
Can 3D Printed Tablets Be Bioequivalent and How to Test It: A PBPK Model Based Virtual Bioequivalence Study for Ropinirole Modified Release Tablets

 , , , , ,
, , , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. 3D Printing and Dissolution Testing
2.2. PBPK Model Building, Verification, and Application
2.3. VBE Study Power Calculations
2.4. Virtual Bioequivalence Trial Design
3. Results
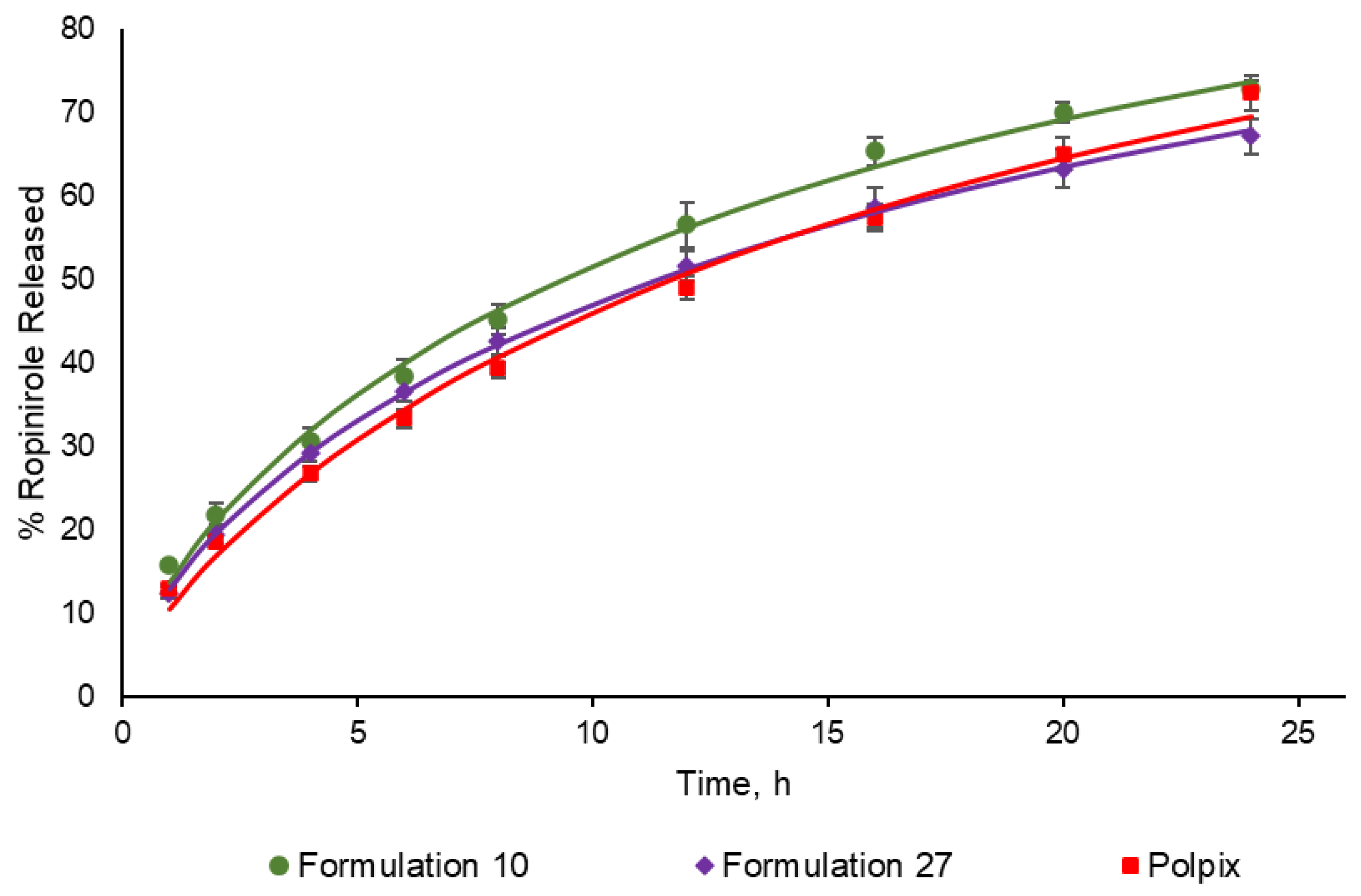
3.1. Dissolution Profiles
3.2. Simulation of Ropinirole Interaction with Ciprofloxacin
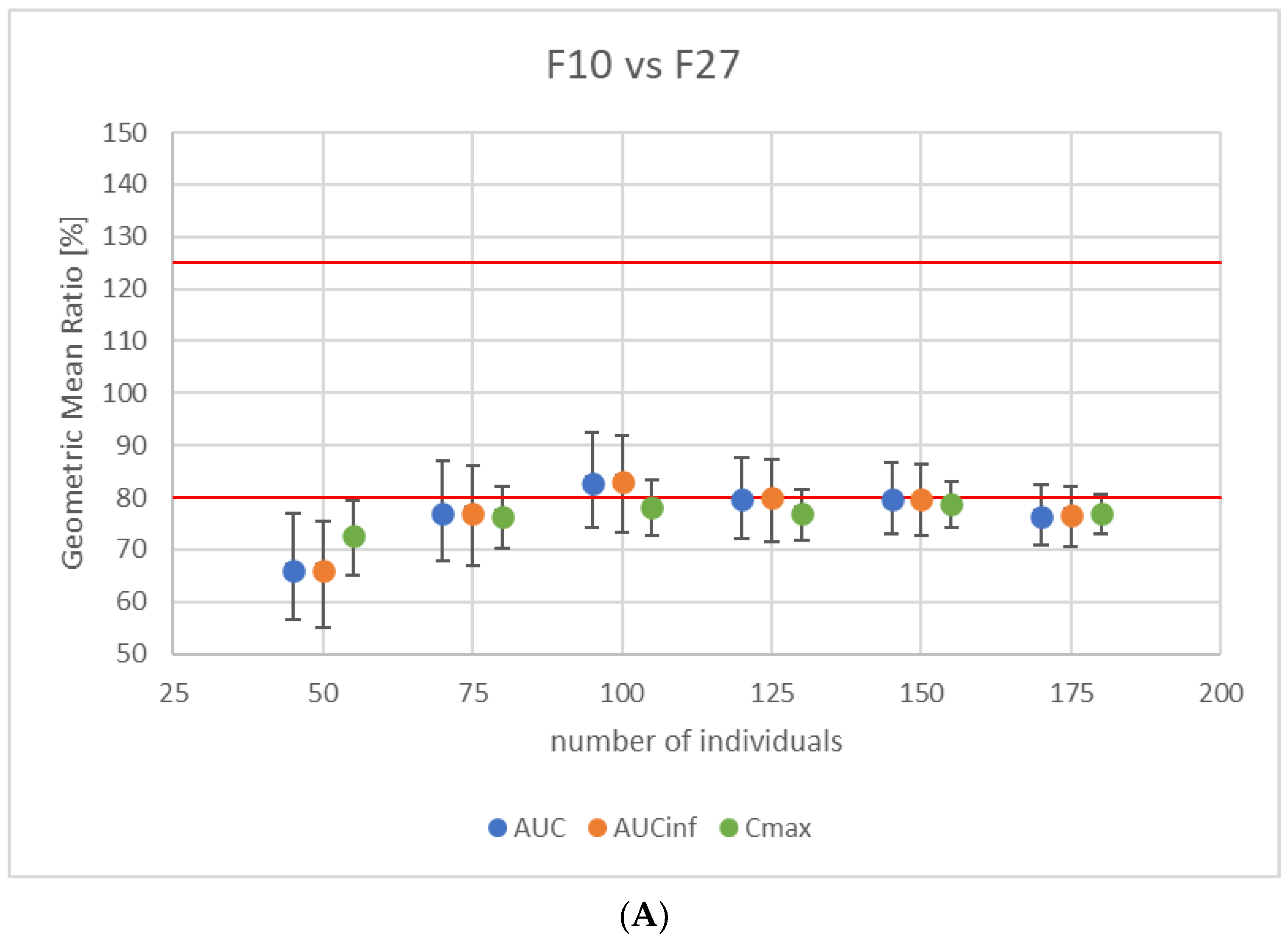
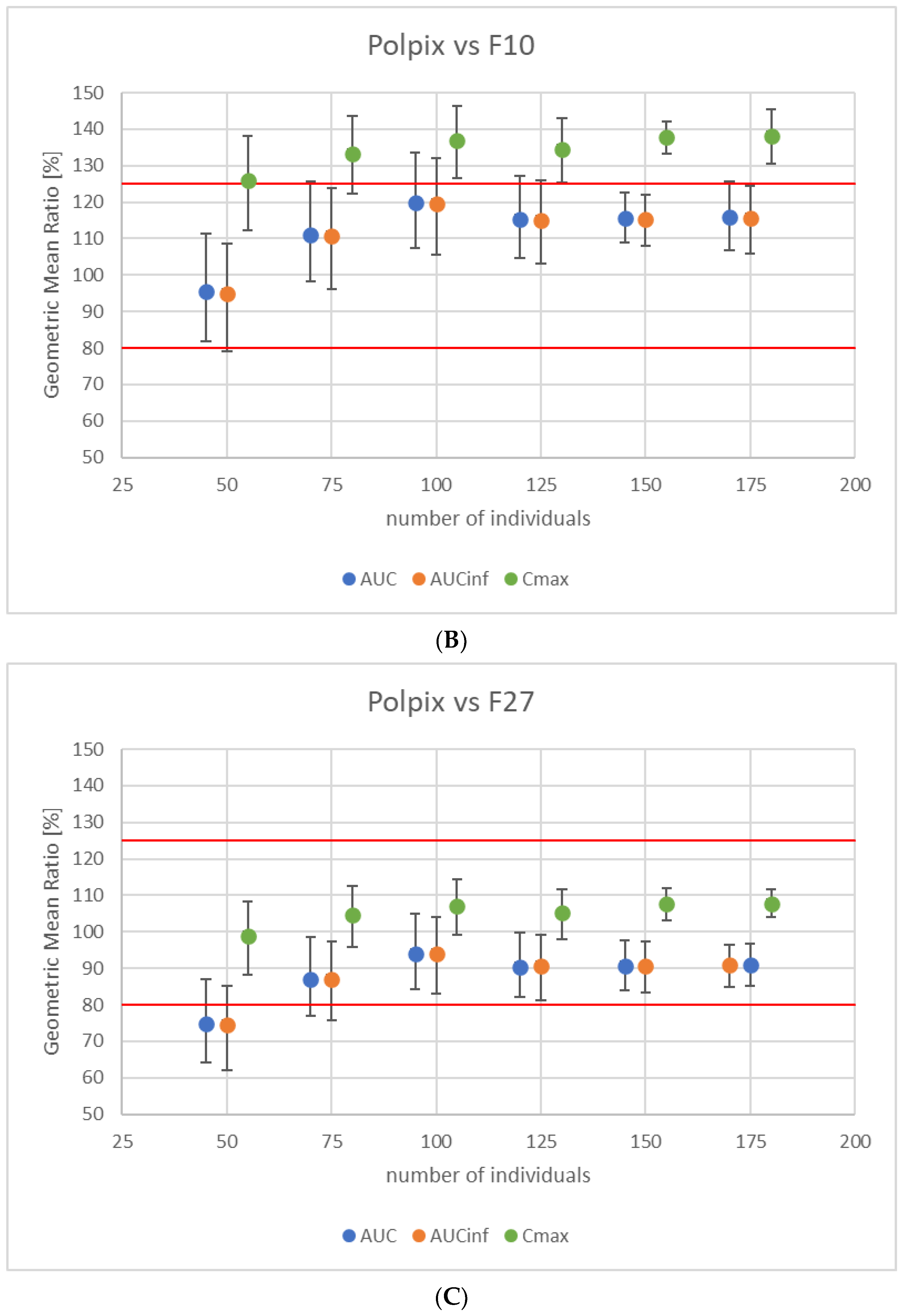
3.3. Study Power Calculation
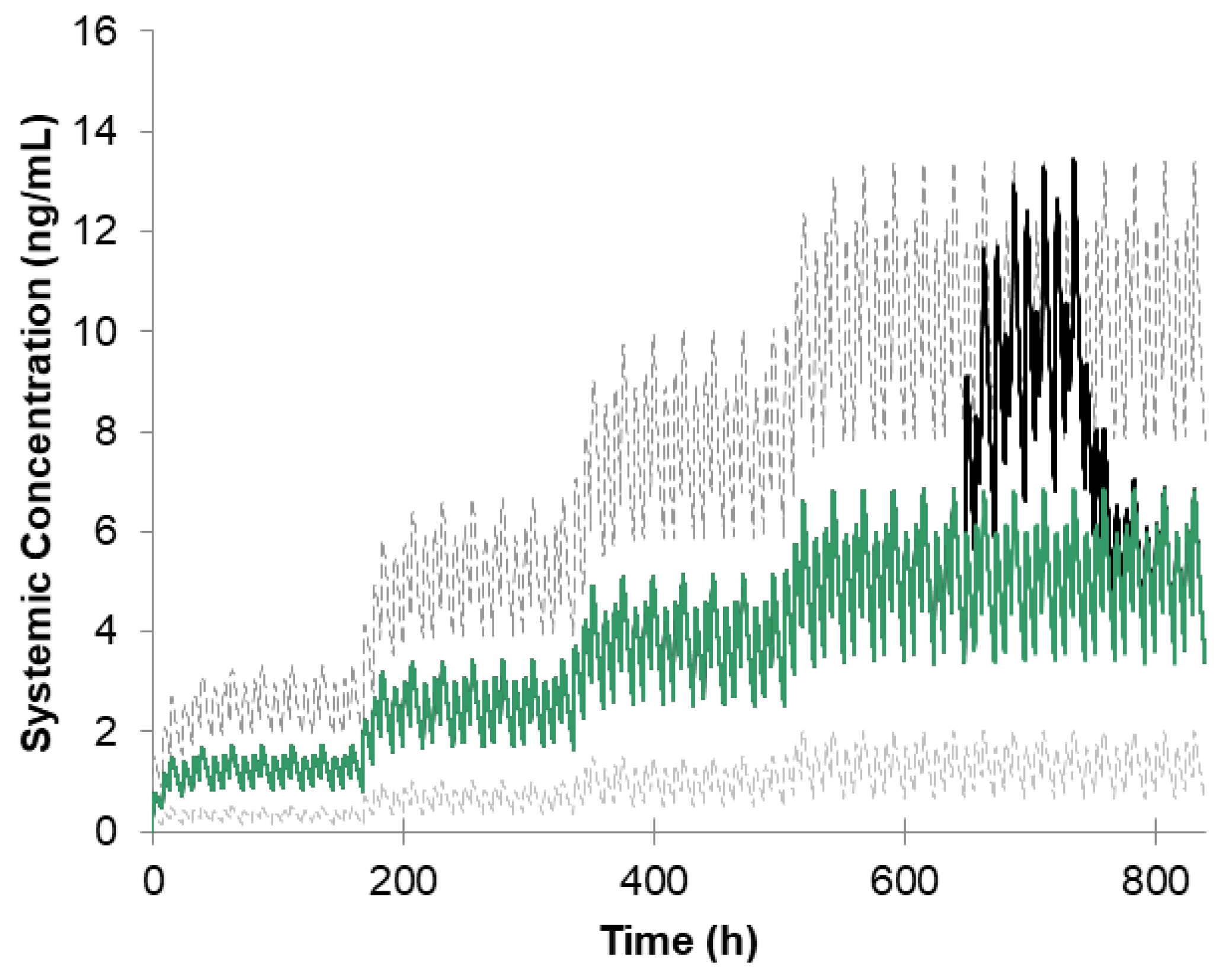
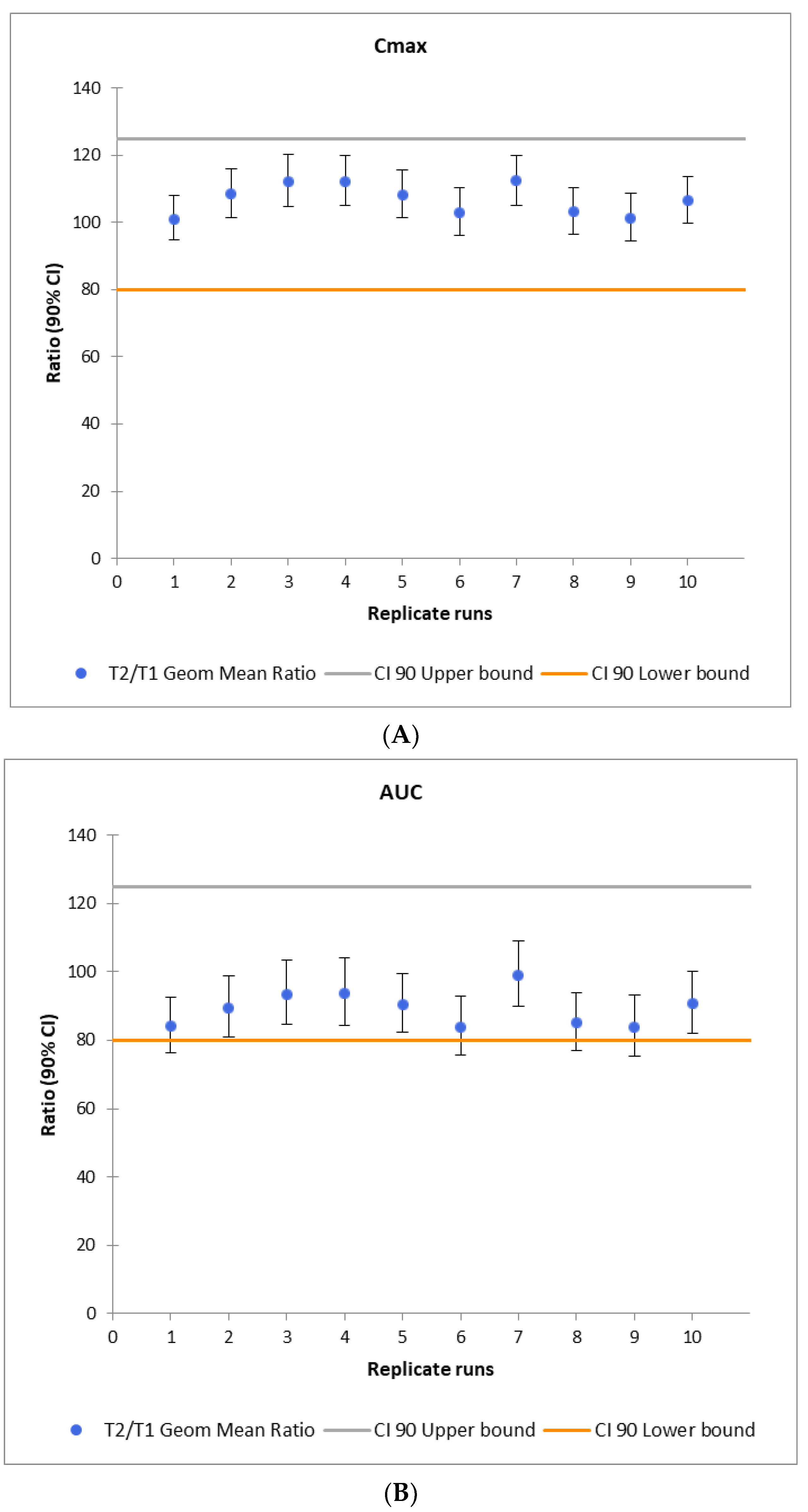
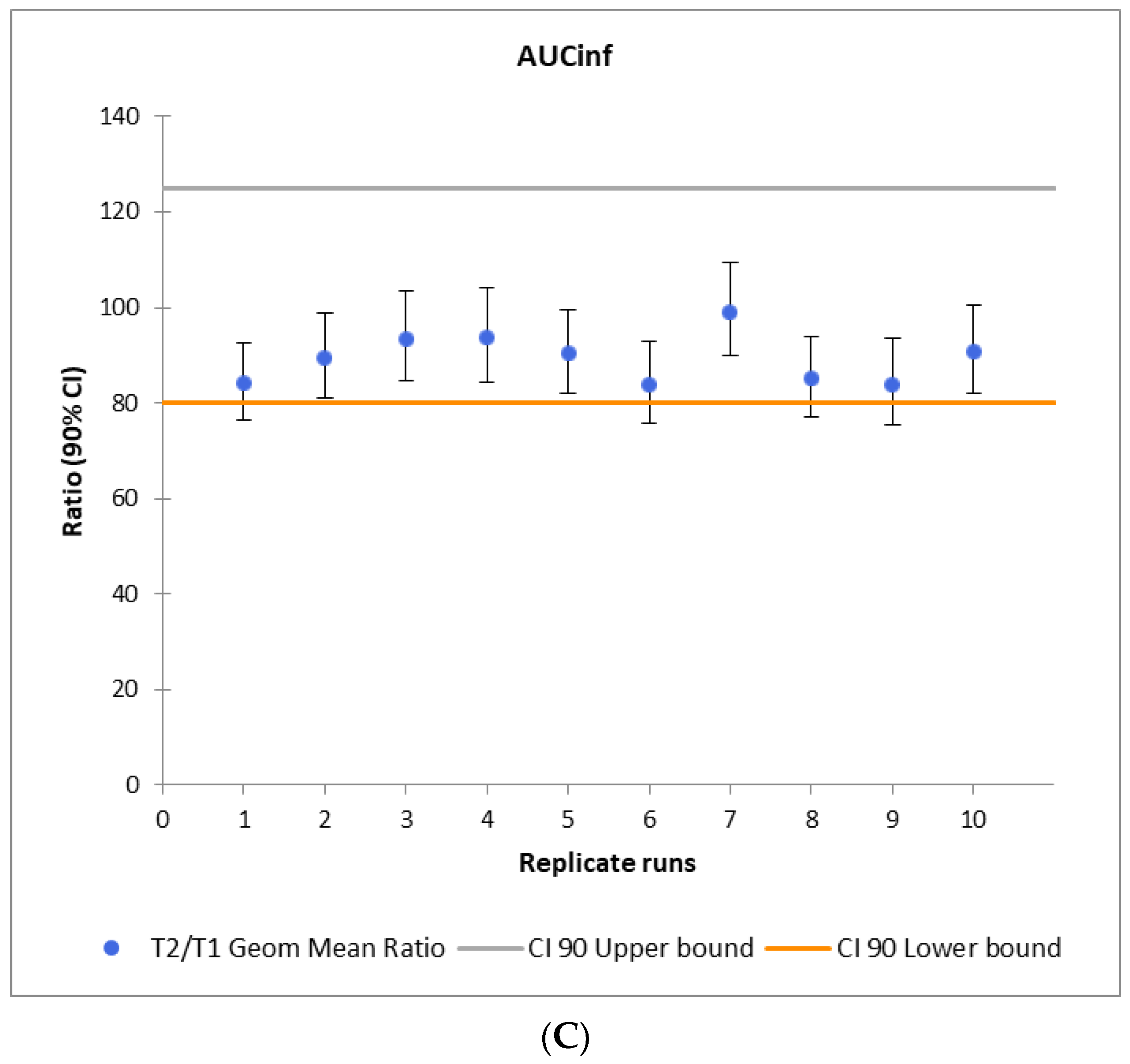
3.4. Virtual Bioequivalence Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chetty, M.; Rose, R.H.; Abduljalil, K.; Patel, N.; Lu, G.; Cain, T.; Jamei, M.; Rostami-Hodjegan, A. Applications