

Triboelectric Charging Properties of the Functional Groups of Common Pharmaceutical Materials Using Density Functional Theory Calculations


Abstract
1. Introduction
2. Theoretical Approach
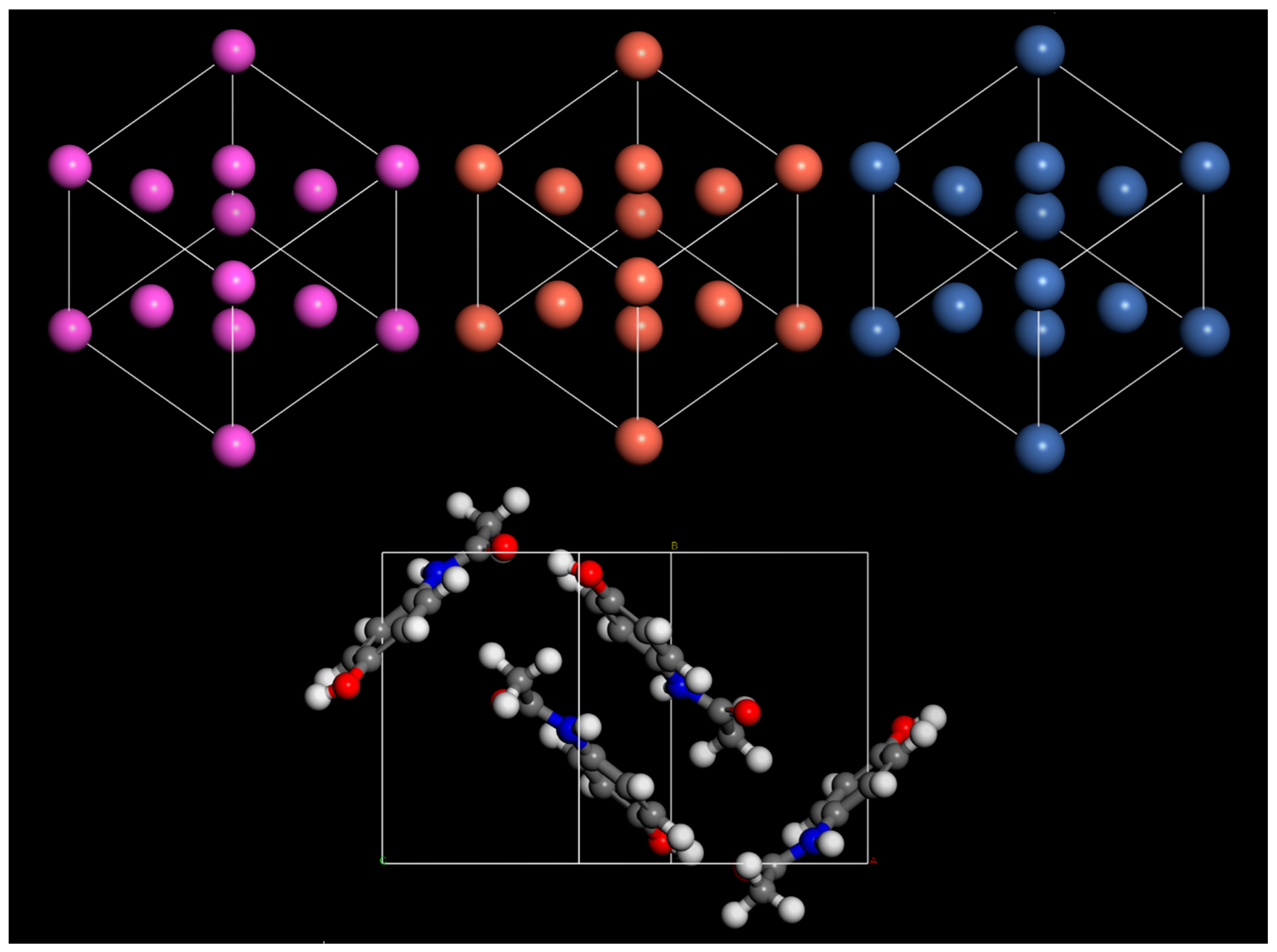
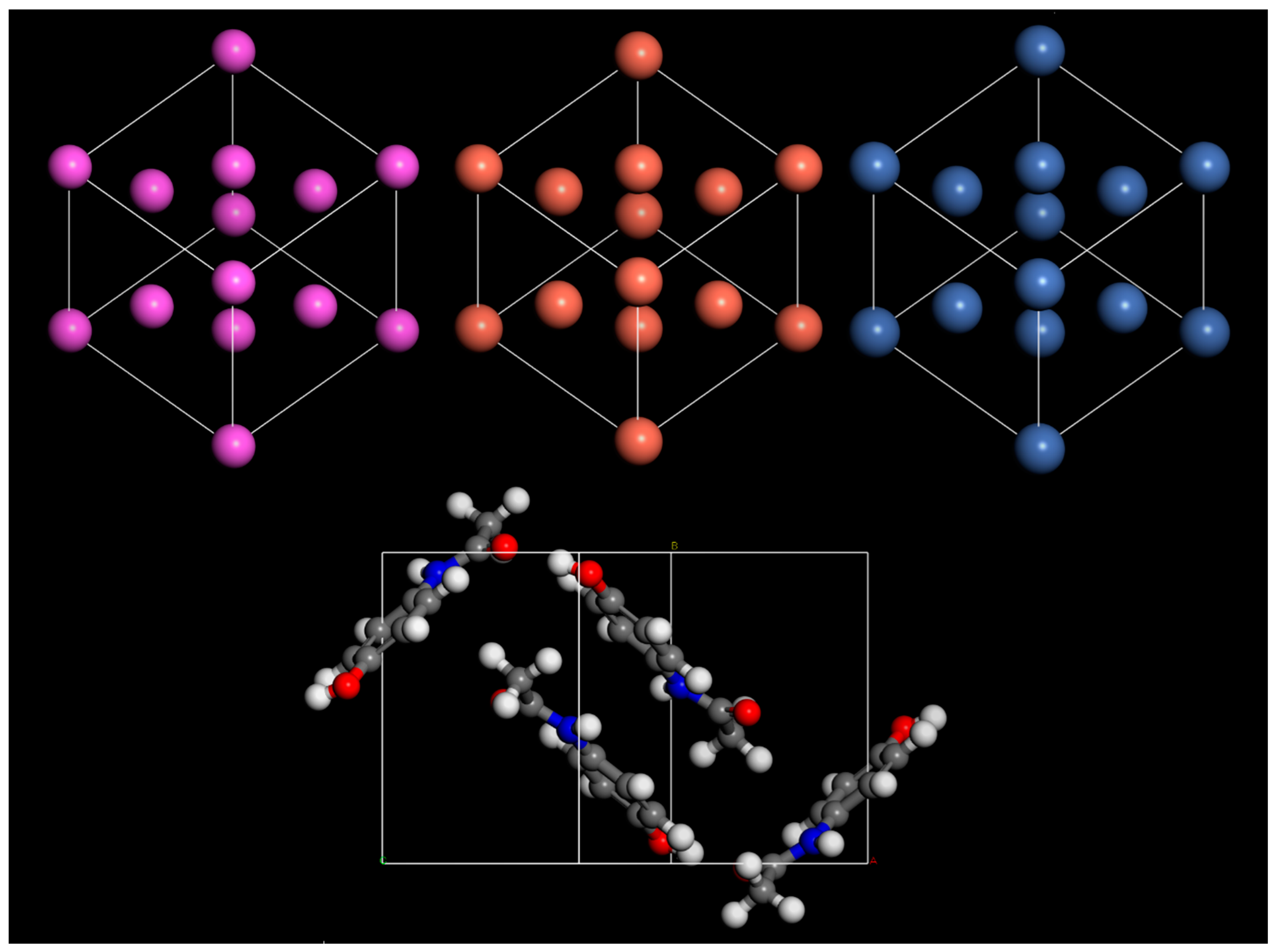
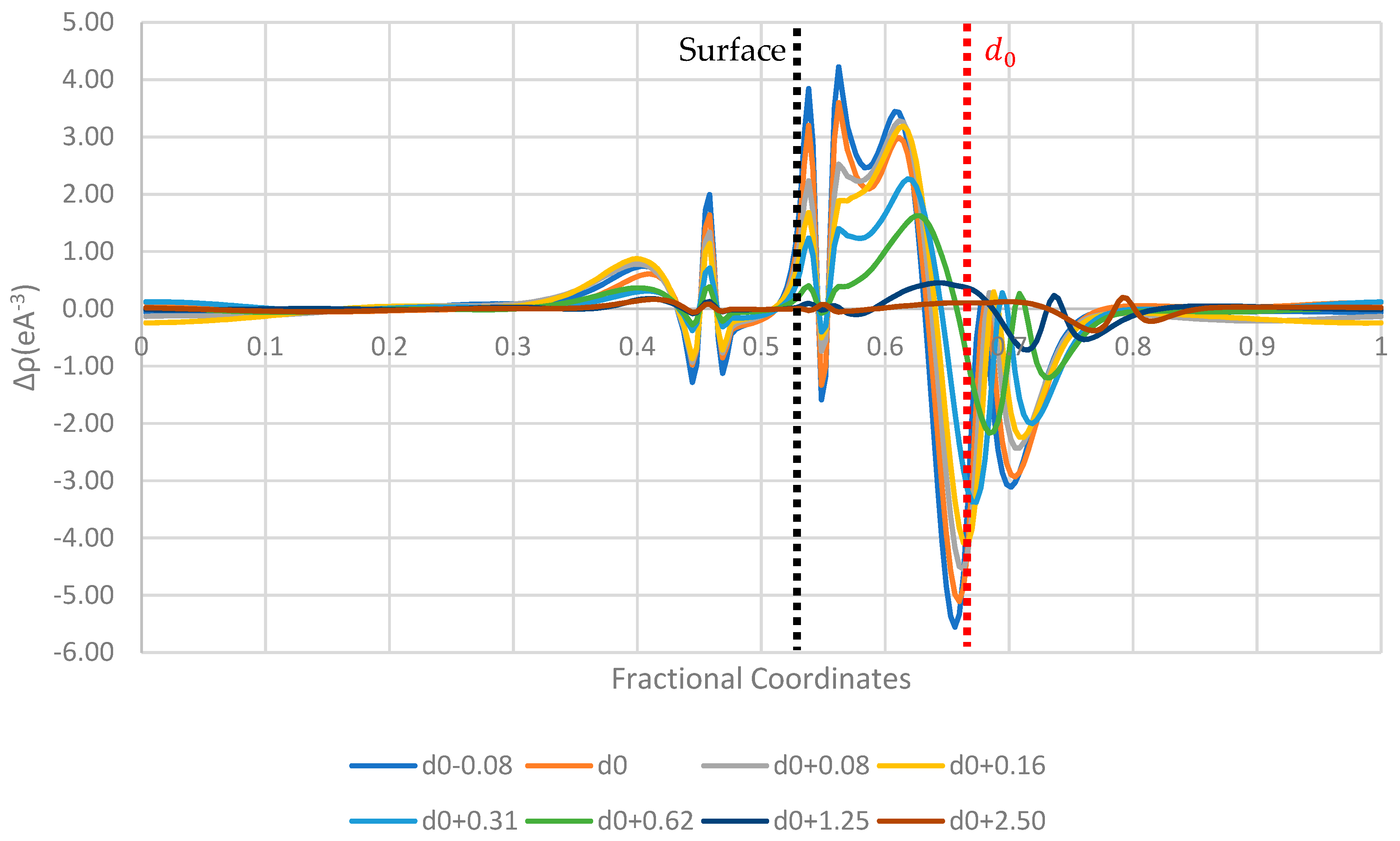
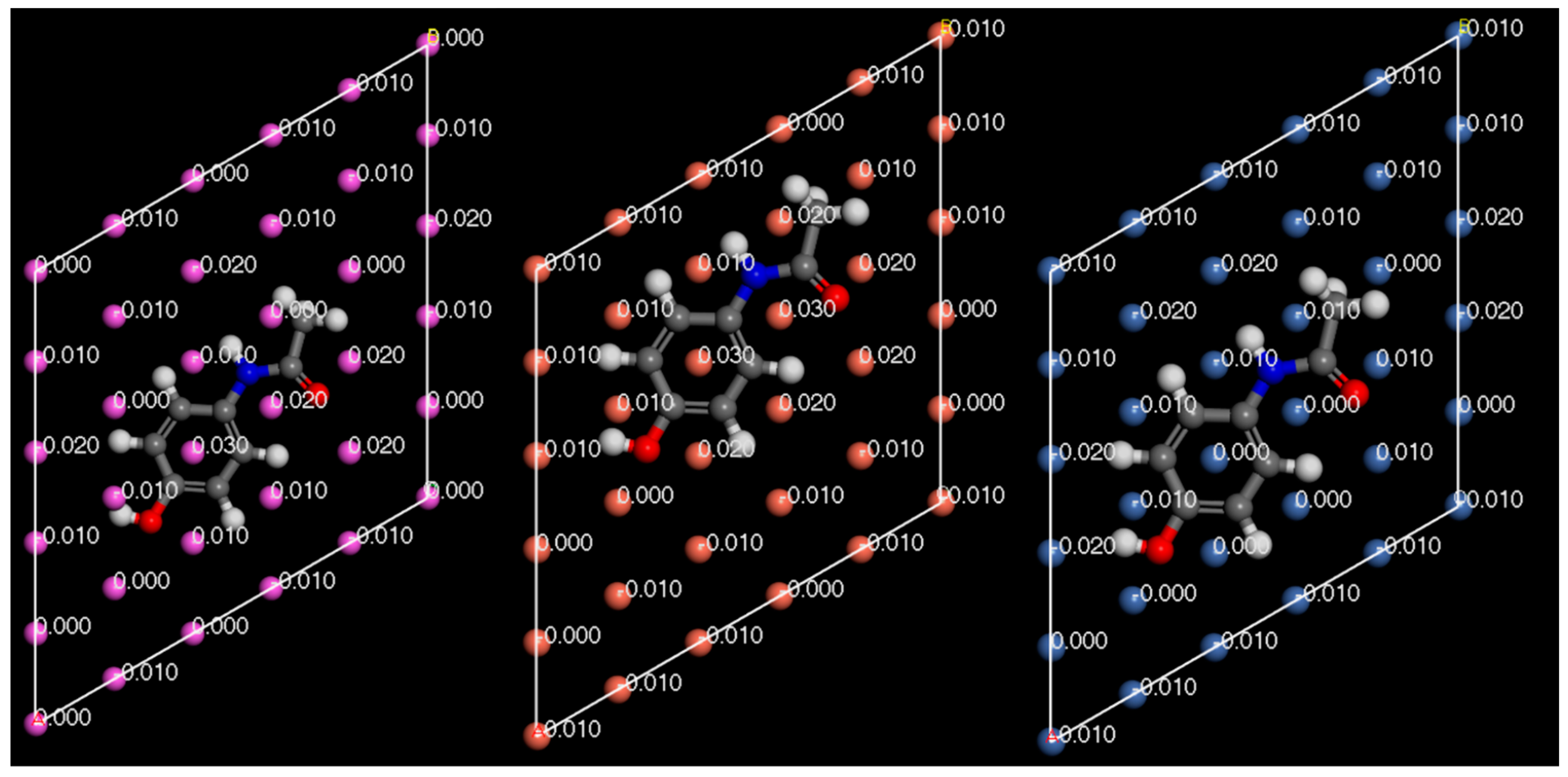
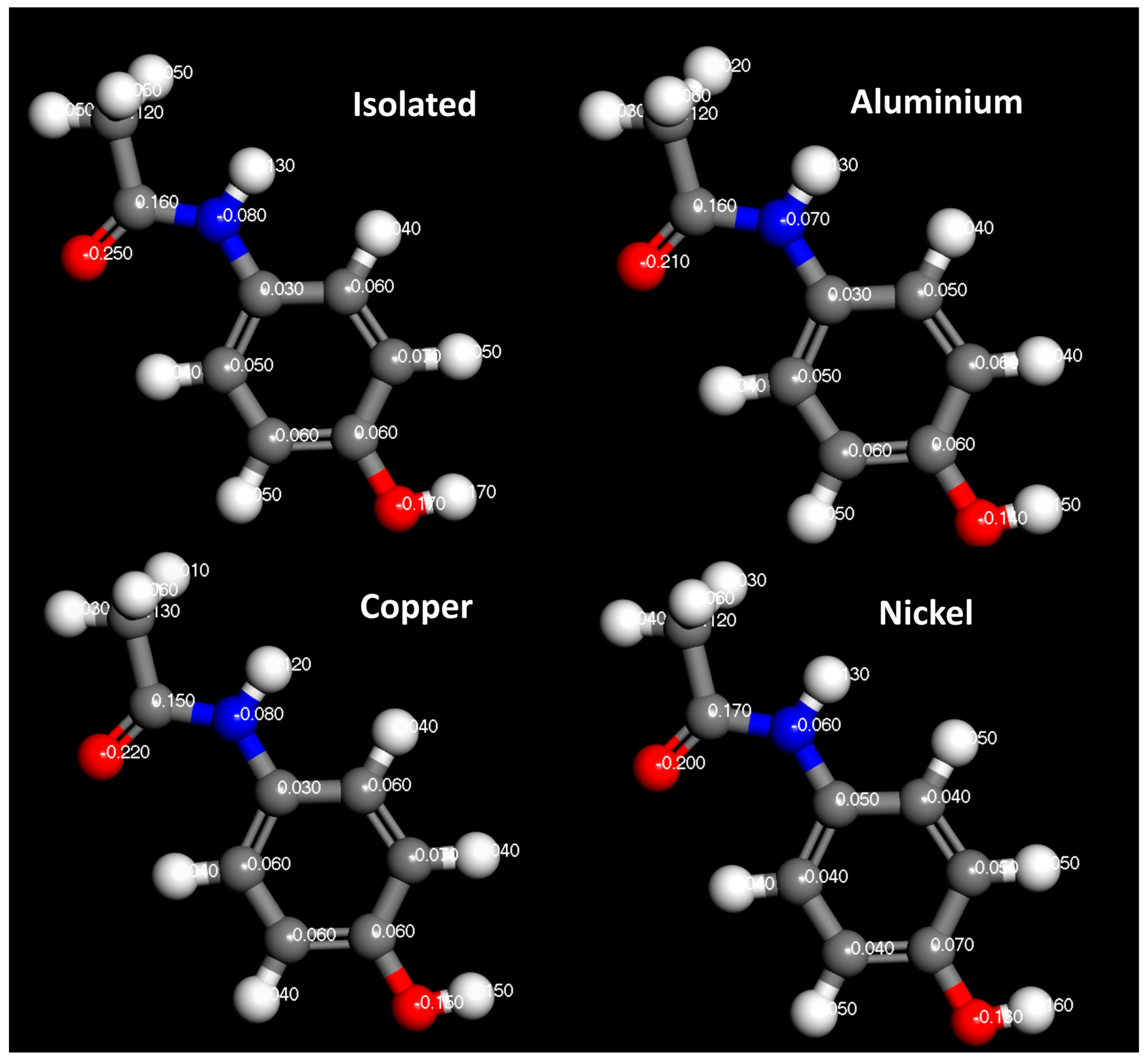
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matsusaka, S.; Maruyama, H.; Matsuyama, T.; Ghadiri, M. Triboelectric Charging of Powders: A Review. Chem. Eng. Sci. 2010, 65, 5781–5807. [Google Scholar] [CrossRef]
- Taghavivand, M.; Sowinski, A.; Mehrani, P. Triboelectric Effects of Continuity Additives and a Silica Catalyst Support on Polyethylene Fluidized Bed Wall Fouling. Chem. Eng. Sci. 2021, 245, 116882. [Google Scholar] [CrossRef]
- Nifuku, M.; Katoh, H. A Study on the Static Electrification of Powders during Pneumatic Transportation and the Ignition of Dust Cloud. Powder Technol. 2003, 135–136, 234–242. [Google Scholar] [CrossRef]
- Pu, Y.; Mazumder, M.; Cooney, C. Effects of Electrostatic Charging on Pharmaceutical Powder Blending Homogeneity. J. Pharm. Sci. 2009, 98, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Karner, S.; Maier, M.; Urbanetz, N.A.; Karner, S.; Maier, M.; Littringer, E.; Urbanetz, N.A. Surface Roughness Effects on the Tribo-Charging and Mixing Homogeneity of Adhesive Mixtures Used in Dry Powder Inhalers. Powder Technol. 2014, 264, 544–549. [Google Scholar] [CrossRef]
