
Determination of the Absolute Configuration of Secondary Alcohols in a Compound Mixture via the Application of Competing Enantioselective Acylation Coupled with LC/MS Analysis



Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Experimental Procedures
2.2. Chemicals and Reagents
2.3. Fungus Material
2.4. Extraction and Isolation
2.5. Competing Enantioselective Acylation (CEA) Coupled with LC/MS Analysis
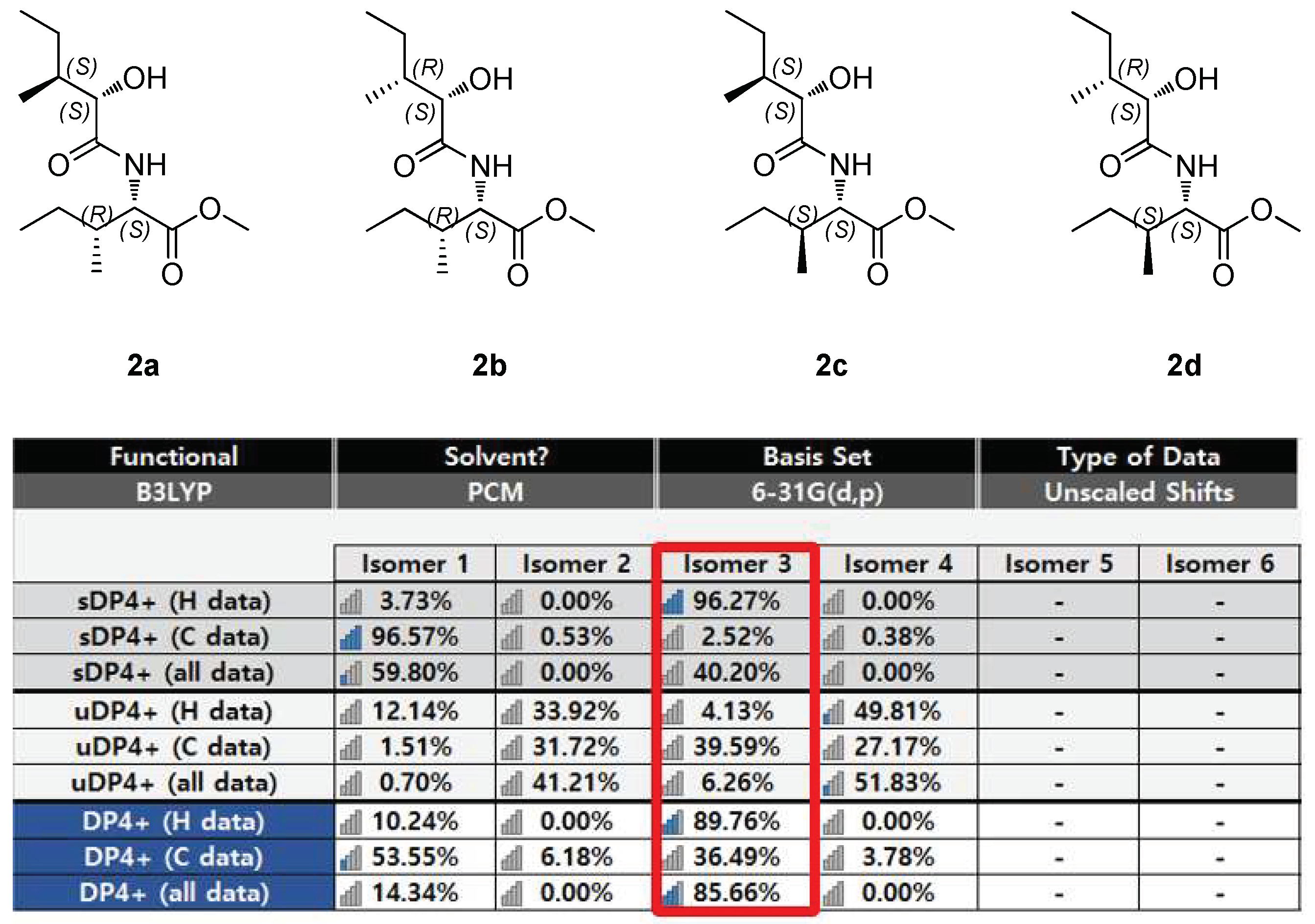
2.6. Computational NMR–Chemical Shift Calculations for DP4+ Analysis
2.7. In Vitro Cytotoxicity Test
3. Results and Discussion
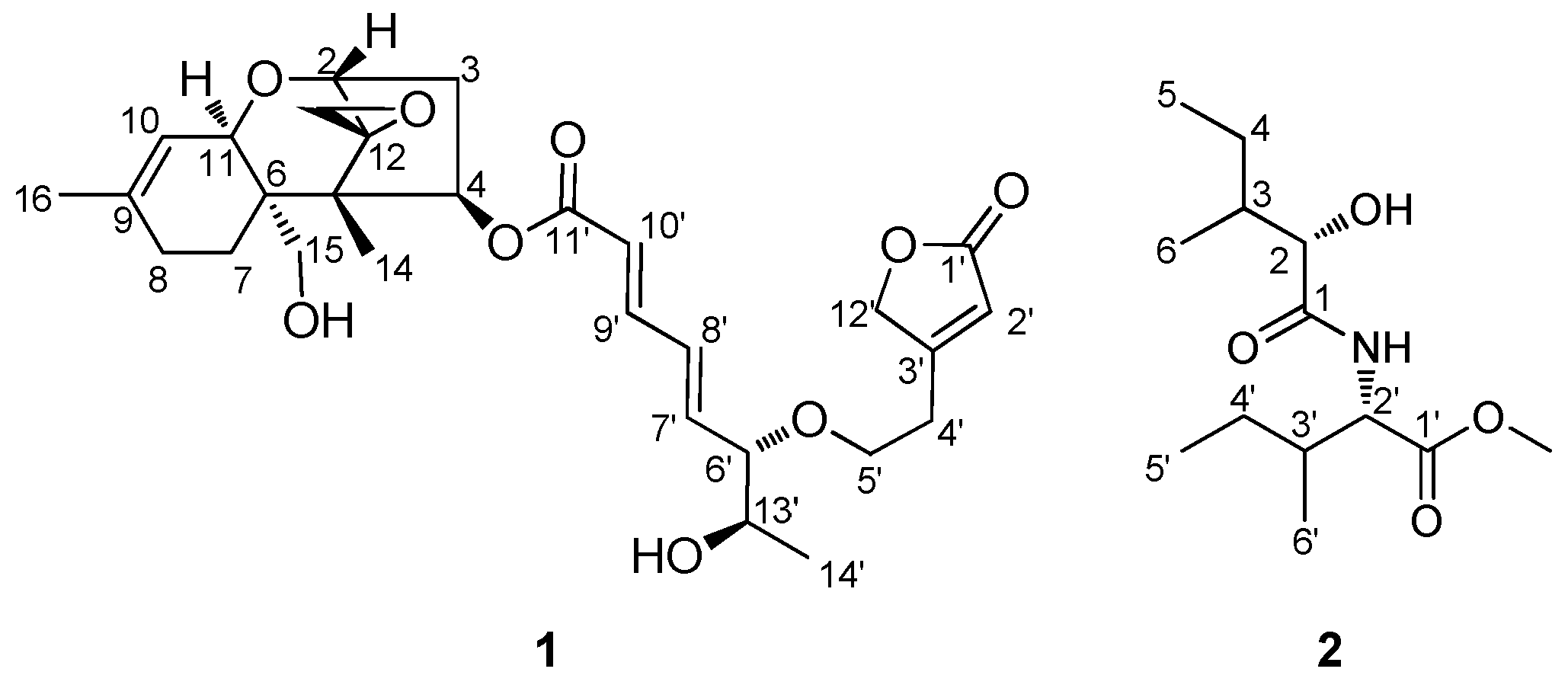
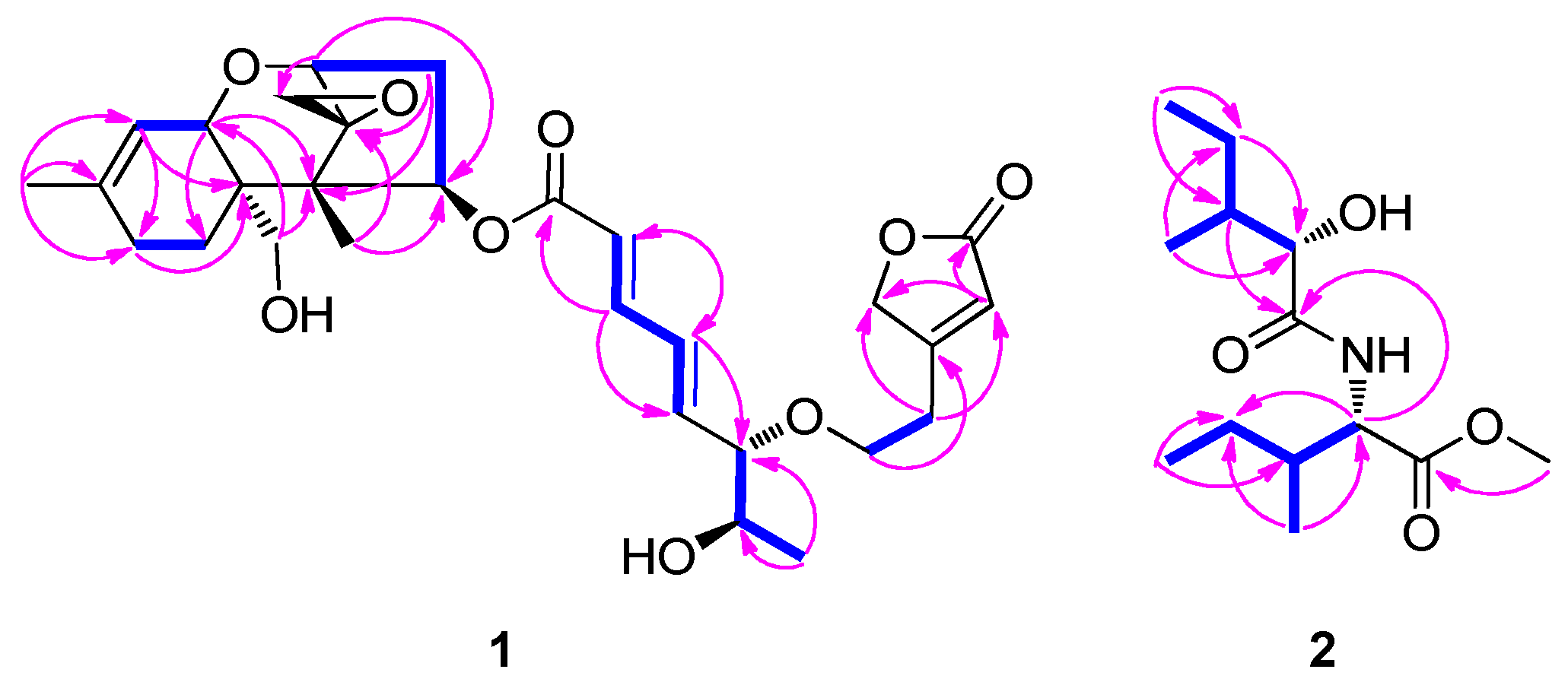
3.1. Planar Structural Elucidation of Compound Mixture
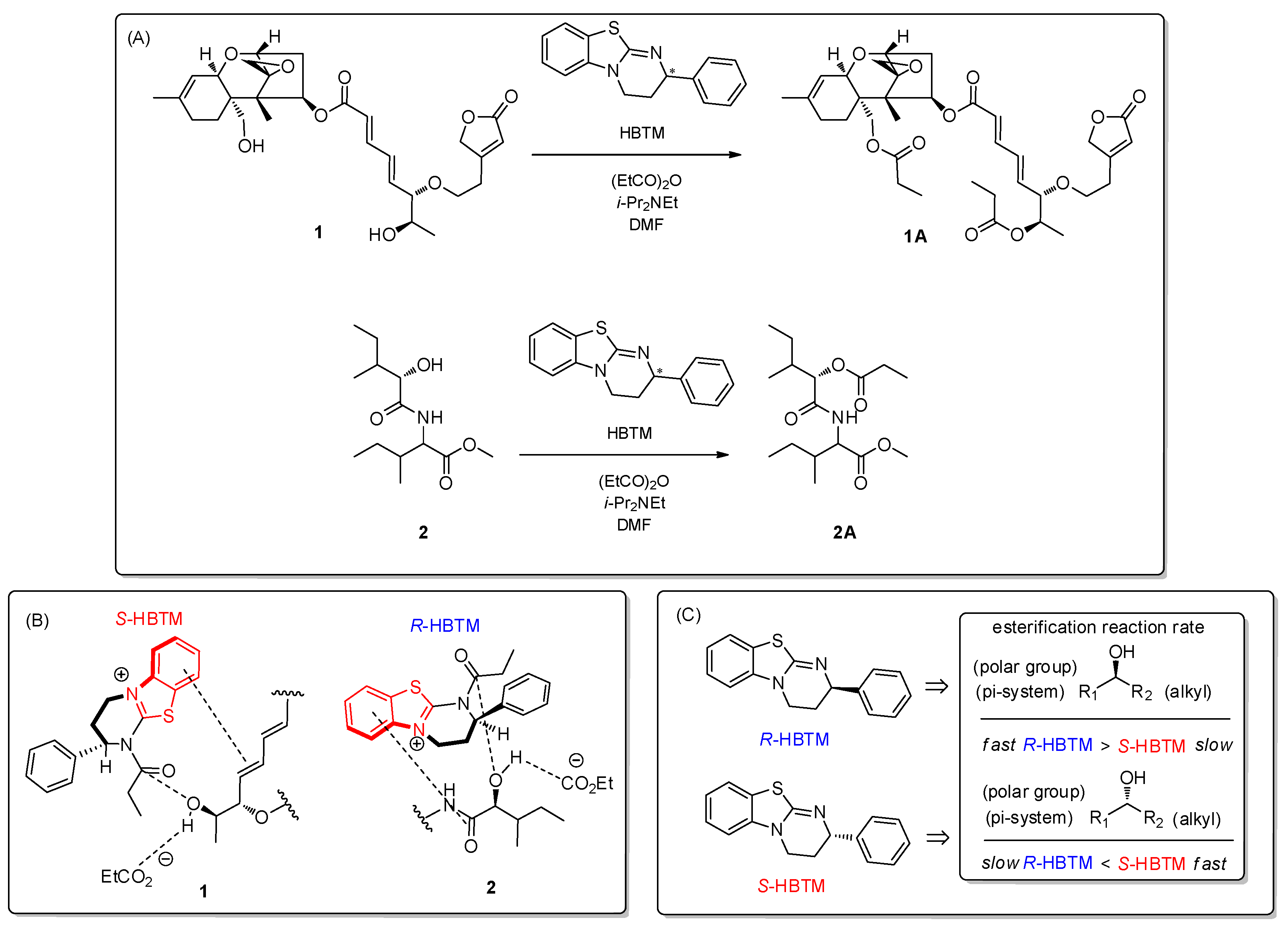
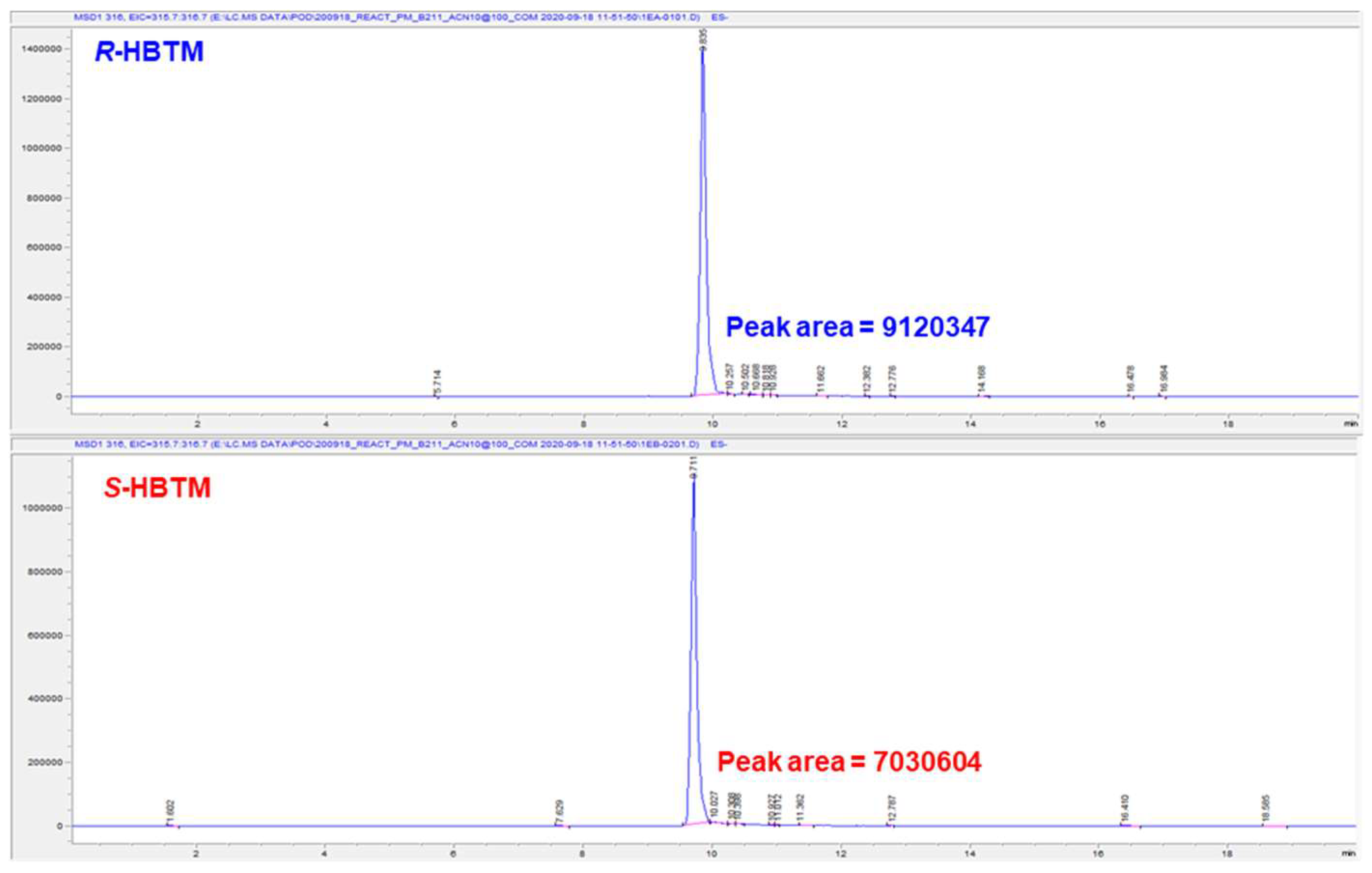
3.2. Determination of Absolute Configuration of Secondary Alcohols in Compound Mixture Using Competing Enantioselective Acylation Coupled with LC/MS
3.3. Evaluation of Cytotoxicity of the Mixture
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scott, K.A.; Ropek, N.; Melillo, B.; Schreiber, S.L.; Cravatt, B.F.; Vinogradova, E.V. Stereochemical diversity as a source of discovery in chemical biology. Curr. Res. Chem. Biol. 2022, 2, 100028. [Google Scholar] [CrossRef]
- Wagner, A.J.; Rychnovsky, S.D. Determination of absolute configuration of secondary alcohols using thin-layer chromatography. J. Org. Chem. 2013, 78, 4594–4598. [Google Scholar] [CrossRef]
- Shujiro, S.; Yutaka, T.; Kazuo, T.; Yohko, Y. Determination of the absolute configuration of a secondary hydroxy group in a chiral secondary alcohol using glycosidation shifts in carbon-13 Nuclear Magnetic Resonance Spectroscopy. J. Am. Chem. Soc. 1978, 100, 3331–3339. [Google Scholar]