of linking PBPK and PD models to predict the impact of genotypic variability, formulation differences, differences in target binding capacity and target site drug concentrations on drug responses and variability. Front. Pharmacol. 2014, 5, 258. [Google Scholar] [CrossRef]
- De Sousa Mendes, M.; Chetty, M. Are Standard Doses of Renally-Excreted Antiretrovirals in Older Patients Appropriate: A PBPK Study Comparing Exposures in the Elderly Population with Those in Renal Impairment. Drugs R D 2019, 19, 339–350. [Google Scholar] [CrossRef]
- Xie, H.-G.; Frueh, F.W. Pharmacogenomics steps toward personalized medicine. Pers. Med. 2005, 2, 325–337. [Google Scholar] [CrossRef]
- Gonzalez, D.; Rao, G.G.; Bailey, S.C.; Brouwer, K.L.R.; Cao, Y.; Crona, D.J.; Kashuba, A.D.M.; Lee, C.R.; Morbitzer, K.; Patterson, J.H.; et al. Precision Dosing: Public Health Need, Proposed Framework, and Anticipated Impact. Clin. Transl. Sci. 2017, 10, 443–454. [Google Scholar] [CrossRef]
- El Aita, I.; Breitkreutz, J.; Quodbach, J. On-demand manufacturing of immediate release levetiracetam tablets using pressure-assisted microsyringe printing. Eur. J. Pharm. Biopharm. 2019, 134, 29–36. [Google Scholar] [CrossRef]
- Skowyra, J.; Pietrzak, K.; Alhnan, M.A. Fabrication of extended-release patient-tailored prednisolone tablets via fused deposition modelling (FDM) 3D printing. Eur. J. Pharm. Sci. 2015, 68, 11–17. [Google Scholar] [CrossRef]
- Lafeber, I.; Tichem, J.M.; Ouwerkerk, N.; van Unen, A.D.; van Uitert, J.J.D.; Bijleveld-Olierook, H.C.M.; Kweekel, D.M.; Zaal, W.M.; Le Brun, P.P.H.; Guchelaar, H.J.; et al. 3D printed furosemide and sildenafil tablets: Innovative production and quality control. Int. J. Pharm. 2021, 603, 120694. [Google Scholar] [CrossRef] [PubMed]