- Lacks, D.J.; Shinbrot, T. Long-Standing and Unresolved Issues in Triboelectric Charging. Nat. Rev. Chem. 2019, 3, 465–476. [Google Scholar] [CrossRef]
- Wang, N.; Zhao, X.B.; Wang, N.; Zhao, X. Atomic Force Microscopy—A Powerful Tool for Studying Contact Electrification. Adv. Mater. Technol. 2023, 8, 2201408. [Google Scholar] [CrossRef]
- Zafar, U.; Alfano, F.; Ghadiri, M. Evaluation of a New Dispersion Technique for Assessing Triboelectric Charging of Powders. Int. J. Pharm. 2018, 543, 151–159. [Google Scholar] [CrossRef]
- Nan, Y.; Shao, J.; Willatzen, M.; Wang, Z.L. Understanding Contact Electrification at Water/Polymer Interface. Res. A Sci. Partn. J. 2022, 2022, 9861463. [Google Scholar] [CrossRef]
- Alfano, F.O.; Di Renzo, A.; Di Maio, F.P.; Ghadiri, M. Computational Analysis of Triboelectrification Due to Aerodynamic Powder Dispersion. Powder Technol. 2021, 382, 491–504. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G.; Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; et al. Current Trends in Computer Aided Drug Design and a Highlight of Drugs Discovered via Computational Techniques: A Review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Rosenkranz, S.; Breitung-Faes, S.; Kwade, A.; Rosenkranz, S.; Breitung-Faes, S.; Kwade, A. Experimental Investigations and Modelling of the Ball Motion in Planetary Ball Mills. Powder Technol. 2011, 212, 224–230. [Google Scholar] [CrossRef]
- Wong, J.; Kwok, P.C.L.; Chan, H.-K. Electrostatics in Pharmaceutical Solids. Chem. Eng. Sci. 2015, 125, 225–237. [Google Scholar] [CrossRef]