- Trost, B.M.; Berlletire, J.L.; Godleski, S.; McDougal, P.G.; Balkovec, J.M. On the use of the O-methylmandelate ester for establishment of absolute configuration of secondary alcohols. J. Org. Chem. 1986, 51, 2370–2374. [Google Scholar] [CrossRef]
- Harada, K.; Shimizu, Y.; Kawakami, A.; Norimoto, M.; Fujii, K. Chromatographic Determination of the Absolute Configuration of an Acyclic Secondary Alcohol Using Difluorodinitrobenzene. Anal. Chem. 2000, 72, 4142–4147. [Google Scholar] [CrossRef] [PubMed]
- Valentová, J.; Lintnerová, L.; Miklášová, N.; Oboňová, B.; Habala, L. Analogues of Anticancer Natural Products: Chiral Aspects. Int. J. Mol. Sci. 2023, 24, 5679. [Google Scholar] [CrossRef] [PubMed]
- Bitchagno, G.T.M.; Nchiozem-Ngnitedem, V.-A.; Melchert, D.; Fobofou, S.A. Demystifying racemic natural products in the homochiral world. Nat. Rev. Chem. 2022, 6, 806–822. [Google Scholar] [CrossRef] [PubMed]
- Finefield, J.M.; Sherman, D.H.; Kreitman, M.; Williams, R.M. Enantiomeric natural products: Occurrence and biogenesis. Angew. Chem. Int. Ed. 2012, 51, 4802–4836. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, A.; Zana, A.; Borsari, C.; Persico, M.; Bova, S.; Tkachuk, O.; Corfu, A.I.; Tamborini, L.; Basilico, N.; Fattorusso, C.; et al. Role of Stereochemistry on the Biological Activity of Nature-Inspired 3-Br-Acivicin Isomers and Derivatives. Molecules 2023, 28, 3172. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.N.L.; Santos, F.M.D.; Batista, J.M.; Cass, Q.B. Enantiomeric Mixtures in Natural Product Chemistry: Separation and Absolute Configuration Assignment. Molecules 2018, 23, 492. [Google Scholar] [CrossRef] [PubMed]
- Snell, T.W.; Carberry, J. Astaxanthin Bioactivity Is Determined by Stereoisomer Composition and Extraction Method. Nutrients 2022, 14, 1522. [Google Scholar] [CrossRef]
