
Delivery Strategies for Colchicine as a Critical Dose Drug: Reducing Toxicity and Enhancing Efficacy
Abstract
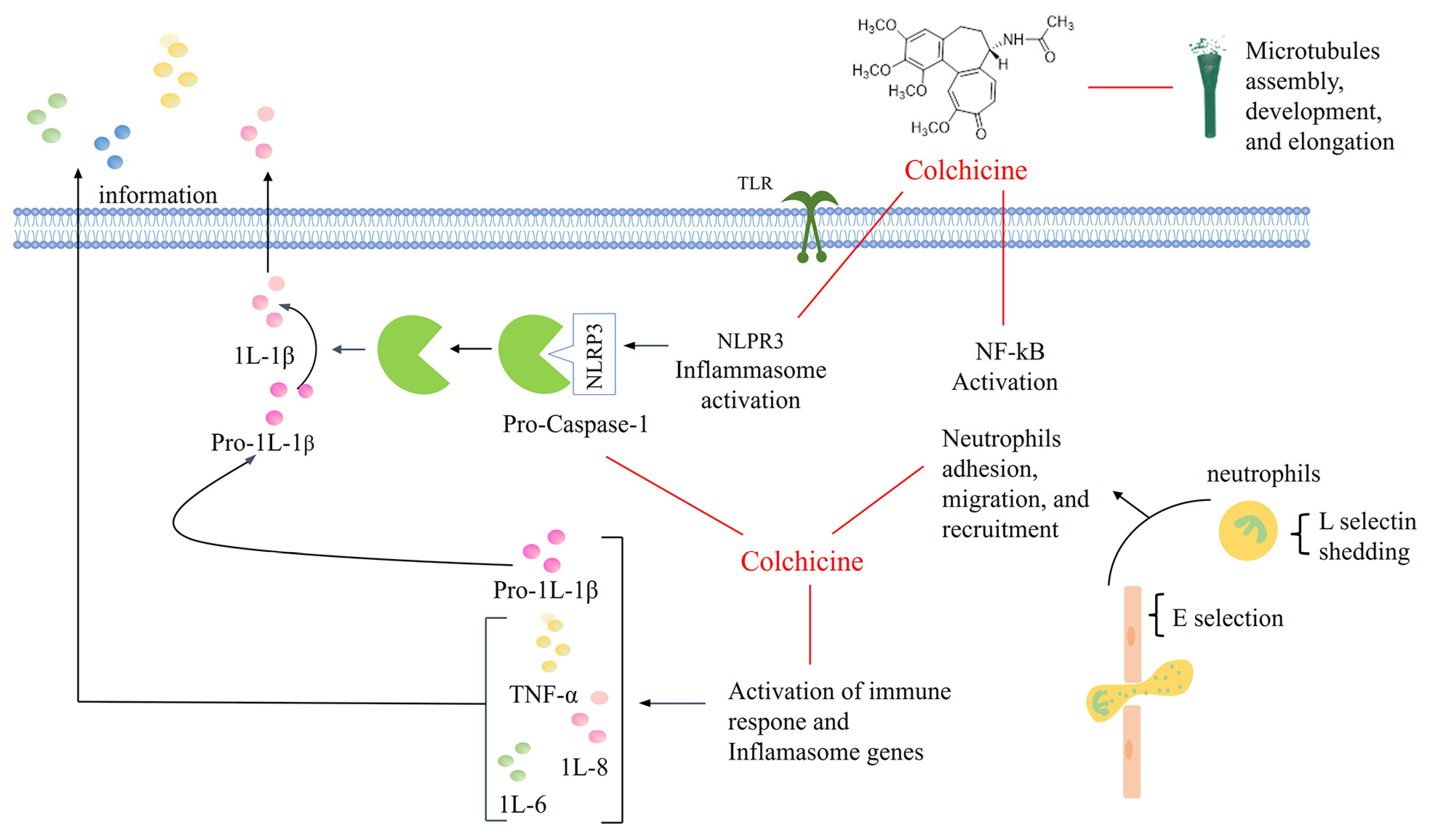
:1. Introduction
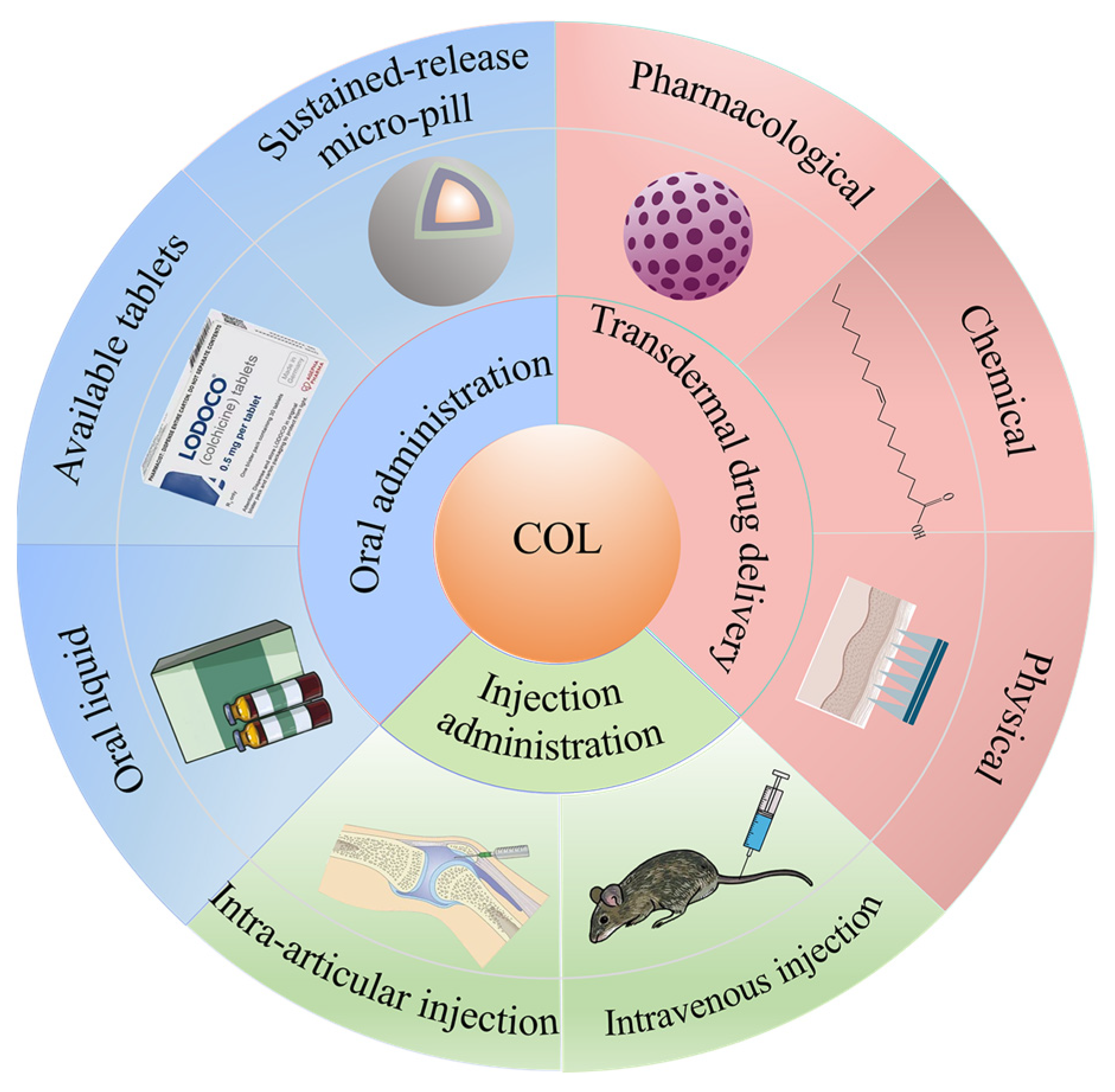
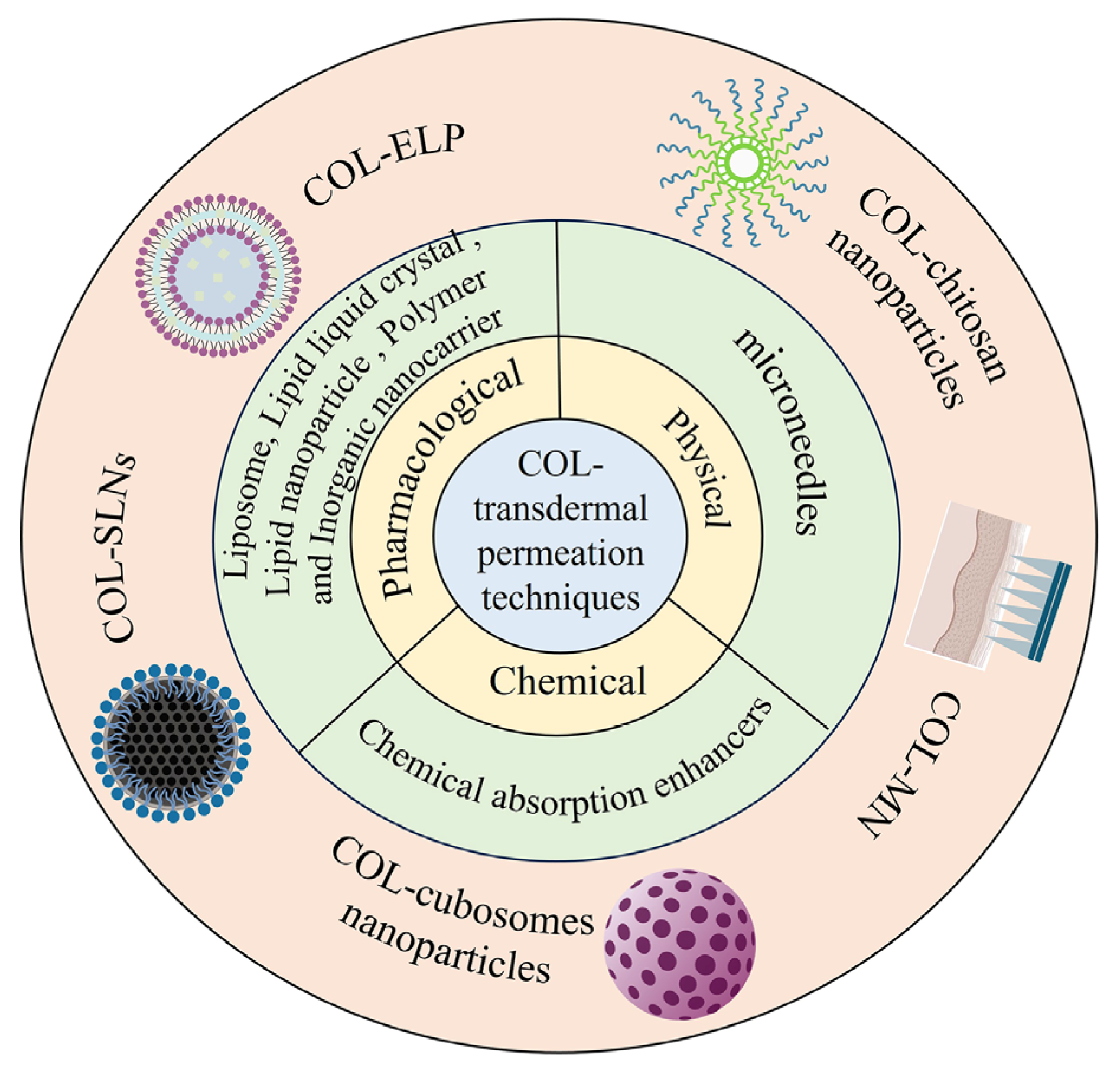
2. Transdermal Drug Delivery
2.1. Pharmacological Permeation Promotion
2.1.1. Liposome Technology
Elastic Liposomes
Alcohol Plasmid
Alcohol Transferosomes
2.1.2. Niosomes Technology
2.1.3. Lipid Liquid Crystal Technology
2.1.4. Lipid Nanoparticle Technology
2.1.5. Natural Nanocarrier Technology
2.1.6. Polymer Nanocarrier Technology
2.1.7. Inorganic Nanocarrier Technology
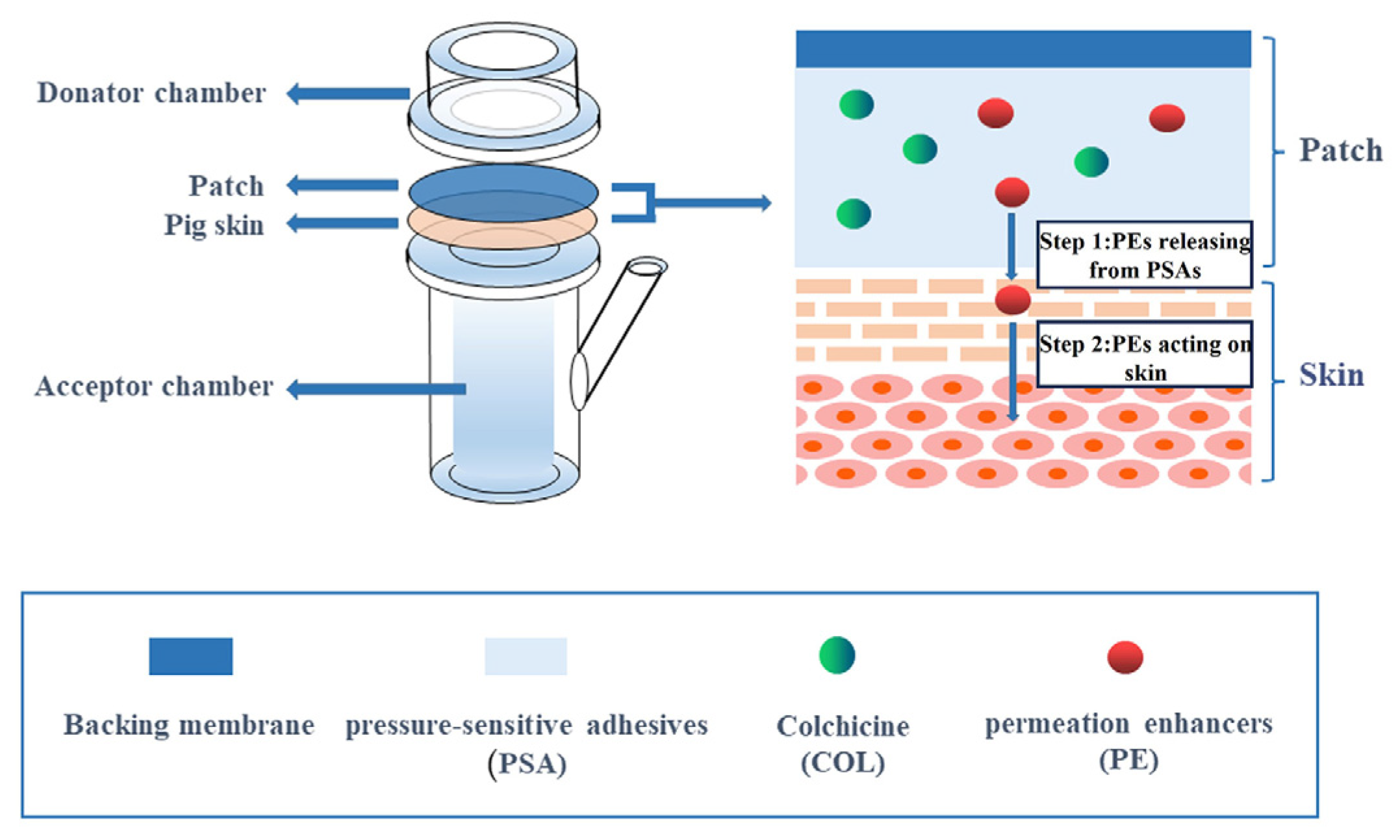
2.2. Chemical Permeation Promotion
2.3. Physical Permeation Promotion
3. Oral Administration
4. Injection Administration
4.1. Intravenous Injection
4.2. Intra-Articular Injection
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karamanou, M.; Tsoucalas, G.; Pantos, K.; Androutsos, G. Isolating Colchicine in 19th Century: An Old Drug Revisited. Curr. Pharm. Des. 2018, 24, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Deftereos, S.G.; Beerkens, F.J.; Shah, B.; Giannopoulos, G.; Vrachatis, D.A.; Giotaki, S.G.; Siasos, G.; Nicolas, J.; Arnott, C.; Patel, S.; et al. Colchicine in Cardiovascular Disease: In-Depth Review. Circulation 2022, 145, 61–78. [Google Scholar]
- Robinson, P.C.; Terkeltaub, R.; Pillinger, M.H.; Shah, B.; Karalis, V.; Karatza, E.; Liew, D.; Imazio, M.; Cornel, J.H.; Thompson, P.L.; et al. Consensus Statement Regarding the Efficacy and Safety of Long-Term Low-Dose Colchicine in Gout and Cardiovascular Disease. Am. J. Med. 2022, 135, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Bouabdallaoui, N.; Tardif, J.C. Repurposing Colchicine for Heart Disease. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Agepha Pharma US. FDA Approves First Anti-Inflammatory Drug for Cardiovascular Disease. Available online: https://us.agephapharma.com/blog/2023/06/20/us-fda-approves-first-anti-inflammatory-drug-for-cardiovascular-disease/ (accessed on 20 June 2023).