- Lee, V.; James, N.M.; Waitukaitis, S.R.; Jaeger, H.M. Collisional Charging of Individual Submillimeter Particles: Using Ultrasonic Levitation to Initiate and Track Charge Transfer. Phys. Rev. Mater. 2018, 2, 35602. [Google Scholar] [CrossRef]
- Biegaj, K.W.; Rowland, M.G.; Lukas, T.M.; Heng, J.Y.Y. Surface Chemistry and Humidity in Powder Electrostatics: A Comparative Study between Tribocharging and Corona Discharge. ACS Omega 2017, 2, 1576–1582. [Google Scholar] [CrossRef]
- Byun, K.-E.; Cho, Y.; Seol, M.; Kim, S.; Kim, S.-W.; Shin, H.-J.; Park, S.; Hwang, S. Control of Triboelectrification by Engineering Surface Dipole and Surface Electronic State. ACS Appl. Mater. Interfaces 2016, 8, 18519–18525. [Google Scholar] [CrossRef]
- Li, L.-H.; Kontsevoi, O.Y.; Freeman, A.J. Atomic-Scale Understanding of the Interaction of Poly(3-Hexylthiophene) with the NiO (100) Surface: A First-Principles Study. J. Phys. Chem. C 2014, 118, 20298–20305. [Google Scholar] [CrossRef]
- Wu, J.; Wang, X.; Li, H.; Wang, F.; Hu, Y. First-Principles Investigations on the Contact Electrification Mechanism between Metal and Amorphous Polymers for Triboelectric Nanogenerators. Nano Energy 2019, 63, 103864. [Google Scholar] [CrossRef]
- Middleton, J.R.; Scott, A.J.; Storey, R.; Marucci, M.; Ghadiri, M. Prediction of the Effective Work Function of Aspirin and Paracetamol Crystals by Density Functional Theory—A First-Principles Study. Cryst. Growth Des. 2023, 23, 6308–6317. [Google Scholar] [CrossRef] [PubMed]