- Wang, R.B.; Ma, S.G.; Jamieson, C.S.; Gao, R.M.; Liu, Y.B.; Li, Y.; Wang, X.J.; Li, Y.H.; Houk, K.N.; Qu, J.; et al. Library construction of stereochemically diverse isomers of spirooliganin: Their total synthesis and antiviral activity. Chem. Sci. 2021, 12, 7003–7011. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.N.L.; Angrisani, B.R.; Lima, M.E.D.; Da Silva, S.M.; Schettini, V.H.; Chagas, H.A.; Santos, F.M.d., Jr.; Batista, J.M., Jr.; Valverde, A.L. Absolute configuration reassignment of natural products: An overview of the last decade. J. Braz. Chem. Soc. 2021, 32, 1499–1518. [Google Scholar] [CrossRef]
- Fujii, K.; Ikai, Y.; Oka, H.; Suzuki, M.; Harada, K.-i. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: Combination of Marfey’s method with mass spectrometry and its practical application. Anal. Chem. 1997, 69, 5146–5151. [Google Scholar] [CrossRef]
- Phyo, Y.Z.; Ribeiro, J.; Fernandes, C.; Kijjoa, A.; Pinto, M.M. Marine natural peptides: Determination of absolute configuration using liquid chromatography methods and evaluation of bioactivities. Molecules 2018, 23, 306. [Google Scholar] [CrossRef] [PubMed]
- Hoye, T.R.; Jeffrey, C.S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2, 2451–2458. [Google Scholar] [CrossRef]
- Kusumi, T.; Yabuuchi, T.; Takahashi, H.; Ooi, T. Chiral anisotropic reagents for determining the absolute configuration of secondary alcohols and carboxylic acids. J. Synth. Org. Chem. 2005, 63, 1102–1114. [Google Scholar] [CrossRef]
- Gruene, T.; Wennmacher, J.T.C.; Zaubitzer, C.; Holstein, J.J.; Heidler, J.; Fecteau-Lefebvre, A.; De Carlo, S.; Müller, E.; Goldie, K.N.; Regeni, I.; et al. Rapid Structure Determination of Microcrystalline Molecular Compounds Using Electron Diffraction. Angew. Chem. Int. Ed. 2018, 57, 16313–16317. [Google Scholar] [CrossRef]
