
GRPR-Antagonists Carrying DOTAGA-Chelator via Positively Charged Linkers: Perspectives for Prostate Cancer Theranostics

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Peptides and Reagents
2.2. Radiolabeling and Radiochemical Studies
2.3. In Vitro Studies
2.3.1. In Vitro Binding Specificity Assay
2.3.2. Affinity Measurements
2.3.3. Cellular Internalization
2.4. In Vivo Studies
2.4.1. In Vivo Stability Experiments
2.4.2. Biodistribution
2.4.3. SPECT/CT Imaging
3. Results
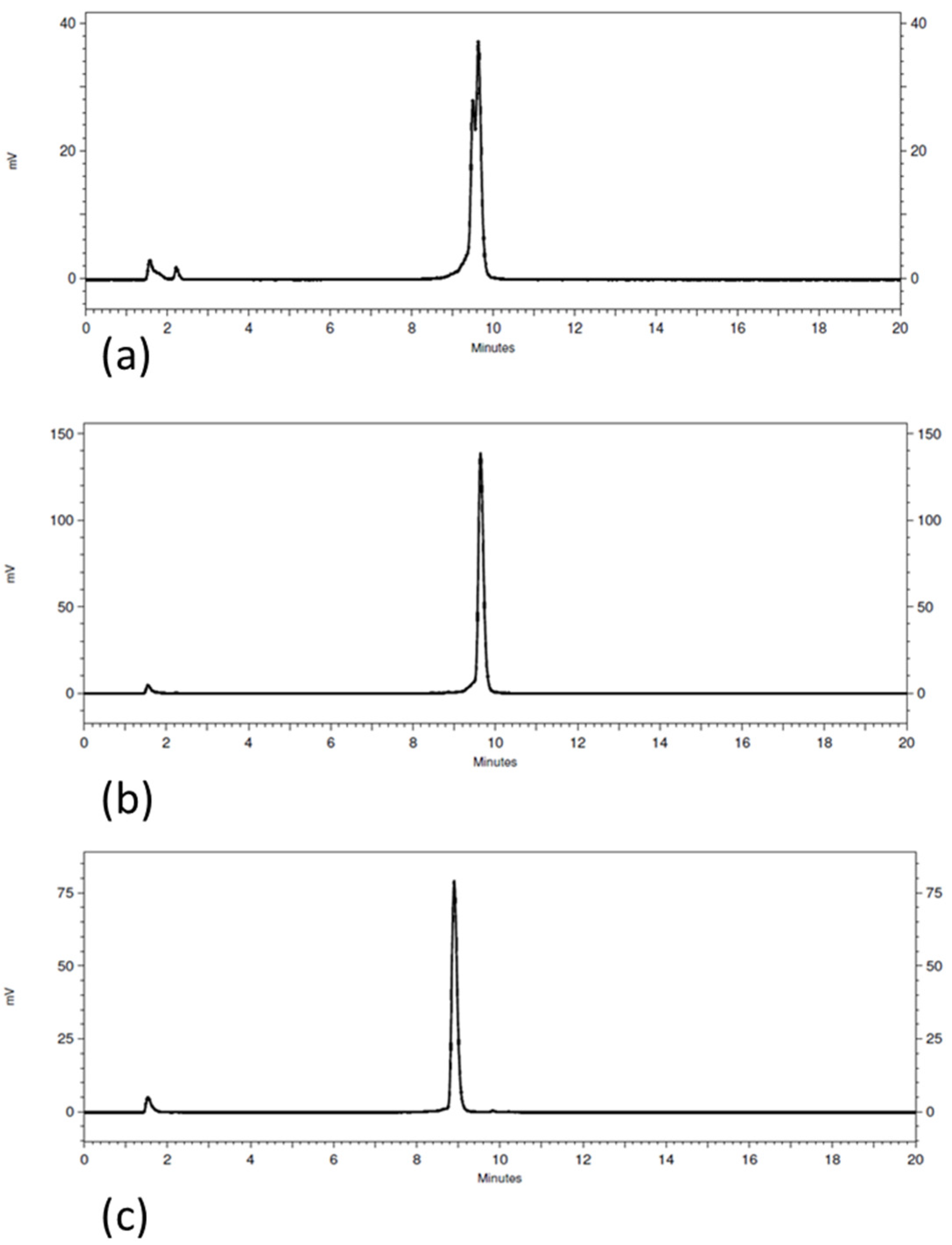
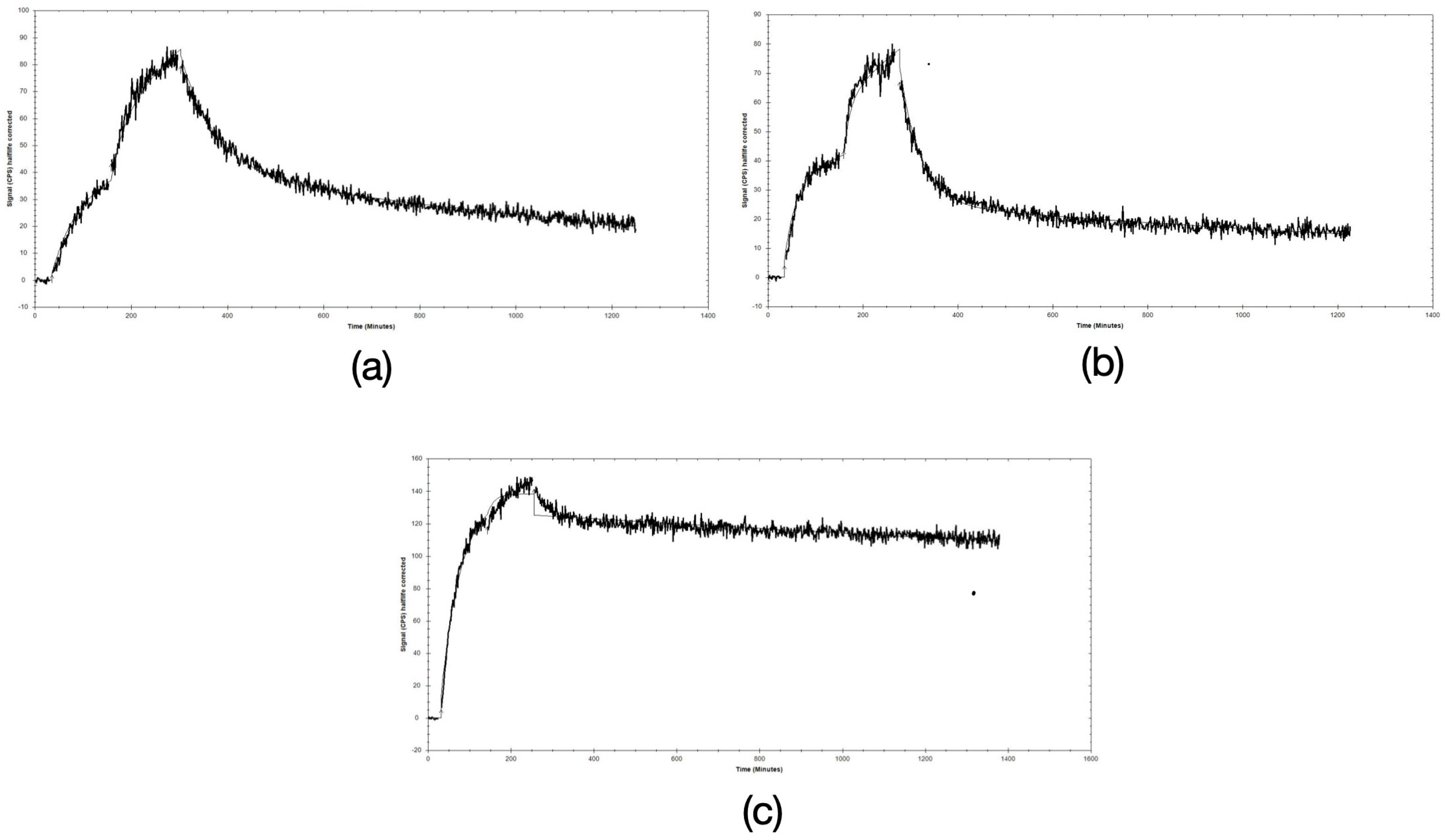
3.1. Radiolabeling and Radiochemical Stability
3.2. In Vitro Studies
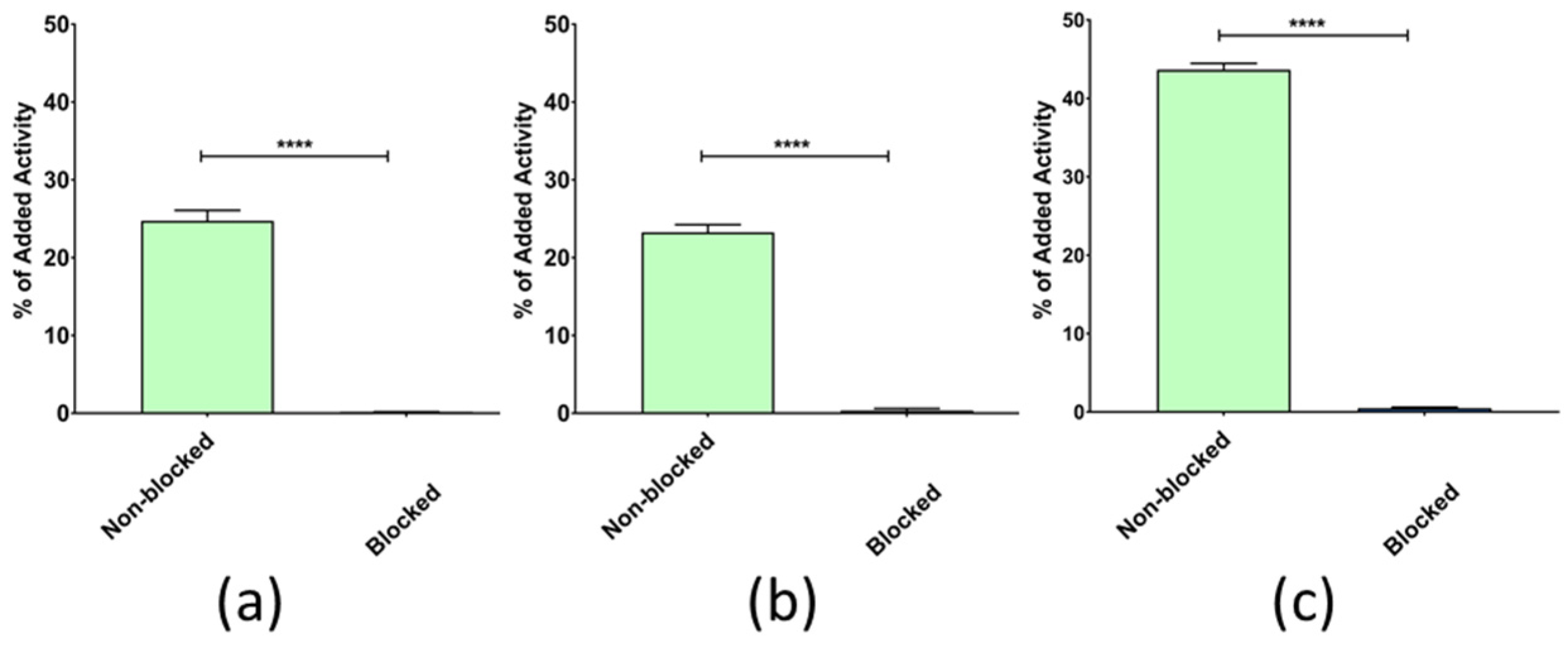
3.2.1. In Vitro GRPR Binding Specificity
3.2.2. GRPR-Affinity Measurements
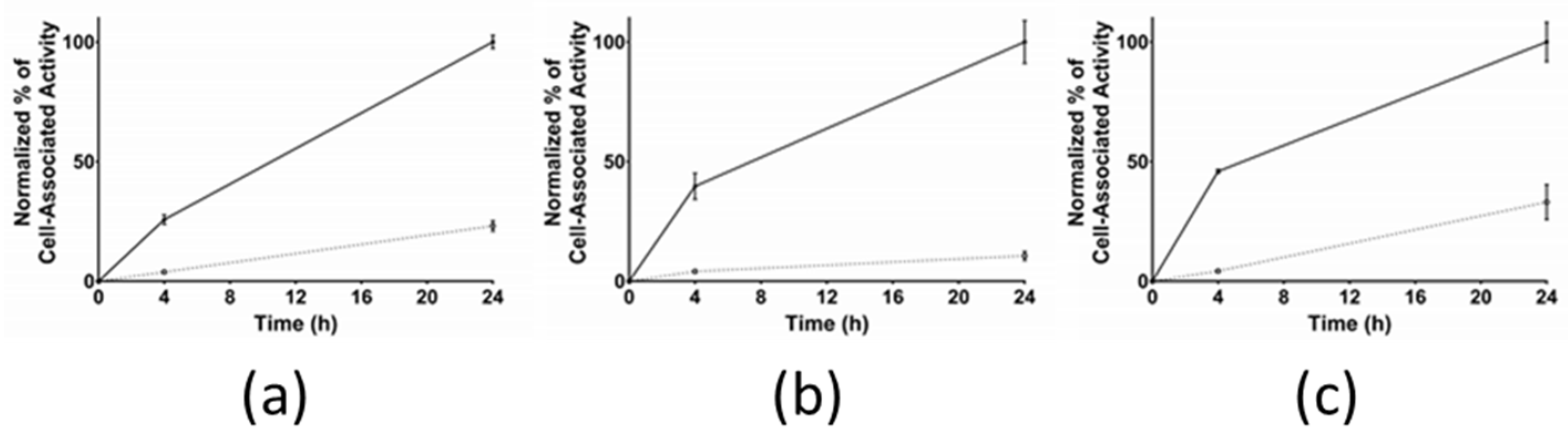
3.2.3. Cellular Internalization
3.3. In Vivo Studies
3.3.1. In Vivo Metabolic Stability
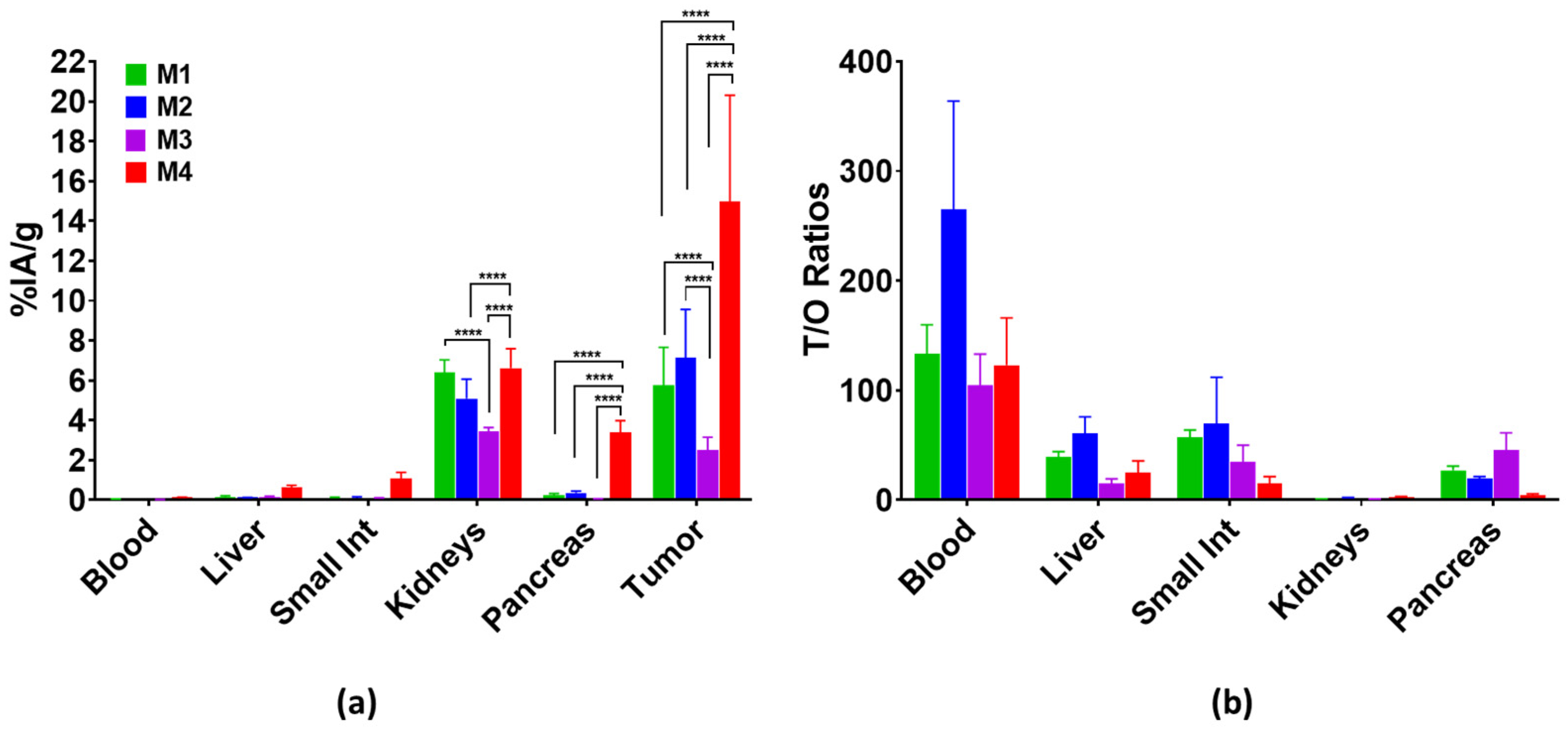
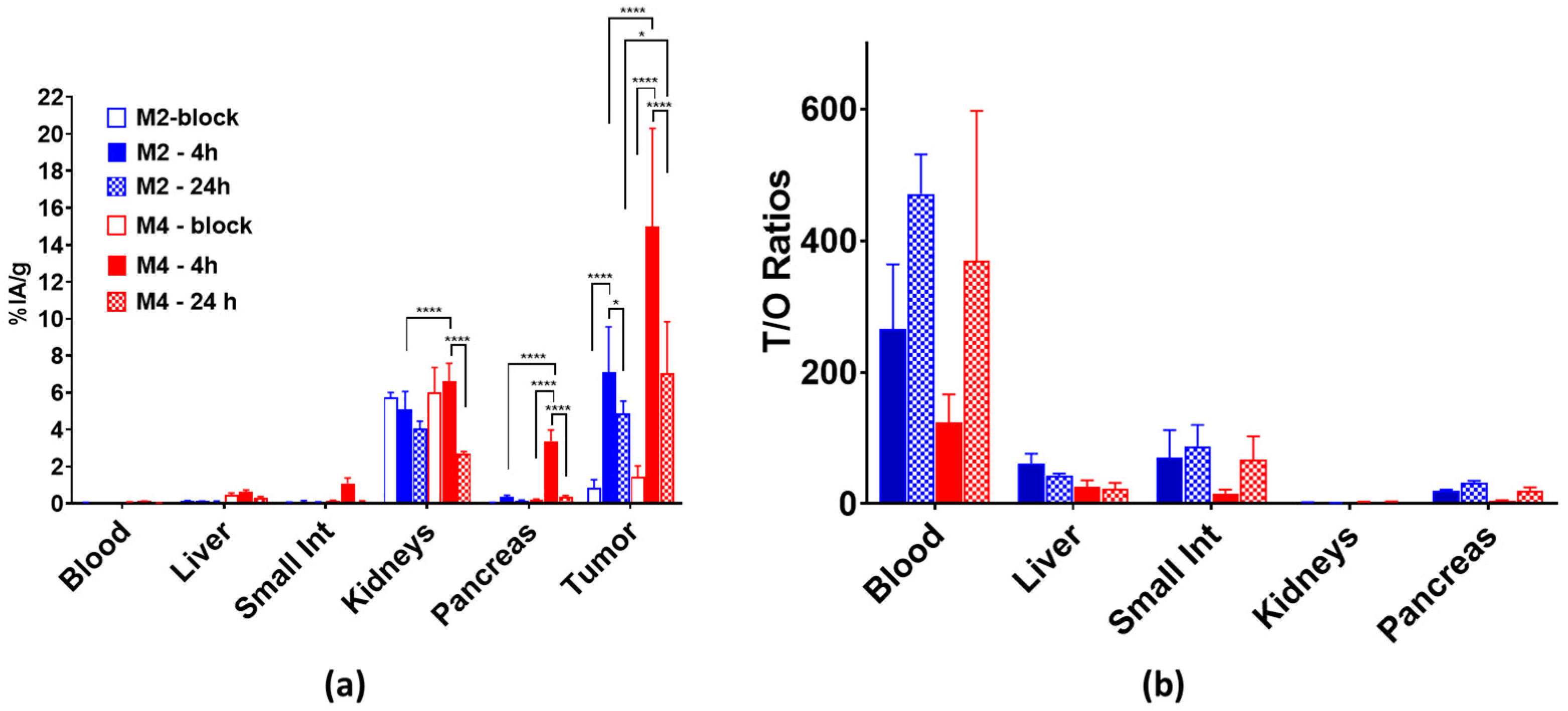
3.3.2. Biodistribution
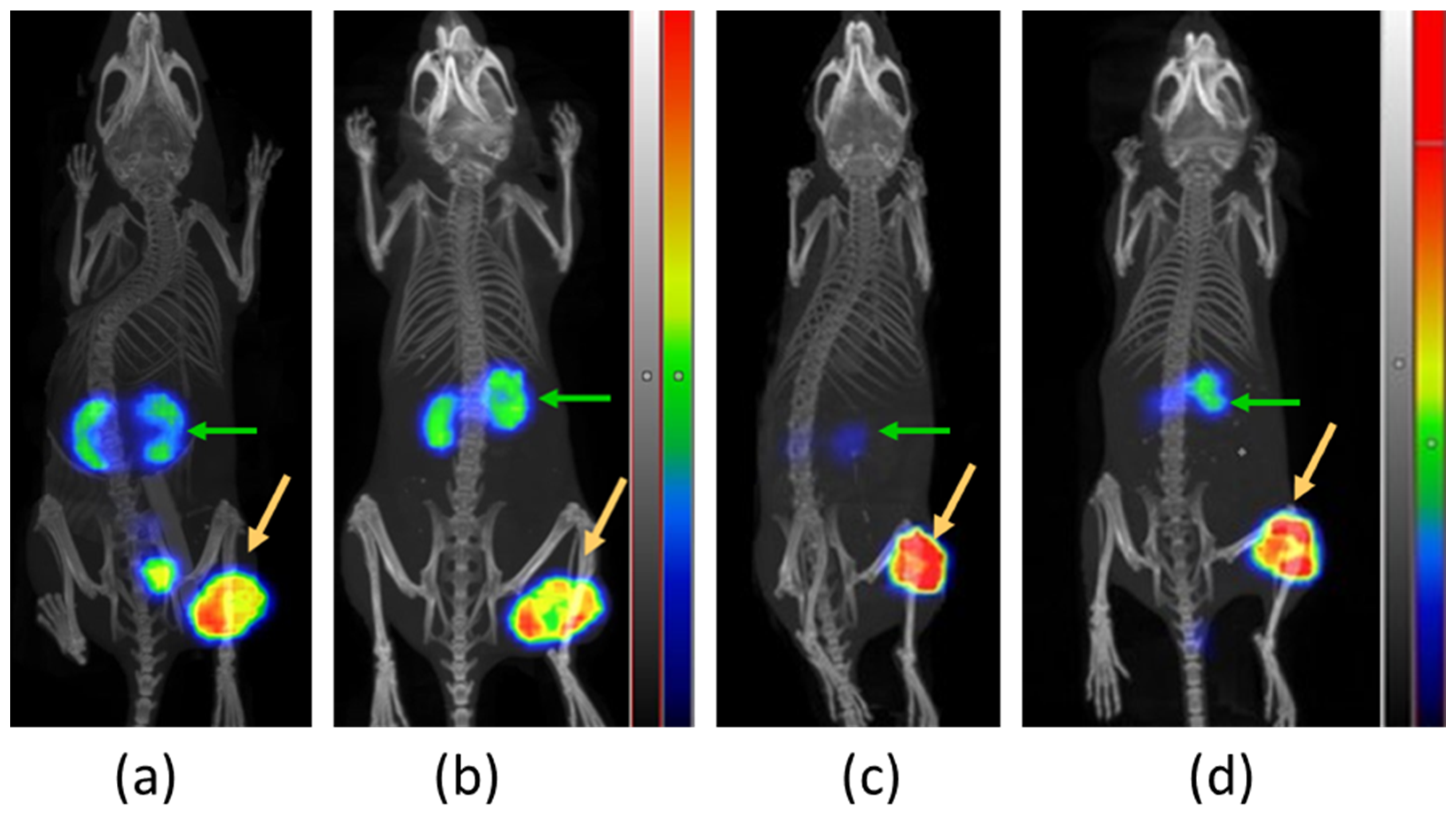
3.3.3. SPECT/CT Imaging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gandaglia, G.; Leni, R.; Bray, F.; Fleshner, N.; Freedland, S.J.; Kibel, A.; Stattin, P.; Van Poppel, H.; La Vecchia, C. Epidemiology and Prevention of Prostate Cancer. Eur. Urol. Oncol. 