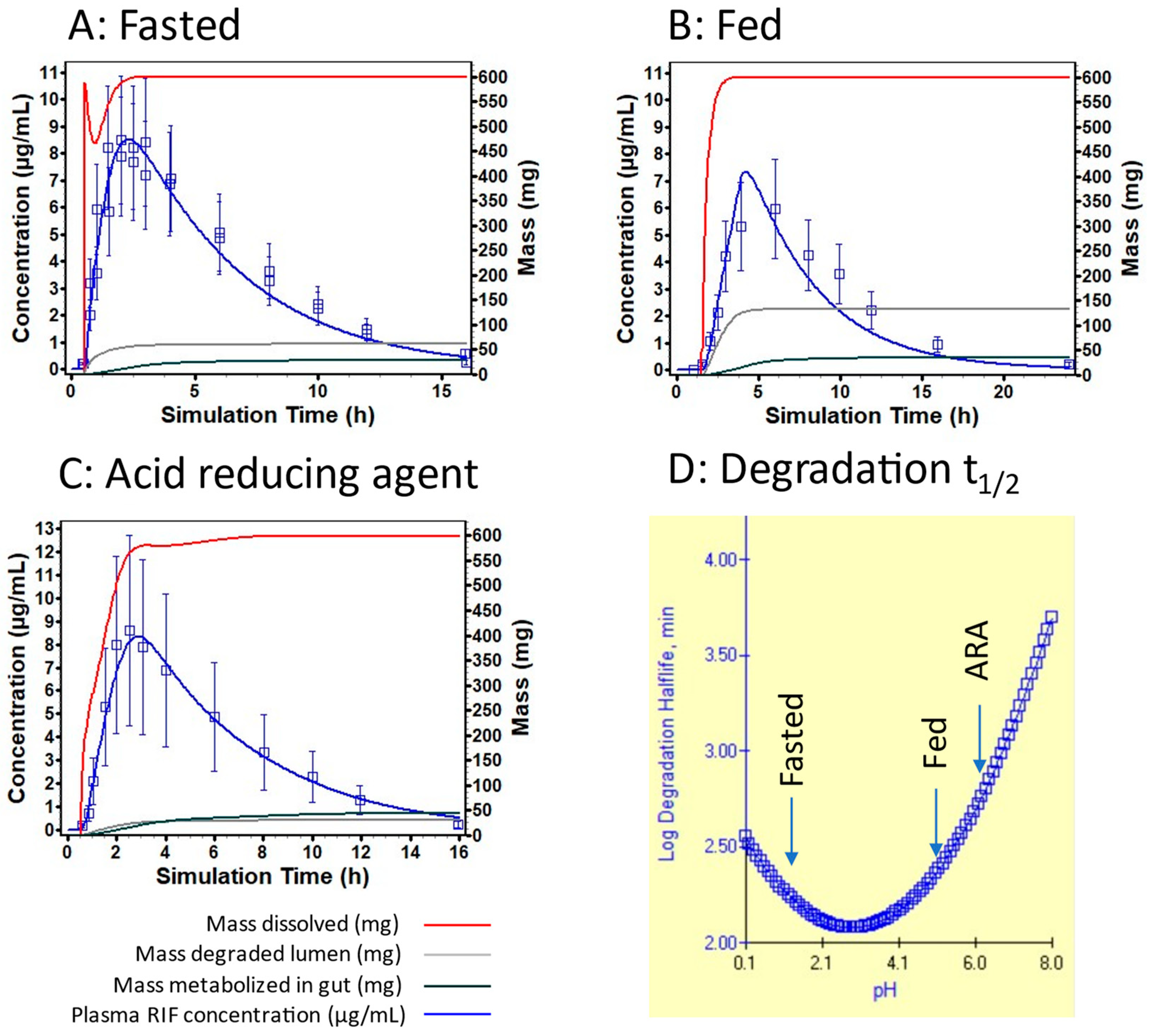
3.3. Examination of Individual Profiles
3.4. Model Application
3.4.1. Model Application to Adult Simulations
3.4.2. Model Application to Pediatric Simulations
4. Discussion
4.1. Isoniazid
4.2. Rifampicin
4.3. Combined View on Safety and Efficacy
4.3.1. Efficacy of INH and RIF
4.3.2. Safety of INH and RIF
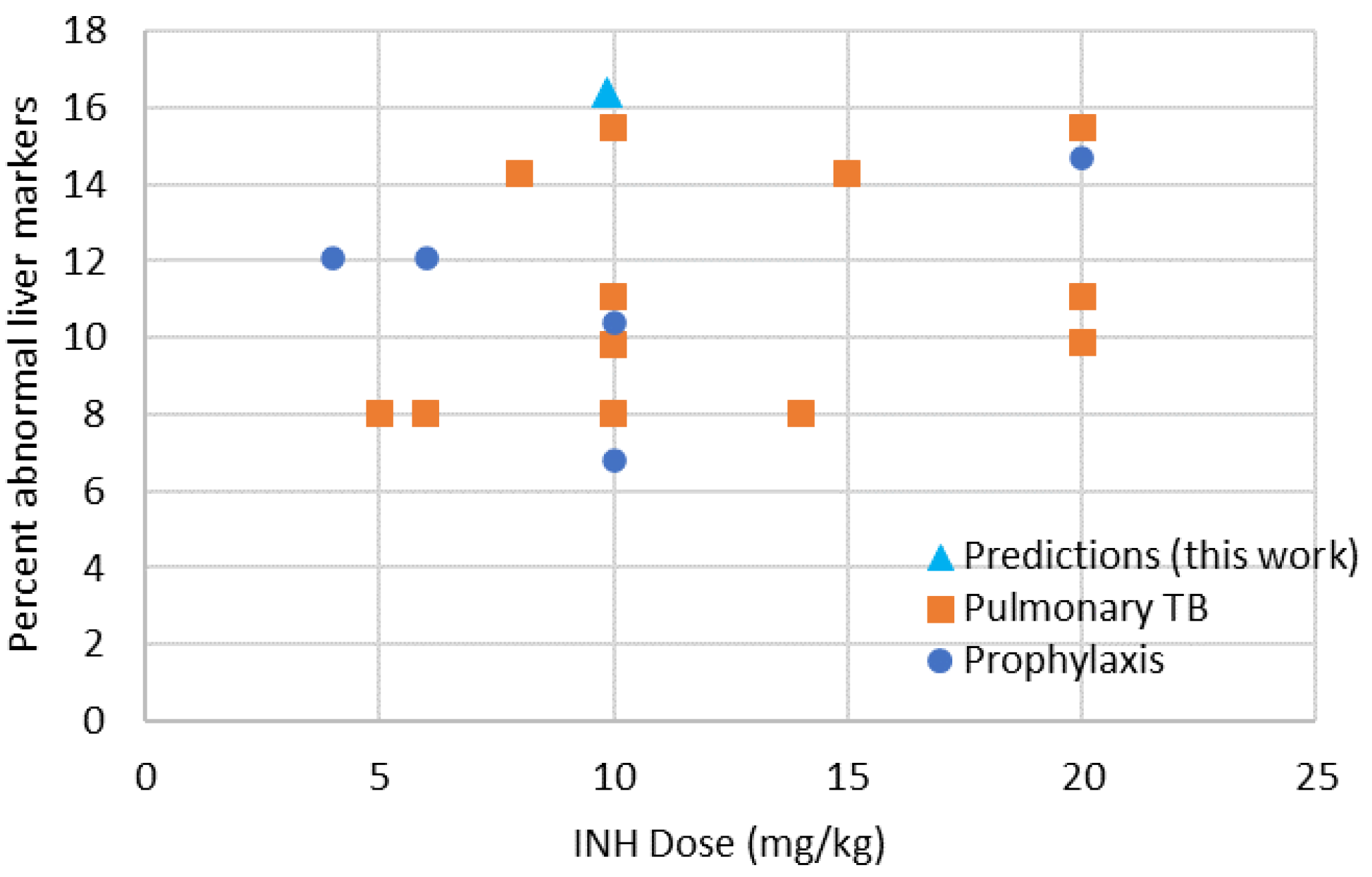
4.3.3. Predicted Safety of INH in Children in Terms of DILI
4.3.4. Predicted Safety of RIF in Children in Terms of DILI and AKI
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brouwer, K.L.; Aleksunes, L.M.; Brandys, B.; Giacoia, G.P.; Knipp, G.; Lukacova, V.; Meibohm, B.; Nigam, S.K.; Rieder, M.; de Wildt, S.N. Human Ontogeny of Drug Transporters: Review and Recommendations of the Pediatric Transporter Working Group. Clin. Pharmacol. Ther. 2015, 98, 266–287. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Baello, S.; Iqbal, M.; Kelly, L.E.; Shannon, P.T.; Chitayat, D.; Matthews, S.G.; Koren, G. The ontogeny of P-glycoprotein in the developing human blood–brain barrier: Implication for opioid toxicity in neonates. Pediatr. Res. 2015, 78, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Holford, N.; Heo, Y.-A.; Anderson, B. A Pharmacokinetic Standard for Babies and Adults. J. Pharm. Sci. 2013, 102, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, A.R.; Gonzalez, D.; Cohen-Wolkowiez, M.; Hornik, C.P.; Edginton, A.N. Improving Pediatric Protein Binding Estimates: An Evaluation of α1-Acid Glycoprotein Maturation in Healthy and Infected Subjects. Clin. Pharmacokinet. 2018, 57, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.E.; Edginton, A.N. Evaluation of models for predicting pediatric fraction unbound in plasma for human health risk assessment. J. Toxicol. Environ. Health. Part A 2021, 84, 67–83. [Google Scholar] [CrossRef]
- Abdulla, A.; Edwina, A.E.; Flint, R.B.; Allegaert, K.; Wildschut, E.D.; Koch, B.C.P.; de Hoog, M. Model-Informed Precision Dosing of Antibiotics in Pediatric Patients: A Narrative Review. Front. Pediatr. 2021, 9, 624639. [Google Scholar] [CrossRef]
- Cristofoletti, R.; Charoo, N.A.; Dressman, J.B. Exploratory Investigation of the Limiting Steps of Oral Absorption of Fluconazole and Ketoconazole in Children Using an In Silico Pediatric Absorption Model. J. Pharm. Sci. 2016, 105, 2794–2803. [Google Scholar] [CrossRef]
- Miao, L.; Mousa, Y.M.; Zhao, L.; Raines, K.; Seo, P.; Wu, F. Using a Physiologically Based Pharmacokinetic Absorption Model to Establish Dissolution Bioequivalence Safe Space for Oseltamivir in Adult and Pediatric Populations. AAPS J. 2020, 22, 107. [Google Scholar] [CrossRef]
- Asaumi, R.; Toshimoto, K.; Tobe, Y.; Hashizume, K.; Nunoya, K.-i.; Imawaka, H.; Lee, W.; Sugiyama, Y. Comprehensive PBPK Model of Rifampicin for Quantitative Prediction of Complex Drug-Drug Interactions: CYP3A/2C9 Induction and OATP Inhibition Effects. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 186–196. [Google Scholar] [CrossRef]
- Asaumi, R.; Nunoya, K.-i.; Yamaura, Y.; Taskar, K.S.; Sugiyama, Y. Robust physiologically based pharmacokinetic model of rifampicin for predicting drug–drug interactions via P-glycoprotein induction and inhibition in the intestine, liver, and kidney. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 919–933. [Google Scholar] [CrossRef]
- Rasool, M.F.; Khalid, S.; Majeed, A.; Saeed, H.; Imran, I.; Mohany, M.; Al-Rejaie, S.S.; Alqahtani, F. Development and Evaluation of Physiologically Based Pharmacokinetic Drug–Disease Models for Predicting Rifampicin Exposure in Tuberculosis and Cirrhosis Populations. Pharmaceutics 2019, 11, 578. [Google Scholar] [CrossRef] [PubMed]
- Balhara, A.; Singh, S. PBPK Analysis to Study the Impact of Genetic Polymorphism of NAT2 on Drug-Drug Interaction Potential of Isoniazid. Pharm. Res. 2021, 38, 1485–1496. [Google Scholar] [CrossRef] [PubMed]