- Jones, C.G.; Martynowycz, M.W.; Hattne, J.; Fulton, T.J.; Stoltz, B.M.; Rodriguez, J.A.; Nelson, H.M.; Gonen, T. The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Cent. Sci. 2018, 4, 1587–1592. [Google Scholar] [CrossRef]
- Park, J.D.; Li, Y.; Moon, K.; Han, E.; Lee, S.R.; Seyedsayamdosy, M. Structural Elucidation of Cryptic Algaecides in Marine Algal-Bacterial Symbioses by NMR Spectroscopy and MicroED. Angew. Chem. Int. Ed. 2022, 61, e202114022. [Google Scholar] [CrossRef]
- Lee, S.R.; Park, H.B.; Kim, K.H. Highly sensitive, simple, and cost-and time-effective method to determine the absolute configuration of a secondary alcohol using competing enantioselective acylation coupled with LC/MS. Anal. Chem. 2018, 90, 13212–13216. [Google Scholar] [CrossRef]
- Lee, S.R.; Yi, S.A.; Nam, K.; Ryoo, R.; Lee, J.; Kim, K.H.J. Pantheric Acids A-C from a Poisonous Mushroom, Amanita pantherina, Promote Lipid Accumulation in Adipocytes. Nat. Prod. 2019, 82, 3489–3493. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Alishir, A.; Zhang, S.; Zhang, Y.; Choi, S.; Pang, C.; Bae, H.Y.; Jung, W.H.; Kim, K.H. Identification of Obscurolide-Type Metabolites and Antifungal Metabolites from the Termite-Associated Streptomyces neopeptinius BYF101. J. Nat. Prod. 2023, 86, 1891–1900. [Google Scholar] [CrossRef]
- Yu, J.S.; Jeong, S.Y.; Li, C.; Oh, T.; Kwon, M.; Ahn, J.S.; Ko, S.-K.; Ko, Y.-J.; Cao, S.; Kim, K.H. New phenalenone derivatives from the Hawaiian volcanic soil-associated fungus Penicillium herquei FT729 and their inhibitory effects on indoleamine 2, 3-dioxygenase 1 (IDO1). Arch. Pharm. Res. 2022, 45, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.E.; Park, K.H.; Hong, J.H.; Kim, S.H.; Park, K.M.; Kim, K.H. Anti-osteoporosis effects of