- Brunsteiner, M.; Zellnitz, S.; Pinto, J.T.; Karrer, J.; Paudel, A. Can We Predict Trends in Tribo-Charging of Pharmaceutical Materials from First Principles? Powder Technol. 2019, 356, 892–898. [Google Scholar] [CrossRef]
- Maurer, R.J.; Freysoldt, C.; Reilly, A.M.; Brandenburg, J.G.; Hofmann, O.T.; Björkman, T.; Lebègue, S.; Tkatchenko, A. Advances in Density-Functional Calculations for Materials Modeling. Annu. Rev. Mater. Res. 2019, 49, 1–30. [Google Scholar] [CrossRef]
- Reilly, A.M.; Cooper, R.I.; Adjiman, C.S.; Bhattacharya, S.; Boese, A.D.; Brandenburg, J.G.; Bygrave, P.J.; Bylsma, R.; Campbell, J.E.; Car, R.; et al. Report on the Sixth Blind Test of Organic Crystal Structure Prediction Methods. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Obot, I.B.; Macdonald, D.D.; Gasem, Z.M.; Obot, I.B.; Macdonald, D.D.; Gasem, Z.M. Density Functional Theory (DFT) as a Powerful Tool for Designing New Organic Corrosion Inhibitors. Part 1: An Overview. Corros. Sci. 2015, 99, 1–30. [Google Scholar] [CrossRef]
- Robinson, R.L.M.; Geatches, D.; Vatvani, D.R.M.; Marchese Robinson, R.L.; Geatches, D.; Morris, C.; Mackenzie, R.; Maloney, A.G.P.; Roberts, K.J.; Moldovan, A.; et al. Evaluation of Force-Field Calculations of Lattice Energies on a Large Public Dataset, Assessment of Pharmaceutical Relevance, and Comparison to Density Functional Theory. J. Chem. Inf. Model. 2019, 59, 4778–4792. [Google Scholar] [CrossRef]