- Pepin, X.J.H.; Suarez-Sharp, S. Effect of Food Composition on the PK of Isoniazid Quantitatively Explained Using Physiologically Based Biopharmaceutics Modeling. AAPS J. 2024, 26, 54. [Google Scholar] [CrossRef]
- De Zwart, L.L.; Rompelberg, C.J.M.; Sips, A.J.A.M.; Welink, J.; Engelen, J.G.M. RIVM Report 623860 010: Anatomical and Physiological Differences Between Various Species Used in Studies on the Pharmacokinetics and Toxicology of Xenobiotics. A Review of Literature; Rijksinstituut voor Volksgezondheid en Milieu RIVM: Bilthoven, The Netherlands, 1999; pp. 1–100. [Google Scholar]
- Brown, R.P.; Delp, M.D.; Lindstedt, S.L.; Rhomberg, L.R.; Beliles, R.P. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 1997, 13, 407–484. [Google Scholar] [CrossRef]
- Snyder, W.S.; Cook, M.J.; Nasset, E.S.; Karhausen, L.R.; Howells, G.P.; Tipton, I.H. Report of the Task Group on reference man (ICRP Publication 23); Elsevier Sci. Inc.: Amsterdam, The Netherlands, 1975; p. 480. [Google Scholar]
- Price, P.S.; Conolly, R.B.; Chaisson, C.F.; Gross, E.A.; Young, J.S.; Mathis, E.T.; Tedder, D.R. Modeling interindividual variation in physiological factors used in PBPK models of humans. Crit. Rev. Toxicol. 2003, 33, 469–503. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, S.; Wada, D.R.; Berling, B.M.; Benoni, G. Prediction of the disposition of midazolam in surgical patients by a physiologically based pharmacokinetic model. J. Pharm. Sci. 2001, 90, 1226–1241. [Google Scholar] [CrossRef]
- Kearns, G.L.; Abdel-Rahman, S.M.; Alander, S.W.; Blowey, D.L.; Leeder, J.S.; Kauffman, R.E. Developmental pharmacology--drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 2003, 349, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.A.; Levey, A.S. Frequently Asked Questions About GFR Estimates; National Kidney Foundation: New York, NY, USA, 2007. [Google Scholar]
- McNamara, P.J.; Alcorn, J. Protein binding predictions in infants. AAPS PharmSci 2002, 4, 3. [Google Scholar] [CrossRef]
- Fredholt, F.; Di Meo, C.; Sloth, S.; Müllertz, A.; Berthelsen, R. Direct visualizing of paracetamol immediate release tablet disintegration in vivo and in vitro. Eur. J. Pharm. Biopharm. 2022, 180, 63–70. [Google Scholar] [CrossRef]
- Stevens, H.; Voelker, M.; Gow, L.; MacDougall, F.; Bieri, G. In-vivo disintegration and absorption of two fast acting aspirin tablet formulations compared to ibuprofen tablets using pharmacoscintigraphy. J. Drug Deliv. Sci. Technol. 2019, 51, 535–541. [Google Scholar] [CrossRef]
- Lenz, J.; Fuest, F.; Finke, J.H.; Bunjes, H.; Kwade, A.; Juhnke, M. Tablet Disintegration and Dispersion under In Vivo-like Hydrodynamic Conditions. Pharmaceutics 2022, 14, 208. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.; O’Mahony, B.; Lindsay, B.; Jones, T.; Grattan, T.J.; Rostami-Hodjegan, A.; Stevens, H.N.E.; Wilson, C.G. Comparison of the Rates of Disintegration, Gastric Emptying, and Drug Absorption Following Administration of a New and a Conventional Paracetamol Formulation, Using γ Scintigraphy. Pharm. Res. 2003, 20, 1668–1673. [Google Scholar] [CrossRef] [PubMed]
- Schiller, C.; Frohlich, C.P.; Giessmann, T.; Siegmund, W.; Monnikes, H.; Hosten, N.; Weitschies, W. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment. Pharmacol. Ther. 2005, 22, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Mudie, D.M.; Murray, K.; Hoad, C.L.; Pritchard, S.E.; Garnett, M.C.; Amidon, G.L.; Gowland, P.A.; Spiller, R.C.; Amidon, G.E.; Marciani, L. Quantification of gastrointestinal liquid volumes and distribution following a 240 mL dose of water in the fasted state. Mol. Pharm. 2014, 11, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Takano, R.; Sugano, K.; Higashida, A.; Hayashi, Y.; Machida, M.; Aso, Y.; Yamashita, S. Oral absorption of poorly water-soluble drugs: Computer simulation of fraction absorbed in humans from a miniscale dissolution test. Pharm. Res. 2006, 23, 1144–1156. [Google Scholar] [CrossRef]