triterpenoids from the fruit of sea buckthorn (Hippophae rhamnoides) through the promotion of osteoblast differentiation in mesenchymal stem cells, C3H10T1/2. Arch. Pharm. Res. 2023, 46, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Saikawa, Y.; Okamoto, H.; Inui, T.; Makabe, M.; Okuno, T.; Suda, T.; Hashimoto, K.; Nakata, M. Toxic principles of a poisonous mushroom Podostroma cornu-damae. Tetrahedron 2001, 57, 8277–8281. [Google Scholar] [CrossRef]
- Kim, H.N.; Do, H.H.; Seo, J.S.; Kim, H.Y. Two cases of incidental Podostroma cornu-damae poisoning. Clin. Exp. Emerg. Med. 2016, 3, 186. [Google Scholar] [CrossRef]
- Lee, S.R.; Seok, S.; Ryoo, R.; Choi, S.U.; Kim, K.H. Macrocyclic trichothecene mycotoxins from a deadly poisonous mushroom, Podostroma cornu-damae. J. Nat. Prod. 2018, 82, 122–128. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Na, M.W.; Park, E.C.; Kim, J.-C.; Kang, D.-M.; Hamishehkar, H.; Ahn, M.-J.; Kim, J.K.; Kim, K.H. Labdane-type Diterpenes from Pinus eldarica Needles and Their Anti-Helicobacter pylori Activity. ACS Omega 2022, 7, 29502–29507. [Google Scholar] [CrossRef]
- Nicolás, G.; María, M.Z.; Ariel, M.S. Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds using Quantum Chemical Calculations of NMR Shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar]
- Zhuang, Y.; Yang, F.; Menon, A.; Song, J.M.; Espinoza, R.V.; Schultz, P.J.; Garner, A.L.; Tripathi, A. An ECD and NMR/DP4+ Computational Pipeline for Structure Revision and Elucidation of Diphenazine-Based Natural Products. J. Nat. Prod. 2023, 86, 1801–1814. [Google Scholar] [CrossRef]