- Akkermans, R.L.C.; Spenley, N.A.; Robertson, S.H. COMPASS III: Automated Fitting Workflows and Extension to Ionic Liquids. Mol. Simul. 2021, 47, 540–551. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Für Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 73005. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, R.W.G. Crystal Structures, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1965; Volume 1. [Google Scholar]
- Bruhn, J.F.; Scapin, G.; Cheng, A.; Mercado, B.Q.; Waterman, D.G.; Ganesh, T.; Dallakyan, S.; Read, B.N.; Nieusma, T.; Lucier, K.W.; et al. Small Molecule Microcrystal Electron Diffraction for the Pharmaceutical Industry—Lessons Learned from Examining over Fifty Samples. Front. Mol. Biosci. 2021, 8, 648603. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theor. Chim. Acta. 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Lowell, J.; Rose-Innes, A.C. Contact Electrification. Adv. Phys. 1980, 29, 947–1023. [Google Scholar] [CrossRef]
- Fatti, G.; Ciniero, A.; Dini, D.; Fatti, G.; Ciniero, A.; Ko, H.; Lee, H.U.; Na, Y.; Jeong, C.K.; Lee, S.; et al. Rational Design Strategy for Triboelectric Nanogenerators Based on Electron Back Flow and Ionic Defects: The Case of Polytetrafluoroethylene. Adv. Electron. Mater. 2023, 9, 2300333. [Google Scholar] [CrossRef]
- Singh-Miller, N.E.; Marzari, N. Surface Energies, Work Functions, and Surface Relaxations of Low-Index Metallic Surfaces from First Principles. Phys. Rev. B 2009, 80, 235407. [Google Scholar] [CrossRef]