2021, 4, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Aboagye, E.O.; Barwick, T.D.; Haberkorn, U. Radiotheranostics in Oncology: Making Precision Medicine Possible. CA Cancer J. Clin. 2023, 73, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Bodei, L.; Herrmann, K.; Schöder, H.; Scott, A.M.; Lewis, J.S. Radiotheranostics in Oncology: Current Challenges and Emerging Opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 534–550. [Google Scholar] [CrossRef] [PubMed]
- Fanti, S.; Briganti, A.; Emmett, L.; Fizazi, K.; Gillessen, S.; Goffin, K.; Hadaschik, B.A.; Herrmann, K.; Kunikowska, J.; Maurer, T.; et al. EAU-EANM Consensus Statements on the Role of Prostate-Specific Membrane Antigen Positron Emission Tomography/Computed Tomography in Patients with Prostate Cancer and with Respect to [177Lu]Lu-PSMA Radioligand Therapy. Eur. Urol. Oncol. 2022, 5, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Baratto, L.; Song, H.; Duan, H.; Hatami, N.; Bagshaw, H.P.; Buyyounouski, M.; Hancock, S.; Shah, S.; Srinivas, S.; Swift, P.; et al. PSMA- and GRPR-Targeted PET: Results from 50 Patients with Biochemically Recurrent Prostate Cancer. J. Nucl. Med. 2021, 62, 1545–1549. [Google Scholar] [CrossRef] [PubMed]
- Mansi, R.; Nock, B.A.; Dalm, S.U.; Busstra, M.B.; van Weerden, W.M.; Maina, T. Radiolabeled Bombesin Analogs. Cancers 2021, 13, 5766. [Google Scholar] [CrossRef] [PubMed]