- Kim, D.H.; Ham, S.L.; Khan, Z.; Kim, S.Y.; Choi, S.U.; Kim, C.S.; Lee, K.R. Terpenoids from Glechoma hederacea var. longituba and their biological activities. Beilstein J. Org. Chem. 2022, 18, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, N.; Fu, P.; Wang, W.; Bian, S.; Zhang, H.; Shen, S.; Han, B. Structure Elucidation of Two Intriguing Neo-Debromoaplysiatoxin Derivatives from Marine Cyanobacterium Lyngbya sp. Showing Strong Inhibition of Kv1.5 Potassium Channel and Differential Cytotoxicity. Molecules 2023, 28, 2786. [Google Scholar] [CrossRef] [PubMed]
- Longhai, J.; Jinping, L.; Shu, W.; Linxian, Z.; Jiannan, L. Evaluation of 20(S)-ginsenoside Rg3 loaded hydrogel for the treatment of perianal ulcer in a rat model. J. Ginseng Res. 2022, 46, 771–779. [Google Scholar]
- Abdullahi, S.; Mangala, G.P.; Mohd, M.; Pitta, V.P.; Gabriel, A.O. Chemical Profile and Cytotoxicity Activity of Stem-bark of Anacardium occidentale. Nat. Prod. Sci. 2022, 28, 62–88. [Google Scholar] [CrossRef]
- Levi, M.; Salaroli, R.; Parenti, F.; De Maria, R.; Zannoni, A.; Bernardini, C.; Gola, C.; Brocco, A.; Marangio, A.; Benazzi, C.; et al. Doxorubicin treatment modulates chemoresistance and affects the cell cycle in two canine mammary tumour cell lines. BMC Vet. Res. 2021, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Vu, M.; Kassouf, N.; Ofili, R.; Lund, T.; Bell, C.; Appiah, S. Doxorubicin selectively induces apoptosis through the inhibition of a novel isoform of Bcl-2 in acute myeloid leukaemia MOLM-13 cells with reduced Beclin 1 expression. Int. J. Oncol. 2020, 57, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Maillet, A.; Tan, K.; Chai, X.; Sadananda, S.N.; Mehta, A.; Ooi, J.; Hayden, M.R.; Pouladi, M.A.; Ghosh, S.; Shim, W.; et al. Modeling Doxorubicin-Induced Cardiotoxicity in Human Pluripotent Stem Cell Derived-Cardiomyocytes. Sci. Rep. 2016, 6, 25333. [Google Scholar] [CrossRef]