- Karalia, D.; Siamidi, A.; Karalis, V.; Vlachou, M. 3D-Printed Oral Dosage Forms: Mechanical Properties, Computational Approaches and Applications. Pharmaceutics 2021, 13, 1401. [Google Scholar] [CrossRef] [PubMed]
- FDA. Guidance for Industry Bioavailability Studies Submitted in NDAs or INDs—General Considerations, U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). 2022. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioavailability-studies-submitted-ndas-or-inds-general-considerations (accessed on 23 August 2023).
- Rowland, M.; Peck, C.; Tucker, G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 45–73. [Google Scholar] [CrossRef] [PubMed]
- Templeton, I.E.; Jones, N.S.; Musib, L. Pediatric Dose Selection and Utility of PBPK in Determining Dose. AAPS J. 2018, 20, 31. [Google Scholar] [CrossRef] [PubMed]
- Heimbach, T.; Chen, Y.; Chen, J.; Dixit, V.; Parrott, N.; Peters, S.A.; Poggesi, I.; Sharma, P.; Snoeys, J.; Shebley, M.; et al. Physiologically-Based Pharmacokinetic Modeling in Renal and Hepatic Impairment Populations: A Pharmaceutical Industry Perspective. Clin. Pharmacol. Ther. 2021, 110, 297–310. [Google Scholar] [CrossRef]
- Ganti, A.; Yu, S.; Sharpnack, D.; Ingalla, E.; De Bruyn, T. Physiologically-based pharmacokinetic/pharmacodynamic modeling to predict tumor growth inhibition and the efficacious dose of selective estrogen receptor degraders in humans. Biopharm. Drug Dispos. 2023, 44, 301–314. [Google Scholar] [CrossRef]
- Doki, K.; Darwich, A.S.; Patel, N.; Rostami-Hodjegan, A. Virtual bioequivalence for achlorhydric subjects: The use of PBPK modelling to assess the formulation-dependent effect of achlorhydria. Eur. J. Pharm. Sci. 2017, 109, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Laisney, M.; Heimbach, T.; Mueller-Zsigmondy, M.; Blumenstein, L.; Costa, R.; Ji, Y. Physiologically Based Biopharmaceutics Modeling to Demonstrate Virtual Bioequivalence and Bioequivalence Safe-space for Ribociclib which has Permeation Rate-controlled Absorption. J. Pharm. Sci. 2022, 111, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Loisios-Konstantinidis, I.; Cristofoletti, R.; Fotaki, N.; Turner, D.B.; Dressman, J. Establishing virtual bioequivalence and clinically relevant specifications using in vitro biorelevant dissolution testing and physiologically-based population pharmacokinetic modeling. case example: Naproxen. Eur. J. Pharm. Sci. 2020, 143, 105170. [Google Scholar] [CrossRef] [PubMed]
- FY2019 GDUFA Research Report: Locally Acting Physiologically Based Pharmacokinetic Modeling: Summary of FY2019 Activities. Available online: https://www.fda.gov/media/135187/download#page=35 (accessed on 28 May 2023).