- Gelber, R.; Jacobsen, P.; Levy, L. A study of the availability of six commercial formulations of isoniazid. Clin. Pharmacol. Ther. 1969, 10, 841–848. [Google Scholar] [CrossRef]
- Agrawal, S.; Panchagnula, R. Dissolution test as a surrogate for quality evaluation of rifampicin containing fixed dose combination formulations. Int. J. Pharm. 2004, 287, 97–112. [Google Scholar] [CrossRef]
- Agrawal, S.; Singh, I.; Kaur, K.J.; Bhade, S.; Kaul, C.L.; Panchagnula, R. Bioequivalence trials of rifampicin containing formulations: Extrinsic and intrinsic factors in the absorption of rifampicin. Pharmacol. Res. 2004, 50, 317–327. [Google Scholar] [CrossRef]
- Pepin, X.; Goetschy, M.; Abrahmsén-Alami, S. Mechanistic models for USP2 dissolution apparatus, including fluid hydrodynamics and sedimentation. J. Pharm. Sci. 2022, 111, 185–196. [Google Scholar] [CrossRef]
- Valentin, J. Basic anatomical and physiological data for use in radiological protection: Reference values. Ann. ICRP 2002, 32, 1–277. [Google Scholar] [CrossRef]
- Becker, C.; Dressman, J.B.; Amidon, G.L.; Junginger, H.E.; Kopp, S.; Midha, K.K.; Shah, V.P.; Stavchansky, S.; Barends, D.M. Biowaiver monographs for immediate release solid oral dosage forms: Isoniazid. J. Pharm. Sci. 2007, 96, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Budha, N.R.; Lee, R.E.; Meibohm, B. Biopharmaceutics, pharmacokinetics and pharmacodynamics of antituberculosis drugs. Curr. Med. Chem. 2008, 15, 809–825. [Google Scholar] [CrossRef] [PubMed]
- Ranaldi, G.; Islam, K.; Sambuy, Y. Epithelial cells in culture as a model for the intestinal transport of antimicrobial agents. Antimicrob. Agents Chemother. 1992, 36, 1374–1381. [Google Scholar] [CrossRef]
- Boxenbaum, H.; Riegelman, S. Pharmacokinetics of isoniazid and some metabolites in man. J. Pharmacokinet. Biopharm. 1976, 4, 287–325. [Google Scholar] [CrossRef]
- Bing, C.; Xiaomeia, C.; Jinhenga, L. Gene dose effect of NAT2 variants on the pharmacokinetics of isoniazid and acetylisoniazid in healthy Chinese subjects. Drug Metab. Drug Interact. 2011, 26, 113–118. [Google Scholar] [CrossRef]
- Ermondi, G.; Vallaro, M.; Saame, J.; Toom, L.; Leito, I.; Ruiz, R.; Caron, G. Rifampicin as an example of beyond-rule-of-5 compound: Ionization beyond water and lipophilicity beyond octanol/water. Eur. J. Pharm. Sci. 2021, 161, 105802. [Google Scholar] [CrossRef]
- Becker, C.; Dressman, J.B.; Junginger, H.E.; Kopp, S.; Midha, K.K.; Shah, V.P.; Stavchansky, S.; Barends, D.M. Biowaiver monographs for immediate release solid oral dosage forms: Rifampicin. J. Pharm. Sci. 2009, 98, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Boman, G.; Ringberger, V.A. Binding of rifampicin by human plasma proteins. Eur. J. Clin. Pharmacol. 1974, 7, 369–373. [Google Scholar] [CrossRef]
- Litjens, C.H.C.; Aarnoutse, R.E.; van Ewijk-Beneken Kolmer, E.W.J.; Svensson, E.M.; Colbers, A.; Burger, D.M.; Boeree, M.J.; Te Brake, L.H.M. Protein binding of rifampicin is not saturated when using high-dose rifampicin. J. Antimicrob. Chemother. 2019, 74, 986–990. [Google Scholar] [CrossRef]
- Biganzoli, E.; Cavenaghi, L.A.; Rossi, R.; Brunati, M.C.; Nolli, M.L. Use of a Caco-2 cell culture model for the characterization of intestinal absorption of antibiotics. Farmaco 1999, 54, 594–599. [Google Scholar] [CrossRef]
- Wasserman, S.; Davis, A.; Stek, C.; Chirehwa, M.; Botha, S.; Daroowala, R.; Bremer, M.; Maxebengula, M.; Koekemoer, S.; Goliath, R. Plasma pharmacokinetics of high-dose oral versus intravenous rifampicin in patients with tuberculous meningitis: A randomized controlled trial. Antimicrob. Agents Chemother. 2021, 65, e00140-00121. [Google Scholar] [CrossRef] [PubMed]