- Kawano, H. Effective Work Functions of the Elements: Database, Most Probable Value, Previously Recommended Value, Polycrystalline Thermionic Contrast, Change at Critical Temperature, Anisotropic Dependence Sequence, Particle Size Dependence. Prog. Surf. Sci. 2022, 97, 100583. [Google Scholar] [CrossRef]
- Antony, A.C.; Thelen, D.; Zhelev, N.; Adib, K.; Manley, R.G. Electronic Charge Transfer during Metal/SiO2 Contact: Insight from Density Functional Theory. J. Appl. Phys. 2021, 129, 65304. [Google Scholar] [CrossRef]
- Tao, J.; Wang, L.; Li, J.; Dai, Z. Contact Electrification and Adhesion between Carbon Nanotube and Graphene on Metal Surfaces: Insights from First-Principles Study. J. Bionic Eng. 2022, 19, 103–112. [Google Scholar] [CrossRef]
- Veregin, R.P.N.; Hawkins, M.S.; Li, Q.; Gusarov, S.; Kovalenko, A.; Veregin, R.P.N.; Hawkins, M.S. Linking the Chemistry and Physics of Electronic Charge Transfer in Insulators: Theory and Experiment. J. Imaging Sci. Technol. 2013, 57, 30401–30412. [Google Scholar] [CrossRef]
- Li, Q.; Gusarov, S.; Kovalenko, A.; Veregin, R.P.N.; Veregin, R.P.N. Interfacial Water and Triboelectric Charging: A Theoretical Study of the Role of Water on the PMMA/Silica Tribopair. J. Imaging Sci. Technol. 2015, 59, 40502–40515. [Google Scholar] [CrossRef]
- Wu, J.; Wang, X.; Li, H.; Wang, F.; Yang, W.; Hu, Y. Insights into the Mechanism of Metal-Polymer Contact Electrification for Triboelectric Nanogenerator via First-Principles Investigations. Nano Energy 2018, 48, 607–616. [Google Scholar] [CrossRef]
- Yoshida, M.; Ii, N.; Shimosaka, A.; Shirakawa, Y.; Hidaka, J. Experimental and Theoretical Approaches to Charging Behavior of Polymer Particles. Chem. Eng. Sci. 2006, 61, 2239–2248. [Google Scholar] [CrossRef]