- Casey, A.; Quinn, S.; McAdam, B.; Kennedy, M.; Sheahan, R. Colchicine-regeneration of an old drug. Ir. J. Med. Sci. 2023, 192, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Angelidis, C.; Kotsialou, Z.; Kossyvakis, C.; Vrettou, A.R.; Zacharoulis, A.; Kolokathis, F.; Kekeris, V.; Giannopoulos, G. Colchicine Pharmacokinetics and Mechanism of Action. Curr. Pharm. Des. 2018, 24, 659–663. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Yao Hui, L.L.; Kraus, V.B. Colchicine–Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef]
- Tong, D.C.; Wilson, A.M.; Layland, J. Colchicine in cardiovascular disease: An ancient drug with modern tricks. Heart 2016, 102, 995–1002. [Google Scholar] [CrossRef]
- Korkmaz, S.; Erturan, I.; Naziroglu, M.; Uguz, A.C.; Cig, B.; Ovey, I.S. Colchicine modulates oxidative stress in serum and neutrophil of patients with Behcet disease through regulation of Ca(2)(+) release and antioxidant system. J. Membr. Biol. 2011, 244, 113–120. [Google Scholar] [CrossRef]
- Nuki, G. Colchicine: Its mechanism of action and efficacy in crystal-induced inflammation. Curr. Rheumatol. Rep. 2008, 10, 218–227. [Google Scholar] [CrossRef]
- Nasr, A.; Lauterio, T.J.; Davis, M.W. Unapproved drugs in the united states and the food and drug administration. Adv. Ther. 2011, 28, 842–856. [Google Scholar] [CrossRef] [PubMed]
- Habet, S. Narrow Therapeutic Index drugs: Clinical pharmacology perspective. J. Pharm. Pharmacol. 2021, 73, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, Y.; Aks, S.E.; Hutson, J.R.; Juurlink, D.N.; Nguyen, P.; Dubnov-Raz, G.; Pollak, U.; Koren, G.; Bentur, Y. Colchicine poisoning: The dark side of an ancient drug. Clin. Toxicol. 2010, 48, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Molad, Y. Update on colchicine and its mechanism of action. Curr. Rheumatol. Rep. 2002, 4, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Stamp, L.K.; Horsley, C.; Te Karu, L.; Dalbeth, N.; Barclay, M. Colchicine: The good, the bad, the ugly and how to minimize the risks. Rheumatology 2023, kead625. [Google Scholar] [CrossRef] [PubMed]
- Prausnitz, M.R.; Langer, R. Transdermal drug delivery. Nat. Biotechnol. 2008, 26, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Dai, J.; Ling, M.; Yu, L.; Yu, Z.; Tang, L. Delivery of triptolide: A combination of traditional Chinese medicine and nanomedicine. J. Nanobiotechnol. 2022, 20, 194. [Google Scholar] [CrossRef]
- Hwa, C.; Bauer, E.A.; Cohen, D.E. Skin biology. Dermatol. Ther. 2011, 24, 464–470. [Google Scholar] [CrossRef]
- Phatale, V.; Vaiphei, K.K.; Jha, S.; Patil, D.; Agrawal, M.; Alexander, A. Overcoming skin barriers through advanced transdermal drug delivery approaches. J. Control Release 2022, 351, 361–380. [Google Scholar] [CrossRef]
- Wiedersberg, S.; Guy, R.H. Transdermal drug delivery: 30+ years of war and still fighting! J. Control Release 2014, 190, 150–156. [Google Scholar] [CrossRef]
- Cheung, K.; Das, D.B. Microneedles for drug delivery: Trends and progress. Drug Deliv. 2016, 23, 2338–2354. [Google Scholar] [CrossRef]
- Singh, H.P.; Utreja, P.; Tiwary, A.K.; Jain, S. Elastic liposomal formulation for sustained delivery of colchicine: In vitro characterization and in vivo evaluation of anti-gout activity. AAPS J. 2009, 11, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, N.; Song, H.; Li, H.; Wen, J.; Tan, X.; Zheng, W. Design, characterization and comparison of transdermal delivery of colchicine via borneol-chemically-modified and borneol-physically-modified ethosome. Drug Deliv. 2019, 26, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Abdulbaqi, I.M.; Darwis, Y.; Assi, R.A.; Khan, N.A.K. Transethosomal gels as carriers for the transdermal delivery of colchicine: Statistical optimization, characterization, and ex vivo evaluation. Drug Des. Devel Ther. 2018, 12, 795–813. [Google Scholar] [CrossRef]
- Elsewedy, H.S.; Younis, N.S.; Shehata, T.M.; Mohamed, M.E.; Soliman, W.E. Enhancement of Anti-Inflammatory Activity of Optimized Niosomal Colchicine Loaded into Jojoba Oil-Based Emulgel Using Response Surface Methodology. Gels 2021, 8, 16. [Google Scholar] [CrossRef]