- Loos, U.; Musch, E.; Jensen, J.C.; Mikus, G.; Schwabe, H.K.; Eichelbaum, M. Pharmacokinetics of oral and intravenous rifampicin during chronic administration. Klin. Wochenschr. 1985, 63, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, V.G.; Ramos, L.M.; Monteiro, H.S.; Barroso, E.C.; Bushen, O.Y.; Façanha, M.C.; Peloquin, C.A.; Guerrant, R.L.; Lima, A.A. Intestinal permeability and malabsorption of rifampin and isoniazid in active pulmonary tuberculosis. Braz. J. Infect. Dis. 2006, 10, 374–379. [Google Scholar] [CrossRef]
- Pepin, X.J.; Flanagan, T.R.; Holt, D.J.; Eidelman, A.; Treacy, D.; Rowlings, C.E. Justification of Drug Product Dissolution Rate and Drug Substance Particle Size Specifications Based on Absorption PBPK Modeling for Lesinurad Immediate Release Tablets. Mol. Pharm. 2016, 13, 3256–3269. [Google Scholar] [CrossRef] [PubMed]
- Andreas, C.J.; Pepin, X.; Markopoulos, C.; Vertzoni, M.; Reppas, C.; Dressman, J. Mechanistic investigation of the negative food effect of modified release zolpidem. Eur. J. Pharm. Sci. 2017, 102, 284–298. [Google Scholar] [CrossRef]
- Pepin, X.J.H.; Moir, A.J.; Mann, J.C.; Sanderson, N.J.; Barker, R.; Meehan, E.; Plumb, A.P.; Bailey, G.R.; Murphy, D.S.; Krejsa, C.M.; et al. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part II. A mechanistic PBPK model for IR formulation comparison, proton pump inhibitor drug interactions, and administration with acidic juices. Eur. J. Pharm. Biopharm. 2019, 142, 435–448. [Google Scholar] [CrossRef]
- Langguth, P.; Lee, K.M.; Spahn-Langguth, H.; Amidon, G.L. Variable gastric emptying and discontinuities in drug absorption profiles: Dependence of rates and extent of cimetidine absorption on motility phase and pH. Biopharm. Drug Dispos. 1994, 15, 719–746. [Google Scholar] [CrossRef] [PubMed]
- Lipka, E.; Lee, I.D.; Langguth, P.; Spahn-Langguth, H.; Mutschler, E.; Amidon, G.L. Celiprolol double-peak occurrence and gastric motility: Nonlinear mixed effects modeling of bioavailability data obtained in dogs. J. Pharmacokinet. Biopharm. 1995, 23, 267–286. [Google Scholar] [CrossRef]
- Weitschies, W.; Wedemeyer, R.S.; Kosch, O.; Fach, K.; Nagel, S.; Soderlind, E.; Trahms, L.; Abrahamsson, B.; Monnikes, H. Impact of the intragastric location of extended release tablets on food interactions. J. Controll. Release 2005, 108, 375–385. [Google Scholar] [CrossRef]
- Weitschies, W.; Friedrich, C.; Wedemeyer, R.S.; Schmidtmann, M.; Kosch, O.; Kinzig, M.; Trahms, L.; Sorgel, F.; Siegmund, W.; Horkovics-Kovats, S.; et al. Bioavailability of amoxicillin and clavulanic acid from extended release tablets depends on intragastric tablet deposition and gastric emptying. Eur. J. Pharm. Biopharm. 2008, 70, 641–648. [Google Scholar] [CrossRef]
- Phaisal, W.; Jantarabenjakul, W.; Wacharachaisurapol, N.; Tawan, M.; Puthanakit, T.; Wittayalertpanya, S.; Chariyavilaskul, P. Pharmacokinetics of isoniazid and rifapentine in young pediatric patients with latent tuberculosis infection. Int. J. Infect. Dis. 2022, 122, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Thee, S.; Seddon, J.A.; Donald, P.R.; Seifart, H.I.; Werely, C.J.; Hesseling, A.C.; Rosenkranz, B.; Roll, S.; Magdorf, K.; Schaaf, H.S. Pharmacokinetics of isoniazid, rifampin, and pyrazinamide in children younger than two years of age with tuberculosis: Evidence for implementation of revised World Health Organization recommendations. Antimicrob. Agents Chemother. 2011, 55, 5560–5567. [Google Scholar] [CrossRef] [PubMed]
- McIlleron, H.; Willemse, M.; Werely, C.J.; Hussey, G.D.; Schaaf, H.S.; Smith, P.J.; Donald, P.R. Isoniazid plasma concentrations in a cohort of South African children with tuberculosis: Implications for international pediatric dosing guidelines. Clin. Infect. Dis. 2009, 48, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Kiser, J.J.; Zhu, R.; D’Argenio, D.Z.; Cotton, M.F.; Bobat, R.; McSherry, G.D.; Madhi, S.A.; Carey, V.J.; Seifart, H.I.; Werely, C.J.; et al. Isoniazid pharmacokinetics, pharmacodynamics, and dosing in South African infants. Ther. Drug Monit. 2012, 34, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Roy, V.; Tekur, U.; Chopra, K. Pharmacokinetics of isoniazid in pulmonary tuberculosis--a comparative study at two dose levels. Indian Pediatr. 1996, 33, 287–291. [Google Scholar] [PubMed]