- Lin, S.; Xu, C.; Xu, L.; Wang, Z.L. The Overlapped Electron-Cloud Model for Electron Transfer in Contact Electrification. Adv. Funct. Mater. 2020, 30, 1909724. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System—Xc Functional | Kinetic Energy Cutoff | MP Grid Sampling (a × b × c) | No. of k-Points |
|---|---|---|---|
| Al—LDA, CA-PZ | 210 | 6 × 6 × 6 | 10 |
| Al—GGA, PBE | 210 | 6 × 6 × 6 | 10 |
| Cu—LDA, CA-PZ | 450 | 8 × 8 × 8 | 20 |
| Cu—GGA, PBE | 450 | 8 × 8 × 8 | 20 |
| Ni—LDA, CA-PZ | 440 | 8 × 8 × 8 | 20 |
| Ni—GGA, PBE | 440 | 8 × 8 × 8 | 20 |
| Paracetamol—LDA, CA-PZ | 630 | 4 × 3 × 2 | 8 |
| Paracetamol—GGA, PBE | 630 | 4 × 3 × 2 | 8 |
| Lattice Parameters (Å) | |||||||
|---|---|---|---|---|---|---|---|
| a | b | c | α | β | γ | ||
| Al | Experimental [29] | 4.05 | - | - | 90 | 90 | 90 |
| DFT—(LDA, CA-PZ) | 3.99 | - | - | 90 | 90 | 90 | |
| DFT—(GGA, PBE) | 4.04 | - | - | 90 | 90 | 90 | |
| MM—COMPASSIII | 4.04 | - | - | 90 | 90 | 90 | |
| Cu | Experimental [29] | 3.60 | - | - | 90 | 90 | 90 |
| DFT—(LDA, CA-PZ) | 3.52 | - | - | 90 | 90 | 90 | |
| DFT—(GGA, PBE) | 3.63 | - | - | 90 | 90 | 90 | |
| MM—COMPASSIII | 3.61 | - | - | 90 | 90 | 90 | |
| Ni | Experimental [29] | 3.54 | - | - | 90 | 90 | 90 |
| DFT—(LDA, CA-PZ) | 3.42 | - | - | 90 | 90 | 90 | |
| DFT—(GGA, PBE) | 3.51 | - | - | 90 | 90 | 90 | |
| MM—COMPASSIII | 3.52 | - | - | 90 | 90 | 90 | |
| Paracet. | Experimental [30] | 7.07 | 9.19 | 11.49 | 90 | 98.64 | 90 |