- Tsakalozou, E.; Babiskin, A.; Zhao, L. Physiologically-based pharmacokinetic modeling to support bioequivalence and approval of generic products: A case for diclofenac sodium topical gel, 1. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Tsakalozou, E.; Alam, K.; Babiskin, A.; Zhao, L. Physiologically-Based Pharmacokinetic Modeling to Support Determination of Bioequivalence for Dermatological Drug Products: Scientific and Regulatory Considerations. Clin. Pharmacol. Ther. 2022, 111, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- FDA Guidance for Industry. SUPAC-MR: Modified Release Solid Oral Dosage Forms Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Dissolution Testing and In Vivo Bioequivalence Documentation 1997. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/supac-mr-modified-release-solid-oral-dosage-forms-scale-and-postapproval-changes-chemistry (accessed on 22 August 2023).
- Ferraiolo, M.; Hermans, E. The complex molecular pharmacology of the dopamine D2 receptor: Implications for pramipexole, ropinirole, and rotigotine. Pharmacol. Ther. 2023, 245, 108392. [Google Scholar] [CrossRef] [PubMed]
- Bloomer, J.C.; Clarke, S.E.; Chenery, R.J. In vitro identification of the P450 enzymes responsible for the metabolism of ropinirole. Drug Metab. Dispos. 1997, 25, 840–844. [Google Scholar]
- Iwasaki, S.; Yamamoto, S.; Sano, N.; Tohyama, K.; Kosugi, Y.; Furuta, A.; Hamada, T.; Igari, T.; Fujioka, Y.; Hirabayashi, H.; et al. Direct Drug Delivery of Low-Permeable Compounds to the Central Nervous System Via Intranasal Administration in Rats and Monkeys. Pharm. Res. 2019, 36, 76. [Google Scholar] [CrossRef]
- Shuklinova, O.; Dorożyński, P.; Kulinowski, P.; Bielecka, Z.; Wiśniowska, B.; Polak, S. Development of physiologically based pharmacokinetic model for the immediate release of ropinirole tablets. Acta Pol. Pharm. Drug Res. 2021, 78, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Hanano, M.; Sugiyama, Y.; Harashima, H.; Iga, T. Prediction of the volumes of distribution of basic drugs in humans based on data from animals. J. Pharmacokinet. Biopharm. 1984, 12, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef] [PubMed]
- Shuklinova, O.; Dorożyński, P.; Kulinowski, P.; Polak, S. Quality control dissolution data is biopredictive for a modified release ropinirole formulation: Virtual experiment with the use of re-developed and verified PBPK model. Pharmaceutics 2022, 14, 1514. [Google Scholar] [CrossRef] [PubMed]
- Abduljalil, K.; Cain, T.; Humphries, H.; Rostami-Hodjegan, A. Deciding on success criteria for predictability of pharmacokinetic parameters from in vitro studies: An analysis based on in vivo observations. Drug Metab. Dispos. 2014, 42, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Kuemmel, C.; Yang, Y.; Zhang, X.; Florian, J.; Zhu, H.; Tegenge, M.; Huang, S.-M.; Wang, Y.; Morrison, T.; Zineh, I. Consideration of a Credibility Assessment Framework in Model-Informed Drug Development: Potential Application to Physiologically-Based Pharmacokinetic Modeling and Simulation. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef] [PubMed]
- GSK—A Study to Investigate the Effect of Repeated Oral Doses of Ciprofloxacin on Steady State Ropinirole Pharmacokinetics in Parkinsonian Patients. Available online: https://www.gsk-studyregister.com/en/trial-details/?id=101468/102 (accessed on 5 June 2021).