- Ruslami, R.; Gafar, F.; Yunivita, V.; Parwati, I.; Ganiem, A.R.; Aarnoutse, R.E.; Wilffert, B.; Alffenaar, J.-W.C.; Nataprawira, H.M. Pharmacokinetics and safety/tolerability of isoniazid, rifampicin and pyrazinamide in children and adolescents treated for tuberculous meningitis. Arch. Dis. Child. 2022, 107, 70–77. [Google Scholar] [CrossRef]
- Bekker, A.; Schaaf, H.S.; Draper, H.R.; Laan, L.v.d.; Murray, S.; Wiesner, L.; Donald, P.R.; McIlleron, H.M.; Hesseling, A.C. Pharmacokinetics of Rifampin, Isoniazid, Pyrazinamide, and Ethambutol in Infants Dosed According to Revised WHO-Recommended Treatment Guidelines. Antimicrob. Agents Chemother. 2016, 60, 2171–2179. [Google Scholar] [CrossRef]
- Rey, E.; Gendrel, D.; Treluyer, J.M.; Tran, A.; Pariente-Khayat, A.; d’Athis, P.; Pons, G. Isoniazid pharmacokinetics in children according to acetylator phenotype. Fundam. Clin. Pharmacol. 2001, 15, 355–359. [Google Scholar] [CrossRef]
- Koup, J.R.; Williams-Warren, J.; Viswanathan, C.T.; Weber, A.; Smith, A.L. Pharmacokinetics of Rifampin in Children II. Oral Bioavailability. Ther. Drug Monit. 1986, 8, 17–22. [Google Scholar] [CrossRef]
- McCracken, G.H., Jr.; Ginsburg, C.M.; Zweighaft, T.C.; Clahsen, J. Pharmacokinetics of Rifampin in Infants and Children: Relevance to Prophylaxis Against Haemophilus influenzae Type b Disease. Pediatrics 1980, 66, 17–21. [Google Scholar] [CrossRef]
- Schaaf, H.S.; Willemse, M.; Cilliers, K.; Labadarios, D.; Maritz, J.S.; Hussey, G.D.; McIlleron, H.; Smith, P.; Donald, P.R. Rifampin pharmacokinetics in children, with and without human immunodeficiency virus infection, hospitalized for the management of severe forms of tuberculosis. BMC Med. 2009, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Thee, S.; Detjen, A.; Wahn, U.; Magdorf, K. Rifampicin serum levels in childhood tuberculosis. Int. J. Tuberc. Lung Dis. 2009, 13, 1106–1111. [Google Scholar] [PubMed]
- Stott, K.E.; Pertinez, H.; Sturkenboom, M.G.G.; Boeree, M.J.; Aarnoutse, R.; Ramachandran, G.; Requena-Méndez, A.; Peloquin, C.; Koegelenberg, C.F.N.; Alffenaar, J.W.C.; et al. Pharmacokinetics of rifampicin in adult TB patients and healthy volunteers: A systematic review and meta-analysis. J. Antimicrob. Chemother. 2018, 73, 2305–2313. [Google Scholar] [CrossRef]
- Smith, P.B.; Cotten, C.M.; Hudak Mark, L.; Sullivan Janice, E.; Poindexter Brenda, B.; Cohen-Wolkowiez, M.; Boakye-Agyeman, F.; Lewandowski, A.; Anand, R.; Benjamin Daniel, K.; et al. Rifampin Pharmacokinetics and Safety in Preterm and Term Infants. Antimicrob. Agents Chemother. 2019, 63, e00284-19. [Google Scholar] [CrossRef]
- Pasipanodya, J.G.; McIlleron, H.; Burger, A.; Wash, P.A.; Smith, P.; Gumbo, T. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J. Infect. Dis. 2013, 208, 1464–1473. [Google Scholar] [CrossRef]
- Zheng, X.; Bao, Z.; Forsman, L.D.; Hu, Y.; Ren, W.; Gao, Y.; Li, X.; Hoffner, S.; Bruchfeld, J.; Alffenaar, J.-W. Drug Exposure and Minimum Inhibitory Concentration Predict Pulmonary Tuberculosis Treatment Response. Clin. Infect. Dis. 2021, 73, e3520–e3528. [Google Scholar] [CrossRef]
- Melander, A.; Danielson, K.; Hanson, A.; Jansson, L.; Rerup, C.; Scherstén, B.; Thulin, T.; Wåhlin, E. Reduction of Isoniazid Bioavailability in Normal Men by Concomitant Intake of Food. Acta Medica Scand. 2009, 200, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Peloquin, C.A.; Namdar, R.; Dodge, A.A.; Nix, D.E. Pharmacokinetics of isoniazid under fasting conditions, with food, and with antacids. Int. J. Tuberc. Lung Dis. 1999, 3, 703–710. [Google Scholar]
- Männisto, P.; Mäntylä, R.; Klinge, E.; Nykänen, S.; Koponen, A.; Lamminsivu, U. Influence of various diets on the bioavailability of isoniazid. J. Antimicrob. Chemother. 1982, 10, 427–434. [Google Scholar] [CrossRef]