- Nasr, M.; Younes, H.; Abdel-Rashid, R.S. Formulation and evaluation of cubosomes containing colchicine for transdermal delivery. Drug Deliv. Transl. Res. 2020, 10, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.A.; Jalalpure, S.S.; Kempwade, A.A.; Peram, M.R. Fabrication and in-vivo evaluation of lipid nanocarriers based transdermal patch of colchicine. J. Drug Deliv. Sci. Technol. 2017, 41, 444–453. [Google Scholar] [CrossRef]
- Parashar, P.; Mazhar, I.; Kanoujia, J.; Yadav, A.; Kumar, P.; Saraf, S.A.; Saha, S. Appraisal of anti-gout potential of colchicine-loaded chitosan nanoparticle gel in uric acid-induced gout animal model. Arch. Physiol. Biochem. 2022, 128, 547–557. [Google Scholar] [CrossRef]
- Sadeghzadeh, F.; Ziaratnia, A.S.; Homayouni Tabrizi, M.; Torshizi, G.H.; Alhajamee, M.; Khademi, D. Nanofabrication of PLGA-PEG-chitosan-folic acid systems for delivery of colchicine to HT-29 cancer cells. J. Biomater. Sci. Polym. Ed. 2023, 34, 1–17. [Google Scholar] [CrossRef]
- Mohamed, A.L.; Elmotasem, H.; Salama, A.A.A. Colchicine mesoporous silica nanoparticles/hydrogel composite loaded cotton patches as a new encapsulator system for transdermal osteoarthritis management. Int. J. Biol. Macromol. 2020, 164, 1149–1163. [Google Scholar] [CrossRef]
- Lei, Y.; Yang, G.; Du, F.; Yi, J.; Quan, L.; Liu, H.; Zhou, X.; Gong, W.; Han, J.; Wang, Y.; et al. Formulation and Evaluation of a Drug-in-Adhesive Patch for Transdermal Delivery of Colchicine. Pharmaceutics 2022, 14, 2245. [Google Scholar] [CrossRef]
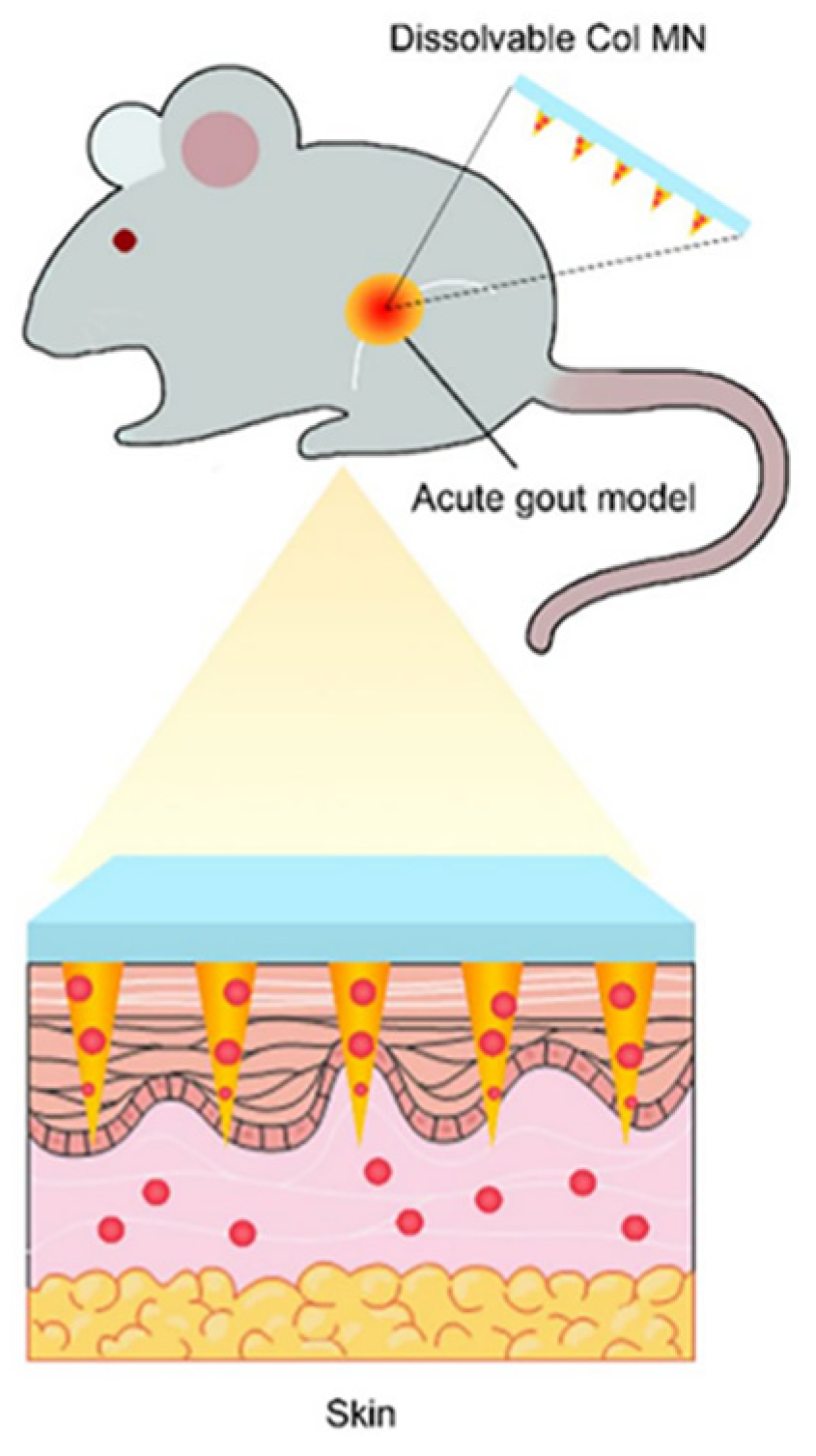
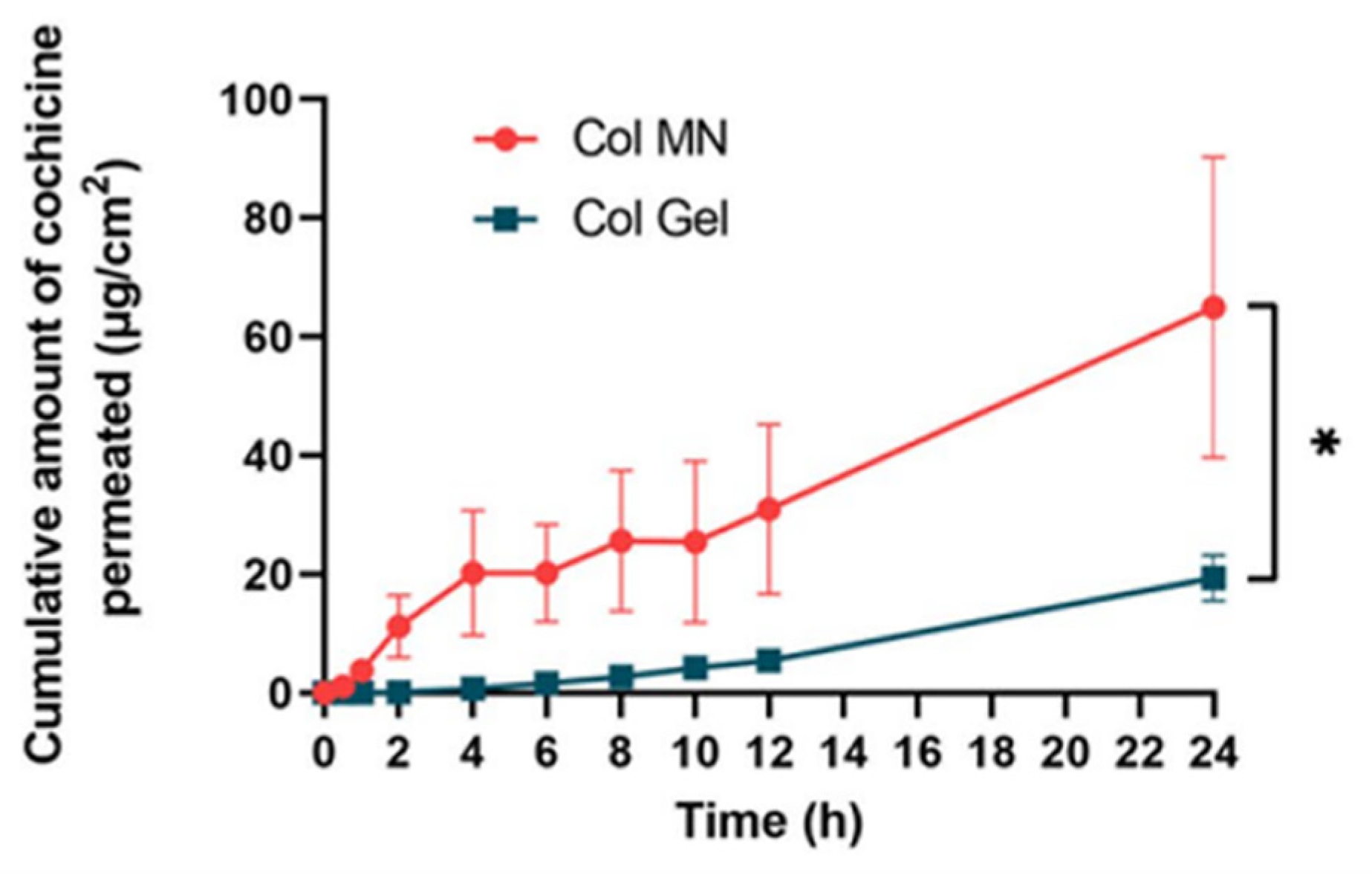
- Liu, Y.; Zhu, X.; Ji, S.; Huang, Z.; Zang, Y.; Ding, Y.; Zhang, J.; Ding, Z. Transdermal delivery of colchicine using dissolvable microneedle arrays for the treatment of acute gout in a rat model. Drug Deliv. 2022, 29, 2984–2994. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Z.; Huang, P.; Lin, J.; Li, J.; Shi, K.; Lin, J.; Hu, J.; Zhao, Z.; Yu, Y.; et al. Rapidly separating dissolving microneedles with sustained-release colchicine and stabilized uricase for simplified long-term gout management. Acta Pharm. Sin. B 2023, 13, 3454–3470. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Wang, W.; Ke, J.; Huang, S.; Wang, J.; Luo, C.; Li, X.; Zhang, K.; Liu, H.; Zheng, W.; et al. A mechanically tough and ultra-swellable microneedle for acute gout arthritis. Biomater. Sci. 2023, 11, 1714–1724. [Google Scholar] [CrossRef]
- Alkilani, A.Z.; Nasereddin, J.; Hamed, R.; Nimrawi, S.; Hussein, G.; Abo-Zour, H.; Donnelly, R.F. Beneath the Skin: A Review of Current Trends and Future Prospects of Transdermal Drug Delivery Systems. Pharmaceutics 2022, 14, 1152. [Google Scholar] [CrossRef] [PubMed]
- Araujo, V.H.S.; Delello Di Filippo, L.; Duarte, J.L.; Sposito, L.; Camargo, B.A.F.; da Silva, P.B.; Chorilli, M. Exploiting solid lipid nanoparticles and nanostructured lipid carriers for drug delivery against cutaneous fungal infections. Crit. Rev. Microbiol. 2021, 47, 79–90. [Google Scholar] [CrossRef]
- Silvestrini, A.V.P.; Caron, A.L.; Viegas, J.; Praca, F.G.; Bentley, M. Advances in lyotropic liquid crystal systems for skin drug delivery. Expert. Opin. Drug Deliv. 2020, 17, 1781–1805. [Google Scholar] [CrossRef]
- Kim, M.H.; Park, D.H.; Yang, J.H.; Choy, Y.B.; Choy, J.H. Drug-inorganic-polymer nanohybrid for transdermal delivery. Int. J. Pharm. 2013, 444, 120–127. [Google Scholar] [CrossRef]
- Carita, A.C.; Eloy, J.O.; Chorilli, M.; Lee, R.J.; Leonardi, G.R. Recent Advances and Perspectives in Liposomes for Cutaneous Drug Delivery. Curr. Med. Chem. 2018, 25, 606–635. [Google Scholar] [CrossRef]
- Sang, R.; Stratton, B.; Engel, A.; Deng, W. Liposome technologies towards colorectal cancer therapeutics. Acta Biomater. 2021, 127, 24–40. [Google Scholar] [CrossRef]
- Abri Aghdam, M.; Bagheri, R.; Mosafer, J.; Baradaran, B.; Hashemzaei, M.; Baghbanzadeh, A.; de la Guardia, M.; Mokhtarzadeh, A. Recent advances on thermosensitive and pH-sensitive liposomes employed in controlled release. J. Control Release 2019, 315, 1–22. [Google Scholar] [CrossRef]
- Firooz, A.; Nafisi, S.; Maibach, H.I. Novel drug delivery strategies for improving econazole antifungal action. Int. J. Pharm. 2015, 495, 599–607. [Google Scholar] [CrossRef]
- Ashtikar, M.; Nagarsekar, K.; Fahr, A. Transdermal delivery from liposomal formulations—Evolution of the technology over the last three decades. J. Control Release 2016, 242, 126–140. [Google Scholar] [CrossRef]