- Ananias, H.J.K.; van den Heuvel, M.C.; Helfrich, W.; de Jong, I.J. Expression of the Gastrin-Releasing Peptide Receptor, the Prostate Stem Cell Antigen and the Prostate-Specific Membrane Antigen in Lymph Node and Bone Metastases of Prostate Cancer. Prostate 2009, 69, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Minamimoto, R.; Sonni, I.; Hancock, S.; Vasanawala, S.; Loening, A.; Gambhir, S.S.; Iagaru, A. Prospective Evaluation of 68 Ga-RM2 PET/MRI in Patients with Biochemical Recurrence of Prostate Cancer and Negative Findings on Conventional Imaging. J. Nucl. Med. 2018, 59, 803–808. [Google Scholar] [CrossRef]
- Reubi, J.C.; Wenger, S.; Schmuckli-Maurer, J.; Schaer, J.-C.; Gugger, M. Bombesin Receptor Subtypes in Human Cancers: Detection with the Universal Radioligand 125I-[D-Tyr6,beta-Ala11,Phe13,Nle14]Bombesin(6-14). Clin. Cancer Res. 2002, 8, 1139–1146. [Google Scholar]
- Reubi, J.C.; Körner, M.; Waser, B.; Mazzucchelli, L.; Guillou, L. High Expression of Peptide Receptors as a Novel Target in Gastrointestinal Stromal Tumours. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 803–810. [Google Scholar] [CrossRef]
- Gugger, M.; Reubi, J.C. Gastrin-Releasing Peptide Receptors in Non-Neoplastic and Neoplastic Human Breast. Am. J. Pathol. 1999, 155, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Baun, C.; Naghavi-Behzad, M.; Hildebrandt, M.G.; Gerke, O.; Thisgaard, H. Gastrin-Releasing Peptide Receptor as a Theranostic Target in Breast Cancer: A