- Kumar, A.K.H.; Chandrasekaran, V.; Kumar, A.K.; Kawaskar, M.; Lavanya, J.; Swaminathan, S.; Ramachandran, G. Food significantly reduces plasma concentrations of first-line anti-tuberculosis drugs. Indian J. Med. Res. 2017, 145, 530–535. [Google Scholar]
- Devani, M.B.; Shishoo, C.J.; Doshi, K.J.; Patel, H.B. Kinetic studies of the interaction between isoniazid and reducing sugars. J. Pharm. Sci. 1985, 74, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.S.; Hardy, J.G.; Fara, J.W. Transit of pharmaceutical dosage forms through the small intestine. Gut 1986, 27, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Keller, G.A.; Fabian, L.; Gomez, M.; Gonzalez, C.D.; Diez, R.A.; Di Girolamo, G. Age-distribution and genotype-phenotype correlation for N-acetyltransferase in Argentine children under isoniazid treatment. Int. J. Clin. Pharmacol. Ther. 2014, 52, 292–302. [Google Scholar] [CrossRef]
- Davit, B.; Chen, M.-L.; Conner, D.; Haidar, S.; Kim, S.; Lee, C.; Lionberger, R.; Makhlouf, F.; Nwakama, P.; Patel, D.; et al. Implementation of a Reference-Scaled Average Bioequivalence Approach for Highly Variable Generic Drug Products by the US Food and Drug Administration. AAPS J. 2012, 14, 915–924. [Google Scholar] [CrossRef]
- Donald, P.R. Antituberculosis drug-induced hepatotoxicity in children. Pediatr. Rep. 2011, 3, e16. [Google Scholar] [CrossRef]
- Wilkins, J.J.; Savic, R.M.; Karlsson, M.O.; Langdon, G.; McIlleron, H.; Pillai, G.; Smith, P.J.; Simonsson, U.S. Population pharmacokinetics of rifampin in pulmonary tuberculosis patients, including a semimechanistic model to describe variable absorption. Antimicrob. Agents Chemother. 2008, 52, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Peloquin, C.A.; Namdar, R.; Singleton, M.D.; Nix, D.E. Pharmacokinetics of Rifampin Under Fasting Conditions, With Food, and With Antacids. Chest 1999, 115, 12–18. [Google Scholar] [CrossRef]
- Diallo, T.; Adjobimey, M.; Ruslami, R.; Trajman, A.; Sow, O.; Obeng Baah, J.; Marks, G.B.; Long, R.; Elwood, K.; Zielinski, D.; et al. Safety and Side Effects of Rifampin versus Isoniazid in Children. N. Engl. J. Med. 2018, 379, 454–463. [Google Scholar] [CrossRef]
- Donald, P.R.; Sirgel, F.A.; Botha, F.J.; Seifart, H.I.; Parkin, D.P.; Vandenplas, M.L.; Van De Wal, B.W.; Maritz, J.S.; Mitchison, D.A. The early bactericidal activity of isoniazid related to its dose size in pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 1997, 156, 895–900. [Google Scholar] [CrossRef]
- Donald, P.R.; Parkin, D.P.; Seifart, H.I.; Schaaf, H.S.; van Helden, P.D.; Werely, C.J.; Sirgel, F.A.; Venter, A.; Maritz, J.S. The influence of dose and N-acetyltransferase-2 (NAT2) genotype and phenotype on the pharmacokinetics and pharmacodynamics of isoniazid. Eur. J. Clin. Pharmacol. 2007, 63, 633–639. [Google Scholar] [CrossRef]
- von Sassen, W.; Castro-Parra, M.; Musch, E.; Eichelbaum, M. Determination of isoniazid, acetylisoniazid, acetylhydrazine and diacetylhydrazine in biological fluids by high-performance liquid chromatography. J. Chromatogr. 1985, 338, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Satyaraddi, A.; Velpandian, T.; Sharma, S.K.; Vishnubhatla, S.; Sharma, A.; Sirohiwal, A.; Makharia, G.K.; Sinha, S.; Biswas, A.; Singh, S. Correlation of plasma anti-tuberculosis drug levels with subsequent development of hepatotoxicity. Int. J. Tuberc. Lung Dis. 2014, 18, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Zanrosso, C.W.; Emerenciano, M.; Gonçalves, B.A.d.A.; Faro, A.; Koifman, S.; Pombo-de-Oliveira, M.S. N -Acetyltransferase 2 Polymorphisms and Susceptibility to Infant Leukemia with Maternal Exposure to Dipyrone during Pregnancy. Cancer Epidemiol. Biomark. Prev. 2010, 19, 3037–3043. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (Years) | Child Weight (kg) | Dose RIF (mg) | Dose INH (mg) | Number of Dispersible Tablets |
|---|---|---|---|---|