- Clarke, J.F.; Thakur, K.; Polak, S. A mechanistic physiologically based model to assess the effect of study design and modified physiology on formulation safe space for virtual bioequivalence of dermatological drug products. Front. Pharmacol. 2022, 13, 1007496. [Google Scholar] [CrossRef]
- Jamei, M.; Marciniak, S.; Feng, K.; Barnett, A.; Tucker, G.; Rostami-Hodjegan, A. The Simcyp population-based ADME simulator. Exp. Opin. Drug Metab. Toxicol. 2009, 5, 211–223. [Google Scholar] [CrossRef]
- Danielak, D.; Paszkowska, J.; Staniszewska, M.; Garbacz, G.; Terlecka, A.; Kubiak, B.; Romański, M. Conjunction of semi-mechanistic in vitro-in vivo modeling and population pharmacokinetics as a tool for virtual bioequivalence analysis—A case study for a BCS class II drug. Eur. J. Pharm. Biopharm. 2023, 186, 132–143. [Google Scholar] [CrossRef]
- Davit, B.; Braddy, A.C.; Conner, D.P.; Yu, L.X. International Guidelines for Bioequivalence of Systemically Available Orally Administered Generic Drug Products: A Survey of Similarities and Differences. AAPS J. 2013, 15, 974–990. [Google Scholar] [CrossRef] [PubMed]
- Wyszogrodzka-Gaweł, G.; Shuklinova, O.; Lisowski, B.; Wiśniowska, B.; Polak, S. 3D printing combined with biopredictive dissolution and PBPK/PD modeling optimization and personalization of pharmacotherapy: Are we there yet? Drug Discov. Today 2023, 28, 103731. [Google Scholar] [CrossRef] [PubMed]
- Trenfield, S.J.; Awad, A.; Goyanes, A.; Gaisford, S.; Basit, A.W. 3D Printing Pharmaceuticals: Drug Development to Frontline Care. Trends Pharmacol. Sci. 2018, 39, 440–451. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation 10 | Formulation 27 | |
|---|---|---|
| Type of PEGDA | 700 | 700 |
| Sodium Alginate concentration [%] | 10 | 0 |
| RH concentration [%] | 2 | 2 |
| Similarity factor (f2) | 57.44 | 71.02 |
| Parameter | Ropinirole Alone, Mean | Ropinirole + Ciprofloxacin, Mean | Geometric Mean Ratio | Observed/ Simulated Ratio |
|---|---|---|---|---|
| AUC0–6 ng.h/mL observed | 30.2 | 57 | 1.84 | 1.17 |
| AUC0–6 ng.h/mL simulated | 31.40 | 66.85 | 2.16 | |
| Cmax (ng/mL) observed | 7.86 | 12.67 | 1.6 | 1.36 |
| Cmax (ng/mL) simulated | 5.98 | 12.70 | 2.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shuklinova, O.; Wyszogrodzka-Gaweł, G.; Baran, E.; Lisowski, B.; Wiśniowska, B.; Dorożyński, P.; Kulinowski, P.; Polak, S. Can 3D Printed Tablets Be Bioequivalent and How to Test It: A PBPK Model Based Virtual Bioequivalence Study for Ropinirole Modified Release Tablets. Pharmaceutics 2024, 16, 259. https://doi.org/10.3390/pharmaceutics16020259
Shuklinova O, Wyszogrodzka-Gaweł G, Baran E, Lisowski B, Wiśniowska B, Dorożyński P, Kulinowski P, Polak S. Can 3D Printed Tablets Be Bioequivalent and How to Test It: A PBPK Model Based Virtual Bioequivalence Study for Ropinirole Modified Release Tablets. Pharmaceutics. 2024; 16(2):259. https://doi.org/10.3390/pharmaceutics16020259
Chicago/Turabian StyleShuklinova, Olha, Gabriela Wyszogrodzka-Gaweł, Ewelina Baran, Bartosz Lisowski, Barbara Wiśniowska, Przemysław Dorożyński, Piotr Kulinowski, and Sebastian Polak. 2024. "Can 3D Printed Tablets Be Bioequivalent and How to Test It: A PBPK Model Based Virtual Bioequivalence Study for Ropinirole Modified Release Tablets" Pharmaceutics 16, no. 2: 259. https://doi.org/10.3390/pharmaceutics16020259
APA StyleShuklinova, O., Wyszogrodzka-Gaweł, G., Baran, E., Lisowski, B., Wiśniowska, B., Dorożyński, P., Kulinowski, P., & Polak, S. (2024). Can 3D Printed Tablets Be Bioequivalent and How to Test It: A PBPK Model Based Virtual Bioequivalence Study for Ropinirole Modified Release Tablets. Pharmaceutics, 16(2), 259. https://doi.org/10.3390/pharmaceutics16020259