Systematic Scoping Review. Semin. Nucl. Med. 2024, 54, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Morgat, C.; MacGrogan, G.; Brouste, V.; Vélasco, V.; Sévenet, N.; Bonnefoi, H.; Fernandez, P.; Debled, M.; Hindié, E. Expression of Gastrin-Releasing Peptide Receptor in Breast Cancer and Its Association with Pathologic, Biologic, and Clinical Parameters: A Study of 1,432 Primary Tumors. J. Nucl. Med. 2017, 58, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Markwalder, R.; Reubi, J.C. Gastrin-Releasing Peptide Receptors in the Human Prostate: Relation to Neoplastic Transformation. Cancer Res. 1999, 59, 1152–1159. [Google Scholar] [PubMed]
- Beer, M.; Montani, M.; Gerhardt, J.; Wild, P.J.; Hany, T.F.; Hermanns, T.; Müntener, M.; Kristiansen, G. Profiling Gastrin-Releasing Peptide Receptor in Prostate Tissues: Clinical Implications and Molecular Correlates. Prostate 2012, 72, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Körner, M.; Waser, B.; Rehmann, R.; Reubi, J.C. Early Over-expression of GRP Receptors in Prostatic Carcinogenesis. Prostate 2014, 74, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Kanellopoulos, P.; Joosten, L.; Mansi, R.; Maina, T. Peptide Radioligands in Cancer Theranostics: Agonists and Antagonists. Pharmaceuticals 2023, 16, 674. [Google Scholar] [CrossRef] [PubMed]