| 0 | 4.6 | 75 | 50 | 1 |
| 1 | 9.2 | 150 | 100 | 2 |
| 2 | 12 | 150 | 100 | 2 |
| 3 | 14 | 225 | 150 | 3 |
| 4 | 16 | 225 | 150 | 3 |
| 5 | 18 | 300 | 200 | 4 |
| 6 | 21 | 300 | 200 | 4 |
| 7 | 23 | 300 | 200 | 4 |
| 8 | 26 | 450 | 300 | 6 |
| 9 | 30 | 450 | 300 | 6 |
| 10 | 34 | 450 | 300 | 6 |
| 11 | 39 | 600 | 300 | Adult tablet |
| Parameter (Unit) | Value for INH | Rationale/Reference(s) |
|---|---|---|
| 1. Physicochemical and Binding Properties | ||
| Molecular mass (g/mol) | 137.14 | From structure |
| Type of drug substance | Crystalline | [34] |
| Log P | 0.64 | [35] |
| pKa | 2.13(B), 3.81(B), 11.03(A) | Measured [35] |
| Intrinsic solubility (mg/mL) | 161 | [34] |
| Human blood-to-plasma ratio (Rbp) | 1.158 | AP v10.3 |
| fu, plasma (%) | 84 | APv10.3 |
| 2. Absorption | ||
| Human effective jejunal permeability (Peff) (×10−4 cm/s) | 6.7 | Calculated from ref. [36] |
| 3. Distribution | ||
| Method | Full body PBPK, Lukacova method of Kp prediction for all tissues | GastroPlus default method |
| 4. Metabolism | ||
| Km,u NAT2 (mg/L) | 5 | Fitted on INH profiles at different doses |
| Vmax NAT2 gut (mg/s), RA | 4.3 × 10−2 | Scaled from liver Vmax values |
| Vmax NAT2 gut (mg/s), IA | 2.72 × 10−2 | Scaled from liver Vmax values |
| Vmax NAT2 gut (mg/s), SA | 1.15 × 10−2 | Scaled from liver Vmax values |
| Vmax NAT2 PBPK (mg/s/mg-enz), RA | 4.94 × 10−3 | Fitted to subject B from ref. [37] |
| Vmax NAT2 PBPK (mg/s/mg-enz), IA | 3.13 × 10−3 | Average of RA and SA values |
| Vmax NAT2 PBPK (mg/s/mg-enz), SA | 1.32 × 10−3 | Fitted to subject B from ref. [37] |
| AcINH/INH | 1 | From structure |
| 5. Elimination | ||
| CLR (L/h/kg) | Given by fu,p × GFR | Default |
| Parameter (Unit) | Value for INH | Rationale/Reference(s) |
|---|---|---|
| 1. Physicochemical and Binding Properties | ||
| Molecular mass (g/mol) | 179.18 | APv10.3 |
| Log P | −0.35 | APv10.3 |
| pKa | 3.26(B), 9.27(A), 10.37(A) | APv10.3 |
| Intrinsic solubility (mg/mL) | 3.73 | APv10.3 |
| Human blood-to-plasma ratio (Rbp) | 0.86 | APv10.3 |
| Fu,plasma (%) | 80.94 | APv10.3 |
| 2. Absorption | ||
| Human effective jejunal permeability (Peff) (×10−4 cm/s) | 2.5 | APv10.3 |
| 3. Distribution | ||
| Method | Full body PBPK, Lukacova method of Kp prediction for all tissues | GastroPlus default method |
| 4. Metabolism | ||
| Vmax, 1A2 (mg/s/mg-enz) | 1.288 × 10−3 | Fitted to data from Bing et al. [38] |
| Km,u 1A2 (mg/L) | 115.4 | AP v10.3 |
| 5. Elimination | ||
| CLR (L/h/kg) | Given by fu,p × GFR | Default |
| Parameter (Unit) | Value for INH | Rationale/Reference(s) |
|---|---|---|
| 1. Physicochemical and Binding Properties | ||
| Molecular mass (g/mol) | 822.96 | From structure |
| Log P | 1.5 | APv10.3 predicted 2.528, close to value reported by Ermondi [39] |
| pKa | 2.97 (A), 7.5 (B) | Measured from [39] |
| Aqueous solubility (mg/mL) | 0.64 @ pH 5.5 | Measured from [40] |
| Human blood-to-plasma ratio (Rbp) | 0.738 | APv10.3 |
| fu, plasma (%) | 13.92 (human) | From [41] not far from 17.4% from APv10.3 or 13.3% measured in [42] |
| 2. Absorption | ||
| Human effective jejunal permeability (Peff) (×10−4 cm/s) | 2.11 | Scaled from Caco2 data from Biganzoli et al. [43] |
| 3. Distribution | ||
| Method | Full body PBPK, Lukacova method of Kp prediction for all tissues | Fitted to IV data from Wasserman et al. [44] |
| 4. Metabolism | ||
| Km,u CYP3A4 (mg/L) | 14.11 | APv10.3 calculated from APv10.3 value of 17.153 μM |
| Vmax CYP3A4 gut (mg/s) | 1.4 × 10−2 | Fitted to oral PK data from Loos [45] |
| Vmax CYP3A4 PBPK (mg/s/mg-enz) | 4.06 × 10−4 | Fitted to IV data from Wasserman et al. [44] |
| Km,u CES2 (mg/L) | 14.11 | Same as CYP3A4 |
| Vmax CES2 PBPK (mg/s/mg-enz) | 2.61 × 10−4 | Fitted to IV data from Wasserman et al. [44] |
| Vmax CES2 gut (mg/s) | 1.4 × 10−2 | Fitted to oral PK data from Loos [45] |
| 5. Elimination | ||
| CLR (L/h/kg) | Given by fu,p × GFR | Default |