| DFT—(GGA, PBE) | 7.02 | 9.04 | 11.76 | 90 | 99.32 | 90 | |
| MM—COMPASSIII | 7.14 | 8.91 | 11.70 | 90 | 97.03 | 90 | |
| System | Fermi Energy (eV) | Vacuum Energy (eV) | Theoretical Work Function (eV) |
|---|---|---|---|
| Isolated Paracetamol | −1.673 | 0.012 | 1.685 |
| Al Surface | −2.215 | 0.073 | 2.288 |
| Cu Surface | −2.879 | 0.073 | 2.952 |
| Ni Surface | −3.403 | 0.057 | 3.460 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Middleton, J.R.; Ghadiri, M.; Scott, A.J. Triboelectric Charging Properties of the Functional Groups of Common Pharmaceutical Materials Using Density Functional Theory Calculations. Pharmaceutics 2024, 16, 433. https://doi.org/10.3390/pharmaceutics16030433
Middleton JR, Ghadiri M, Scott AJ. Triboelectric Charging Properties of the Functional Groups of Common Pharmaceutical Materials Using Density Functional Theory Calculations. Pharmaceutics. 2024; 16(3):433. https://doi.org/10.3390/pharmaceutics16030433
Chicago/Turabian StyleMiddleton, James R., Mojtaba Ghadiri, and Andrew J. Scott. 2024. "Triboelectric Charging Properties of the Functional Groups of Common Pharmaceutical Materials Using Density Functional Theory Calculations" Pharmaceutics 16, no. 3: 433. https://doi.org/10.3390/pharmaceutics16030433
APA StyleMiddleton, J. R., Ghadiri, M., & Scott, A. J. (2024). Triboelectric Charging Properties of the Functional Groups of Common Pharmaceutical Materials Using Density Functional Theory Calculations. Pharmaceutics, 16(3), 433. https://doi.org/10.3390/pharmaceutics16030433