- Maina, T.; Nock, B.A.; Kulkarni, H.; Singh, A.; Baum, R.P. Theranostic Prospects of Gastrin-Releasing Peptide Receptor–Radioantagonists in Oncology. PET Clin. 2017, 12, 297–309. [Google Scholar] [CrossRef]
- Jensen, R.T.; Battey, J.F.; Spindel, E.R.; Benya, R.V. International Union of Pharmacology. LXVIII. Mammalian Bombesin Receptors: Nomenclature, Distribution, Pharmacology, Signaling, and Functions in Normal and Disease States. Pharmacol. Rev. 2008, 60, 1–42. [Google Scholar] [CrossRef]
- de Castiglione, R.; Gozzini, L. Bombesin Receptor Antagonists. Crit. Rev. Oncol. Hematol. 1996, 24, 117–151. [Google Scholar] [CrossRef] [PubMed]
- Azay, J.; Nagain, C.; Llinares, M.; Devin, C.; Fehrentz, J.; Bernad, N.; Roze, C.; Martinez, J. Comparative Study of In Vitro and In Vivo Activities of Bombesin Pseudopeptide Analogs Modified on the C-Terminal Dipeptide Fragment. Peptides 1998, 19, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Mansi, R.; Wang, X.; Forrer, F.; Waser, B.; Cescato, R.; Graham, K.; Borkowski, S.; Reubi, J.C.; Maecke, H.R. Development of a Potent DOTA-Conjugated Bombesin Antagonist for Targeting GRPr-Positive Tumours. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Price, E.W.; Orvig, C. Matching Chelators to Radiometals for Radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Mitran, B.; Rinne, S.S.; Konijnenberg, M.W.; Maina, T.; Nock, B.A.; Altai, M.; Vorobyeva, A.; Larhed, M.; Tolmachev, V.; de