- Sala, M.; Diab, R.; Elaissari, A.; Fessi, H. Lipid nanocarriers as skin drug delivery systems: Properties, mechanisms of skin interactions and medical applications. Int. J. Pharm. 2018, 535, 1–17. [Google Scholar] [CrossRef]
- Sguizzato, M.; Esposito, E.; Cortesi, R. Lipid-Based Nanosystems as a Tool to Overcome Skin Barrier. Int. J. Mol. Sci. 2021, 22, 8319. [Google Scholar] [CrossRef] [PubMed]
- El Maghraby, G.M.; Williams, A.C.; Barry, B.W. Skin delivery of oestradiol from lipid vesicles: Importance of liposome structure. Int. J. Pharm. 2000, 204, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Souto, E.B.; Macedo, A.S.; Dias-Ferreira, J.; Cano, A.; Zielinska, A.; Matos, C.M. Elastic and Ultradeformable Liposomes for Transdermal Delivery of Active Pharmaceutical Ingredients (APIs). Int. J. Mol. Sci. 2021, 22, 9743. [Google Scholar] [CrossRef]
- Nayak, D.; Tippavajhala, V.K. A Comprehensive Review on Preparation, Evaluation and Applications of Deformable Liposomes. Iran. J. Pharm. Res. 2021, 20, 186–205. [Google Scholar] [CrossRef]
- Cevc, G.; Gebauer, D. Hydration-driven transport of deformable lipid vesicles through fine pores and the skin barrier. Biophys. J. 2003, 84, 1010–1024. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Singh, S.; Sharma, D.; Webster, T.J.; Shafaat, K.; Faruk, A. Elastic liposomes as novel carriers: Recent advances in drug delivery. Int. J. Nanomed. 2017, 12, 5087–5108. [Google Scholar] [CrossRef]
- Sharma, V.K.; Sarwa, K.K.; Mazumder, B. Fluidity enhancement: A critical factor for performance of liposomal transdermal drug delivery system. J. Liposome Res. 2014, 24, 83–89. [Google Scholar] [CrossRef]
- Amnuaikit, T.; Limsuwan, T.; Khongkow, P.; Boonme, P. Vesicular carriers containing phenylethyl resorcinol for topical delivery system; liposomes, transfersomes and invasomes. Asian J. Pharm. Sci. 2018, 13, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Jafari, A.; Daneshamouz, S.; Ghasemiyeh, P.; Mohammadi-Samani, S. Ethosomes as dermal/transdermal drug delivery systems: Applications, preparation and characterization. J. Liposome Res. 2023, 33, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Ita, K. Current Status of Ethosomes and Elastic Liposomes in Dermal and Transdermal Drug Delivery. Curr. Pharm. Des. 2016, 22, 5120–5126. [Google Scholar] [CrossRef] [PubMed]
- Abdulbaqi, I.M.; Darwis, Y.; Khan, N.A.; Assi, R.A.; Khan, A.A. Ethosomal nanocarriers: The impact of constituents and formulation techniques on ethosomal properties, in vivo studies, and clinical trials. Int. J. Nanomed. 2016, 11, 2279–2304. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Singh, H.; Bimbrawh, S.; Singh, S.K.; Gulati, M.; Vaidya, Y.; Kaur, P. Ethosomes and Transfersomes: Principles, Perspectives and Practices. Curr. Drug Deliv. 2017, 14, 613–633. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Xu, R.; Wang, Y.; Liu, J.; Wang, Z.; Zhai, G. Ethosomes for skin delivery of ropivacaine: Preparation, characterization and ex vivo penetration properties. J. Liposome Res. 2015, 25, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Opatha, S.A.T.; Titapiwatanakun, V.; Chutoprapat, R. Transfersomes: A Promising Nanoencapsulation Technique for Transdermal Drug Delivery. Pharmaceutics 2020, 12, 855. [Google Scholar] [CrossRef]
- Song, C.K.; Balakrishnan, P.; Shim, C.K.; Chung, S.J.; Chong, S.; Kim, D.D. A novel vesicular carrier, transethosome, for enhanced skin delivery of voriconazole: Characterization and in vitro/in vivo evaluation. Colloids Surf. B Biointerfaces 2012, 92, 299–304. [Google Scholar] [CrossRef]
- Adnan, M.; Afzal, O.; Altamimi, A.S.A.; Alamri, M.A.; Haider, T.; Faheem Haider, M. Development and optimization of transethosomal gel of apigenin for topical delivery: In-vitro, ex-vivo and cell line assessment. Int. J. Pharm. 2023, 631, 122506. [Google Scholar] [CrossRef]
- Garg, V.; Singh, H.; Bhatia, A.; Raza, K.; Singh, S.K.; Singh, B.; Beg, S. Systematic Development of Transethosomal Gel System of Piroxicam: Formulation Optimization, In Vitro Evaluation, and Ex Vivo Assessment. AAPS PharmSciTech 2017, 18, 58–71. [Google Scholar] [CrossRef]
- Chen, S.; Hanning, S.; Falconer, J.; Locke, M.; Wen, J. Recent advances in non-ionic surfactant vesicles (niosomes): Fabrication, characterization, pharmaceutical and cosmetic applications. Eur. J. Pharm. Biopharm. 2019, 144, 18–39. [Google Scholar] [CrossRef] [PubMed]