- Lakornwong, W.; Kanokmedhakul, K.; Soytong, K.; Unartngam, A.; Tontapha, S.; Amornkitbamrung, V.; Kanokmedhakul, S. Types A and D Trichothecene mycotoxins from the fungus Myrothecium roridum. Planta Med. 2019, 85, 774–780. [Google Scholar] [CrossRef]
- Schalk, F.; Um, S.; Guo, H.; Kreuzenbeck, N.B.; Görls, H.; De Beer, Z.W.; Beemelmanns, C. Targeted discovery of tetrapeptides and cyclic polyketide-peptide hybrids from a fungal antagonist of farming termites. Chem. Bio. Chem. 2020, 21, 2991–2996. [Google Scholar] [CrossRef]
- Kwon, Y.; Byun, W.S.; Kim, B.Y.; Song, M.C.; Bae, M.; Yoon, Y.J.; Shin, J.; Lee, S.K.; Oh, D.C. Depsidomycins B and C: New Cyclic Peptides from a Ginseng Farm Soil-Derived Actinomycete. Molecules 2018, 23, 1266. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | ||||
|---|---|---|---|---|---|
| Position | δC | δH (J in Hz) | Position | δC | δH (J in Hz) |
| 2 | 78.8 CH | 3.85, m | 1 | 172.5 C | |
| 3a | 35.7 CH2 | 2.11, m | 2 | 74.3 CH | 4.16, m |
| 3b | 2.51, m | 3 | 37.8 CH | 1.93, m | |
| 4 | 75.5 CH | 6.15, dd (8.5, 4.0) | 4a | 26.1 CH2 | 1.35, m |
| 5 | 48.7 C | 4b | 1.47, m | ||
| 6 | 43.9 C | 5 | 11.7 CH3 | 0.97, t (7.0) | |
| 7a | 21.1 CH2 | 1.55, m | 6 | 12.6 CH3 | 0.85, d (7.0) |
| 7b | 1.98, m | ||||
| 8 | 27.7 CH2 | 2.00, m | 1′ | 172.5 C | |
| 9 | 140.0 C | 2′ | 56.0 CH | 4.63, dd (9.0, 5.0) | |
| 10 | 118.7 CH | 5.50, d (5.5) | 3′ | 37.8 CH | 1.93, m |
| 11 | 66.6 CH | 3.94, d (5.5) | 4′a | 25.3 CH2 | 1.20, m |
| 12 | 65.6 C | 4′b | 1.46, m | ||
| 13a | 48.1 CH2 | 2.83, d (4.0) | 5′ | 11.3 CH3 | 0.93, t (7.0) |
| 13b | 3.15, d (4.0) | 6′ | 15.4 CH3 | 0.93, d (7.0) | |
| 14 | 6.3 CH3 | 0.84, s | OCH3 | 51.5 CH3 | 3.75, s |
| 15a | 62.8 CH2 | 3.84, m | NH | 6.95, d (9.0) | |
| 15b | 3.68, m | ||||
| 16 | 23.1 CH3 | 1.73, s | |||
| 1′ | 173.7 C | ||||
| 2′ | 116.6 CH | 5.92, brs | |||
| 3′ | 166.9 C | ||||