Jong, M.; et al. Trastuzumab Cotreatment Improves Survival of Mice with PC-3 Prostate Cancer Xenografts Treated with the GRPR Antagonist 177Lu-DOTAGA-PEG 2-RM26. Int. J. Cancer 2019, 145, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Abouzayed, A.; Kanellopoulos, P.; Gorislav, A.; Tolmachev, V.; Maina, T.; Nock, B.A.; Orlova, A. Preclinical Characterization of a Stabilized Gastrin-Releasing Peptide Receptor Antagonist for Targeted Cancer Theranostics. Biomolecules 2023, 13, 1134. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Del Pozzo, L.; Abiraj, K.; Mansi, R.; Tamma, M.L.; Cescato, R.; Waser, B.; Weber, W.A.; Reubi, J.C.; Maecke, H.R. PET of Somatostatin Receptor–Positive Tumors Using 64Cu- and 68Ga-Somatostatin Antagonists: The Chelate Makes the Difference. J. Nucl. Med. 2011, 52, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Schreck, M.-V.; Burgard, C.; Schmidtke, A.; Hierlmeier, I.; Stemler, T.; Maus, S.; Rosar, F.; Jung, M.; Speicher, A.; Ezziddin, S.; et al. Radiometal Complexes as Pharmacokinetic Modifiers: A Potent 68Ga-Labeled Gastrin-Releasing Peptide Receptor Antagonist Based on the Macrocyclic Metal Chelator NODIA-Me. Mol. Pharm. 2023, 20, 6463–6473. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, M.; van Eerd-Vismale, J.; Oyen, W.J.G.; de Jong, M.; Zhang, H.; Rolleman, E.; Maecke, H.R.; Behe, M.; Boerman, O. Indication for Different Mechanisms of Kidney Uptake of Radiolabeled Peptides. J. Nucl. Med. 2007, 48, 596–601. [Google Scholar] [CrossRef]