- Rajabalaya, R.; Musa, M.N.; Kifli, N.; David, S.R. Oral and transdermal drug delivery systems: Role of lipid-based lyotropic liquid crystals. Drug Des. Devel Ther. 2017, 11, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Lancelot, A.; Sierra, T.; Serrano, J.L. Nanostructured liquid-crystalline particles for drug delivery. Expert. Opin. Drug Deliv. 2014, 11, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Shan, Q.Q.; Jiang, X.J.; Wang, F.Y.; Shu, Z.X.; Gui, S.Y. Cubic and hexagonal liquid crystals as drug carriers for the transdermal delivery of triptolide. Drug Deliv. 2019, 26, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Wang, X.; Tian, C.; Liu, L.; Xia, M.; Jiang, J.; Gui, S. Dual drug-loaded cubic liquid crystal gels for transdermal delivery: Inner structure and percutaneous mechanism evaluations. Drug Dev. Ind. Pharm. 2019, 45, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Kozaka, S.; Wakabayashi, R.; Kamiya, N.; Goto, M. Design of Swollen Lipidic Cubic Phase to Increase Transcutaneous Penetration of Biomacromolecules. ACS Appl. Mater. Interfaces 2021, 13, 54753–54761. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Han, K.; Peng, X.; Yang, Z.; Qin, L.; Zhu, C.; Huang, X.; Shi, X.; Dian, L.; Lu, M.; et al. Nanostructured cubosomes as advanced drug delivery system. Curr. Pharm. Des. 2013, 19, 6290–6297. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.H.; Radtke, M.; Wissing, S.A. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv. Drug Deliv. Rev. 2002, 54 (Suppl. 1), S131–S155. [Google Scholar] [CrossRef]
- Carvajal-Vidal, P.; Fabrega, M.J.; Espina, M.; Calpena, A.C.; Garcia, M.L. Development of Halobetasol-loaded nanostructured lipid carrier for dermal administration: Optimization, physicochemical and biopharmaceutical behavior, and therapeutic efficacy. Nanomedicine 2019, 20, 102026. [Google Scholar] [CrossRef]
- Bagde, A.; Patel, K.; Kutlehria, S.; Chowdhury, N.; Singh, M. Formulation of topical ibuprofen solid lipid nanoparticle (SLN) gel using hot melt extrusion technique (HME) and determining its anti-inflammatory strength. Drug Deliv. Transl. Res. 2019, 9, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Tenchov, R.; Sasso, J.M.; Wang, X.; Liaw, W.-S.; Chen, C.-A.; Zhou, Q.A. Exosomes─Nature’s Lipid Nanoparticles, a Rising Star in Drug Delivery and Diagnostics. ACS Nano 2022, 16, 17802–17846. [Google Scholar] [CrossRef] [PubMed]
- Dad, H.A.; Gu, T.-W.; Zhu, A.-Q.; Huang, L.-Q.; Peng, L.-H. Plant Exosome-like Nanovesicles: Emerging Therapeutics and Drug Delivery Nanoplatforms. Mol. Ther. 2021, 29, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Giovannelli, L.; Bilia, A.R.; Margheri, F. Exosomes: Potential Next-Generation Nanocarriers for the Therapy of Inflammatory Diseases. Pharmaceutics 2023, 15, 2276. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhuang, X.; Deng, Z.-B.; Jiang, H.; Mu, J.; Wang, Q.; Xiang, X.; Guo, H.; Zhang, L.; Dryden, G.; et al. Targeted Drug Delivery to Intestinal Macrophages by Bioactive Nanovesicles Released from Grapefruit. Mol. Therapy 2014, 22, 522–534. [Google Scholar] [CrossRef]
- Cao, M.; Diao, N.; Cai, X.; Chen, X.; Xiao, Y.; Guo, C.; Chen, D.; Zhang, X. Plant exosome nanovesicles (PENs): Green delivery platforms. Mater. Horiz. 2023, 10, 3879–3894. [Google Scholar] [CrossRef] [PubMed]
- Banik, B.L.; Fattahi, P.; Brown, J.L. Polymeric nanoparticles: The future of nanomedicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 271–299. [Google Scholar] [CrossRef]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef]
- Mikusova, V.; Mikus, P. Advances in Chitosan-Based Nanoparticles for Drug Delivery. Int. J. Mol. Sci. 2021, 22, 9652. [Google Scholar] [CrossRef]
- Niu, J.; Yuan, M.; Chen, C.; Wang, L.; Tang, Z.; Fan, Y.; Liu, X.; Ma, Y.J.; Gan, Y. Berberine-Loaded Thiolated Pluronic F127 Polymeric Micelles for Improving Skin Permeation and Retention. Int. J. Nanomed. 2020, 15, 9987–10005. [Google Scholar] [CrossRef]
- Zorkina, Y.; Abramova, O.; Ushakova, V.; Morozova, A.; Zubkov, E.; Valikhov, M.; Melnikov, P.; Majouga, A.; Chekhonin, V. Nano Carrier Drug Delivery Systems for the Treatment of Neuropsychiatric Disorders: Advantages and Limitations. Molecules 2020, 25, 5294. [Google Scholar] [CrossRef]