| Analyte | Population | PK Parameter | PE (%) | AFE |
|---|---|---|---|---|
| INH | Adult | AUC | 3.8 | 0.97 |
| INH | Pediatric | AUC | 9.4 | 1.09 |
| INH | Adult | Cmax | 5.1 | 1.03 |
| INH | Pediatric | Cmax | 7.3 | 1.08 |
| RIF | Adult | AUC | 11.7 | 1.10 |
| RIF | Adult | Cmax | 10 | 1.15 |
| RIF | Pediatric | Plasma concentrations | 28.5 | 1.14 |
| Population | Analyte | PK Parameter | GMR | 90% CI | VBE Conclusion |
|---|---|---|---|---|---|
| SA | INH | AUCinf | 104.3 | 99.0–109.8 | Passed |
| IA | INH | AUCinf | 108.9 | 100–118.7 | Passed |
| RA | INH | AUCinf | 103.3 | 94.3–113.2 | Passed |
| SA | INH | Cmax | 111.7 | 105.6–118.1 | Passed |
| IA | INH | Cmax | 115.4 | 106.6–124.9 | Passed |
| RA | INH | Cmax | 108.4 | 98.5–119.2 | Passed |
| - | RIF | AUCinf | 99.5 | 87.3–113.4 | Passed |
| - | RIF | Cmax | 102 | 91.4–113.9 | Passed |
| Parameter | Description | Value |
|---|---|---|
| A | Probability to be over Cmax threshold 6.6 μg/mL 1 | 0.8 |
| B | Probability of positive 2-month sputum culture above threshold 2 | 0.01 |
| C | Probability of positive 2-month sputum culture under threshold 2 | 0.19 |
| D | Overall probability of positive 2-month sputum culture 3 | 0.046 |
| Parameter | Description | Value |
|---|---|---|
| A | Probability to be over AUC threshold 13 μg.h/mL 1 | 0.95 |
| B | Probability of poor therapy outcome above threshold 2 | 0.12 |
| C | Probability of poor therapy outcome under threshold 2 | 0.33 |
| D | Overall probability of poor therapy outcome 3 | 0.131 |
| Parameter | Description | Value for Brazil | ||
|---|---|---|---|---|
| A | Genotype | SA | IA | RA |
| B | Frequency of genotype 1 | 0.34 | 0.55 | 0.11 |
| C | Probability to be over AUC threshold 21.78 μg.h/mL 2 | 0.73 | 0.12 | 0 |
| D | Probability of DILI above threshold 3 | 0.324 | 0.324 | 0.324 |
| E | Probability of DILI under threshold 3 | 0.09 | 0.09 | 0.09 |
| F | Overall DILI in pure genotype 4 | 0.261 | 0.118 | 0.09 |
| G | Overall DILI risk in population 5 | 0.164 | ||
| Parameter | Description | Value |
|---|---|---|
| A | Probability to be over AUC threshold 64.49 μg.h/mL 1 | 0.057 |
| B | Probability of DILI above threshold 2 | 0.155 |
| C | Probability of DILI under threshold 2 | 0.0039 |
| D | Overall DILI risk in population 3 | 0.013 |
| Parameter | Description | Value |
|---|---|---|
| A | Probability to be over AUC threshold 82.01 μg.h/mL 1 | 0.023 |
| B | Probability of AKI above threshold 2 | 0.579 |
| C | Probability of AKI under threshold 2 | 0.081 |
| D | Overall AKI risk in population 3 | 0.092 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pepin, X.J.H.; Johansson Soares Medeiros, J.; Deris Prado, L.; Suarez Sharp, S. The Development of an Age-Appropriate Fixed Dose Combination for Tuberculosis Using Physiologically-Based Pharmacokinetic Modeling (PBBM) and Risk Assessment. Pharmaceutics 2024, 16, 1587. https://doi.org/10.3390/pharmaceutics16121587
Pepin XJH, Johansson Soares Medeiros J, Deris Prado L, Suarez Sharp S. The Development of an Age-Appropriate Fixed Dose Combination for Tuberculosis Using Physiologically-Based Pharmacokinetic Modeling (PBBM) and Risk Assessment. Pharmaceutics. 2024; 16(12):1587. https://doi.org/10.3390/pharmaceutics16121587
Chicago/Turabian StylePepin, Xavier J. H., Juliana Johansson Soares Medeiros, Livia Deris Prado, and Sandra Suarez Sharp. 2024. "The Development of an Age-Appropriate Fixed Dose Combination for Tuberculosis Using Physiologically-Based Pharmacokinetic Modeling (PBBM) and Risk Assessment" Pharmaceutics 16, no. 12: 1587. https://doi.org/10.3390/pharmaceutics16121587
APA StylePepin, X. J. H., Johansson Soares Medeiros, J., Deris Prado, L., & Suarez Sharp, S. (2024). The Development of an Age-Appropriate Fixed Dose Combination for Tuberculosis Using Physiologically-Based Pharmacokinetic Modeling (PBBM) and Risk Assessment. Pharmaceutics, 16(12), 1587. https://doi.org/10.3390/pharmaceutics16121587