- Reile, H.; Armatis, P.E.; Schally, A.V. Characterization of High-affinity Receptors for Bombesin/Gastrin Releasing Peptide on the Human Prostate Cancer Cell Lines PC-3 and DU-145: Internalization of Receptor Bound 125I-(Tyr4)Bombesin by Tumor Cells. Prostate 1994, 25, 29–38. [Google Scholar] [CrossRef]
- Gu, J.; Noe, A.; Chandra, P.; Al-Fayoumi, S.; Ligueros-Saylan, M.; Sarangapani, R.; Maahs, S.; Ksander, G.; Rigel, D.F.; Jeng, A.Y.; et al. Pharmacokinetics and Pharmacodynamics of LCZ696, a Novel Dual-Acting Angiotensin Receptor—Neprilysin Inhibitor (ARNi). J. Clin. Pharmacol. 2010, 50, 401–414. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Packer, M.; Solomon, S.D. Neprilysin Inhibition for Heart Failure. N. Engl. J. Med. 2014, 371, 2336–2337. [Google Scholar] [CrossRef] [PubMed]
- Ayalasomayajula, S.; Langenickel, T.; Pal, P.; Boggarapu, S.; Sunkara, G. Clinical Pharmacokinetics of Sacubitril/Valsartan (LCZ696): A Novel Angiotensin Receptor-Neprilysin Inhibitor. Clin. Pharmacokinet. 2017, 56, 1461–1478. [Google Scholar] [CrossRef] [PubMed]
- Kanellopoulos, P.; Kaloudi, A.; Rouchota, M.; Loudos, G.; de Jong, M.; Krenning, E.P.; Nock, B.A.; Maina, T. One Step Closer to Clinical Translation: Enhanced Tumor Targeting of [99mTc]Tc-DB4 and [111In]In-SG4 in Mice Treated with Entresto. Pharmaceutics 2020, 12, 1145. [Google Scholar] [CrossRef] [PubMed]