- Ghosh, B.; Biswas, S. Polymeric micelles in cancer therapy: State of the art. J. Control Release 2021, 332, 127–147. [Google Scholar] [CrossRef]
- Zhang, S.; Gao, H.; Bao, G. Physical Principles of Nanoparticle Cellular Endocytosis. ACS Nano 2015, 9, 8655–8671. [Google Scholar] [CrossRef]
- Zhao, N.; Yan, L.; Zhao, X.; Chen, X.; Li, A.; Zheng, D.; Zhou, X.; Dai, X.; Xu, F.J. Versatile Types of Organic/Inorganic Nanohybrids: From Strategic Design to Biomedical Applications. Chem. Rev. 2019, 119, 1666–1762. [Google Scholar] [CrossRef]
- Sapino, S.; Oliaro-Bosso, S.; Zonari, D.; Zattoni, A.; Ugazio, E. Mesoporous silica nanoparticles as a promising skin delivery system for methotrexate. Int. J. Pharm. 2017, 530, 239–248. [Google Scholar] [CrossRef]
- Tang, F.; Li, L.; Chen, D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv. Mater. 2012, 24, 1504–1534. [Google Scholar] [CrossRef] [PubMed]
- Sapino, S.; Ugazio, E.; Gastaldi, L.; Miletto, I.; Berlier, G.; Zonari, D.; Oliaro-Bosso, S. Mesoporous silica as topical nanocarriers for quercetin: Characterization and in vitro studies. Eur. J. Pharm. Biopharm. 2015, 89, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, W.; Xue, C.; Zhou, S.; Lan, F.; Bi, L.; Xu, H.; Yang, X.; Zeng, F.D. Toxicity and penetration of TiO2 nanoparticles in hairless mice and porcine skin after subchronic dermal exposure. Toxicol. Lett. 2009, 191, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Huang, F.; Zhang, Q.; Liu, J.; Quan, D.; Song, W. Molecular perspective of efficiency and safety problems of chemical enhancers: Bottlenecks and recent advances. Drug Deliv. Transl. Res. 2022, 12, 1376–1394. [Google Scholar] [CrossRef] [PubMed]
- Kovacik, A.; Kopecna, M.; Vavrova, K. Permeation enhancers in transdermal drug delivery: Benefits and limitations. Expert. Opin. Drug Deliv. 2020, 17, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Tang, X.; Jia, Y.; Ho, C.T.; Huang, Q. Applications and delivery mechanisms of hyaluronic acid used for topical/transdermal delivery—A review. Int. J. Pharm. 2020, 578, 119127. [Google Scholar] [CrossRef]
- Moniz, T.; Costa Lima, S.A.; Reis, S. Marine polymeric microneedles for transdermal drug delivery. Carbohydr. Polym. 2021, 266, 118098. [Google Scholar] [CrossRef]
- Yang, D.; Chen, M.; Sun, Y.; Jin, Y.; Lu, C.; Pan, X.; Quan, G.; Wu, C. Microneedle-mediated transdermal drug delivery for treating diverse skin diseases. Acta Biomater. 2021, 121, 119–133. [Google Scholar] [CrossRef]
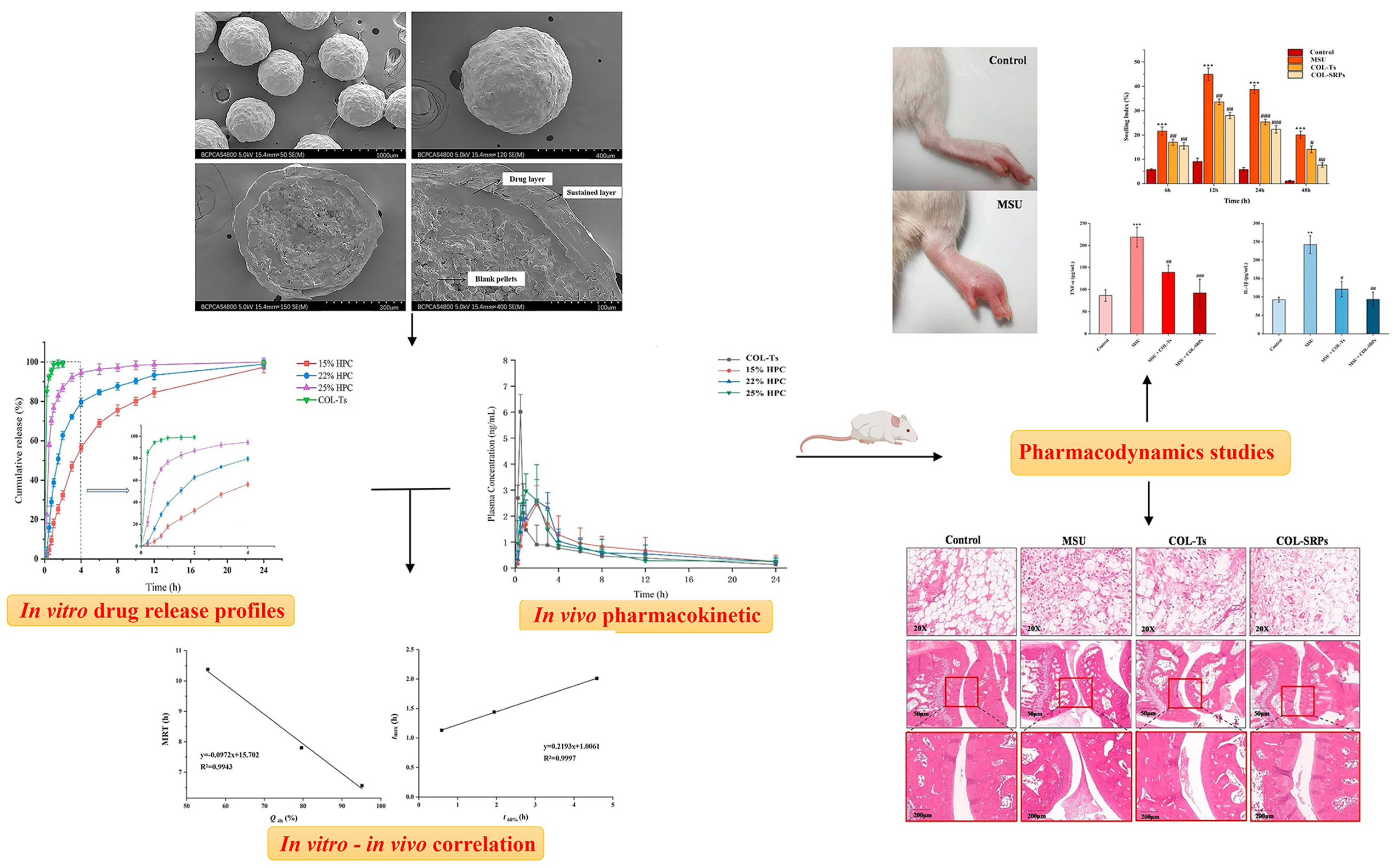
- Lv, B.; Yang, G.; Wei, Y.; Lei, Y.; Ding, Y.; Gong, W.; Wang, Y.; Gao, C.; Han, C. A pharmacokinetic and pharmacodynamic evaluation of colchicine sustained-release pellets for preventing gout. J. Drug Deliv. Sci. Technol. 2022, 67, 103051. [Google Scholar] [CrossRef]
- AbouAitah, K.; Hassan, H.A.; Swiderska-Sroda, A.; Gohar, L.; Shaker, O.G.; Wojnarowicz, J.; Opalinska, A.; Smalc-Koziorowska, J.; Gierlotka, S.; Lojkowski, W. Targeted Nano-Drug Delivery of Colchicine against Colon Cancer Cells by Means of Mesoporous Silica Nanoparticles. Cancers 2020, 12, 144. [Google Scholar] [CrossRef] [PubMed]
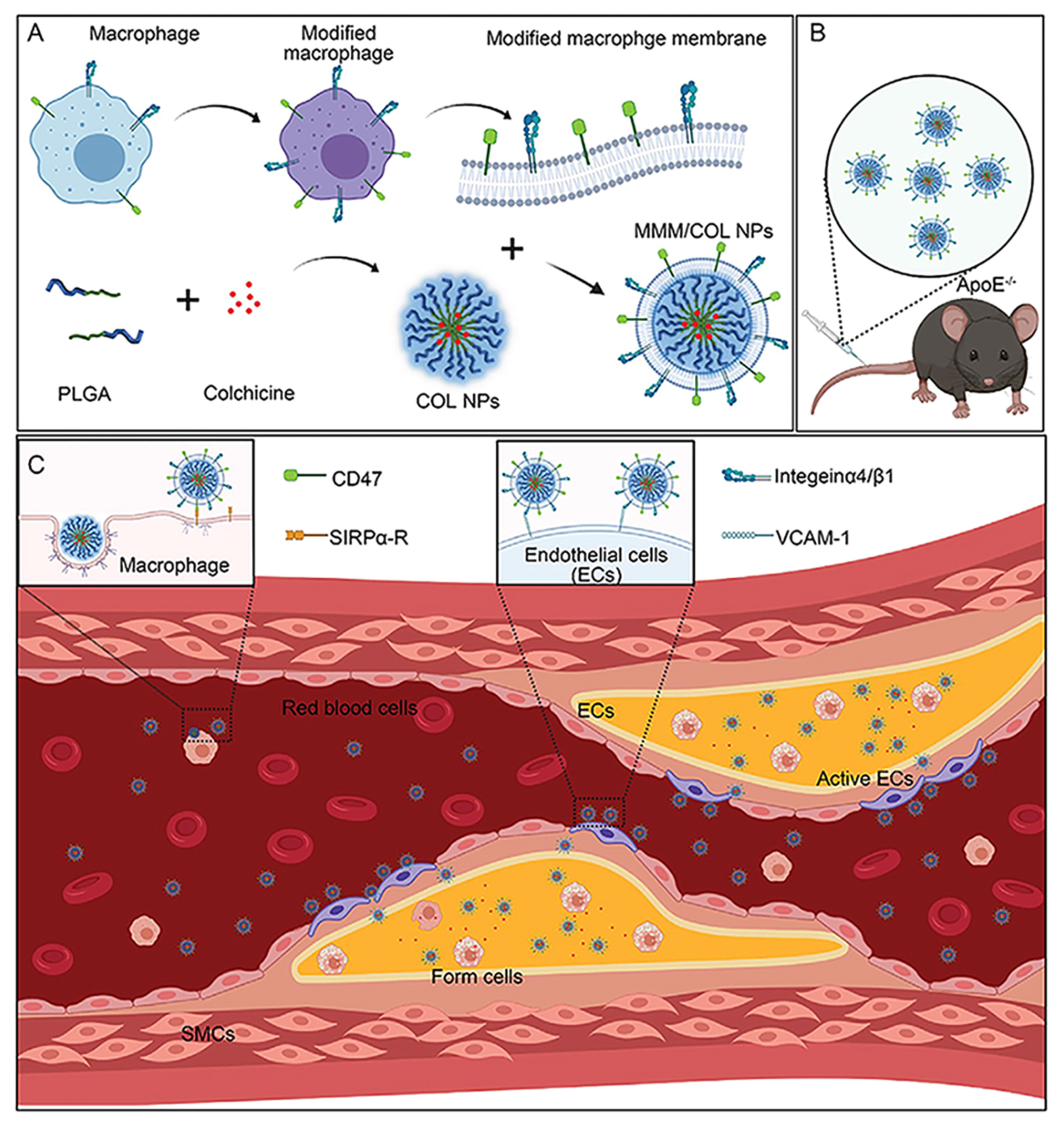
- Li, Y.; Che, J.; Chang, L.; Guo, M.; Bao, X.; Mu, D.; Sun, X.; Zhang, X.; Lu, W.; Xie, J. CD47- and Integrin alpha4/beta1-Comodified-Macrophage-Membrane-Coated Nanoparticles Enable Delivery of Colchicine to Atherosclerotic Plaque. Adv. Healthc. Mater. 2022, 11, e2101788. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef]
- Maduro, A.T.; Luís, C.; Soares, R. Ageing, cellular senescence and the impact of diet: An overview. Porto Biomed. J. 2021, 6, e120. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Khan, D.; Hussain, S.M.; Gerdes, N.; Hagenbeck, C.; Rana, M.; Cornelius, J.F.; Muhammad, S. Colchicine prevents oxidative stress-induced endothelial cell senescence via blocking NF-κB and MAPKs: Implications in vascular diseases. J. Inflamm. 2023, 20, 41. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Du, Q.; Yang, Y.; Wang, J.; Dou, S.; Liu, C.; Duan, J. The protective effect of Luteolin on myocardial ischemia/reperfusion (I/R) injury through TLR4/NF-kappaB/NLRP3 inflammasome pathway. Biomed. Pharmacother. 2017, 91, 1042–1052. [Google Scholar] [CrossRef]