| 4′ | 29.0 CH2 | 2.72, dd (11.0, 5.5) | |||
| 5′a | 66.0 CH2 | 3.56, m | |||
| 5′b | 3.74, m | ||||
| 6′ | 85.8 CH | 3.58, m | |||
| 7′ | 138.5 CH | 5.94, dd (15.5, 8.0) | |||
| 8′ | 132.3 CH | 7.30, dd (15.5, 11.0) | |||
| 9′ | 143.5 CH | 6.38, dd (15.5, 11.0) | |||
| 10′ | 122.4 CH | 6.00, d (15.5) | |||
| 11′ | 167.4 C | ||||
| 12′ | 72.9 CH2 | 4.79, s | |||
| 13′ | 69.4 CH | 3.70, m | |||
| 14′ | 18.0 CH3 | 1.13, d (6.5) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, B.S.; Kim, H.; Baek, J.; Ryoo, R.; Lee, S.R.; Kim, K.H. Determination of the Absolute Configuration of Secondary Alcohols in a Compound Mixture via the Application of Competing Enantioselective Acylation Coupled with LC/MS Analysis. Pharmaceutics 2024, 16, 364. https://doi.org/10.3390/pharmaceutics16030364
Lee BS, Kim H, Baek J, Ryoo R, Lee SR, Kim KH. Determination of the Absolute Configuration of Secondary Alcohols in a Compound Mixture via the Application of Competing Enantioselective Acylation Coupled with LC/MS Analysis. Pharmaceutics. 2024; 16(3):364. https://doi.org/10.3390/pharmaceutics16030364
Chicago/Turabian StyleLee, Bum Soo, Hoon Kim, Jiwon Baek, Rhim Ryoo, Seoung Rak Lee, and Ki Hyun Kim. 2024. "Determination of the Absolute Configuration of Secondary Alcohols in a Compound Mixture via the Application of Competing Enantioselective Acylation Coupled with LC/MS Analysis" Pharmaceutics 16, no. 3: 364. https://doi.org/10.3390/pharmaceutics16030364
APA StyleLee, B. S., Kim, H., Baek, J., Ryoo, R., Lee, S. R., & Kim, K. H. (2024). Determination of the Absolute Configuration of Secondary Alcohols in a Compound Mixture via the Application of Competing Enantioselective Acylation Coupled with LC/MS Analysis. Pharmaceutics, 16(3), 364. https://doi.org/10.3390/pharmaceutics16030364