- Wang, L.; Peng, Y.; Song, L.; Xia, D.; Li, C.; Li, Z.; Li, Q.; Yu, A.; Lu, C.; Wang, Y. Colchicine-Containing Nanoparticles Attenuates Acute Myocardial Infarction Injury by Inhibiting Inflammation. Cardiovasc. Drugs Ther. 2022, 36, 1075–1089. [Google Scholar] [CrossRef]
- Petersen, S.K.; Hansen, I.; Andreasen, R.A. Low frequency of septic arthritis after arthrocentesis and intra-articular glucocorticoid injection. Scand. J. Rheumatol. 2019, 48, 393–397. [Google Scholar] [CrossRef]
- Migliore, A.; Paoletta, M.; Moretti, A.; Liguori, S.; Iolascon, G. The perspectives of intra-articular therapy in the management of osteoarthritis. Expert. Opin. Drug Deliv. 2020, 17, 1213–1226. [Google Scholar] [CrossRef]
- De Zordo, T.; Mur, E.; Bellmann-Weiler, R.; Sailer-Hock, M.; Chhem, R.; Feuchtner, G.M.; Jaschke, W.; Klauser, A.S. US guided injections in arthritis. Eur. J. Radiol. 2009, 71, 197–203. [Google Scholar] [CrossRef]
- Mello, S.B.; Tavares, E.R.; Bulgarelli, A.; Bonfa, E.; Maranhao, R.C. Intra-articular methotrexate associated to lipid nanoemulsions: Anti-inflammatory effect upon antigen-induced arthritis. Int. J. Nanomed. 2013, 8, 443–449. [Google Scholar] [CrossRef]
- Zoghebi, K.A.; Bousoik, E.; Parang, K.; Elsaid, K.A. Design and Biological Evaluation of Colchicine-CD44-Targeted Peptide Conjugate in an In Vitro Model of Crystal Induced Inflammation. Molecules 2019, 25, 46. [Google Scholar] [CrossRef] [PubMed]
- Aboumanei, M.H.; Fayez, H. Intra-articular formulation of colchicine loaded nanoemulsion systems for enhanced locoregional drug delivery: In vitro characterization, (99m)Tc coupling and in vivo biodistribution studies. Drug Dev. Ind. Pharm. 2021, 47, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z.; Xie, K.; Zhao, X.; Jiang, X.; Chen, B.; Han, Y.; Lu, Y.; Huang, L.; Zhang, W.; et al. High-Efficiency Cellular Reprogramming by Nanoscale Puncturing. Nano Lett. 2020, 20, 5473–5481. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transdermal Osmotic Technology | Types | Benefits | Carriers | Reference |
|---|---|---|---|---|
| Pharmacological permeation promotion technology | Liposome technology | Favorable skin affinity and enhancing stratum corneum hydration | COL-ELP | [23] |
| COL-βCD-ELP | [23] | |||
| COL-alcohol plasmids | [24] | |||
| COL-alcohol transporters | [25] | |||
| Niosomes technology | Easily accessible and high stability | COL-vesicles | [26] | |
| Lipid liquid crystal technology | Multi-drug loading with high capacity | COL-cubosomes nanoparticles | [27] | |
| Lipid nanoparticle technology | Strong lipophilicity and small particle size | COL-SLNs | [28] | |
| Polymer nanocarrier technology | Solubilizes and improving release rate | COL-chitosan nanoparticles | [29] | |
| COL-PPCF-NPs | [30] | |||
| Inorganic nanocarrier technology | High drug loading capacity and controllability | COL-MSNs | [31] | |
| Chemical permeation promotion technology | Chemical absorption enhancers | Simple structure and high compliance for PSA | COL-DIA patch | [32] |
| Physical permeation promotion technology | microneedles | Expanding the range of drugs and reducing side effects | COL-MN | [33] |
| UAO-LPO/COL-MN | [34] | |||
| COL-HMNs | [35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, Y.; Yang, Y.; Yang, G.; Li, A.; Yang, Y.; Wang, Y.; Gao, C. Delivery Strategies for Colchicine as a Critical Dose Drug: Reducing Toxicity and Enhancing Efficacy. Pharmaceutics 2024, 16, 222. https://doi.org/10.3390/pharmaceutics16020222
Lei Y, Yang Y, Yang G, Li A, Yang Y, Wang Y, Gao C. Delivery Strategies for Colchicine as a Critical Dose Drug: Reducing Toxicity and Enhancing Efficacy. Pharmaceutics. 2024; 16(2):222. https://doi.org/10.3390/pharmaceutics16020222
Chicago/Turabian StyleLei, Yaran, Yulu Yang, Guobao Yang, Ao Li, Yang Yang, Yuli Wang, and Chunsheng Gao. 2024. "Delivery Strategies for Colchicine as a Critical Dose Drug: Reducing Toxicity and Enhancing Efficacy" Pharmaceutics 16, no. 2: 222. https://doi.org/10.3390/pharmaceutics16020222
APA StyleLei, Y., Yang, Y., Yang, G., Li, A., Yang, Y., Wang, Y., & Gao, C. (2024). Delivery Strategies for Colchicine as a Critical Dose Drug: Reducing Toxicity and Enhancing Efficacy. Pharmaceutics, 16(2), 222. https://doi.org/10.3390/pharmaceutics16020222