- Chatalic, K.L.S.; Kwekkeboom, D.J.; de Jong, M. Radiopeptides for Imaging and Therapy: A Radiant Future. J. Nucl. Med. 2015, 56, 1809–1812. [Google Scholar] [CrossRef] [PubMed]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide Chemistry Toolbox—Transforming Natural Peptides into Peptide Therapeutics. Bioorg. Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic Therapeutic Peptides: Science and Market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Adessi, C.; Soto, C. Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Curr. Med. Chem. 2002, 9, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Maina, T.; Krenning, E.P.; de Jong, M. “To Serve and Protect”: Enzyme Inhibitors as Radiopeptide Escorts Promote Tumor Targeting. J. Nucl. Med. 2014, 55, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Roques, B.P.; Noble, F.; Daugé, V.; Fournié-Zaluski, M.C.; Beaumont, A. Neutral Endopeptidase 24.11: Structure, Inhibition, and Experimental and Clinical Pharmacology. Pharmacol. Rev. 1993, 45, 87–146. [Google Scholar]
- Roques, B.P. Zinc Metallopeptidases: Active Site Structure and Design of Selective and Mixed Inhibitors: New Approaches in the Search for Analgesics and Anti-Hypertensives. Biochem. Soc. Trans. 1993, 21, 678–685. [Google Scholar] [CrossRef] [PubMed]
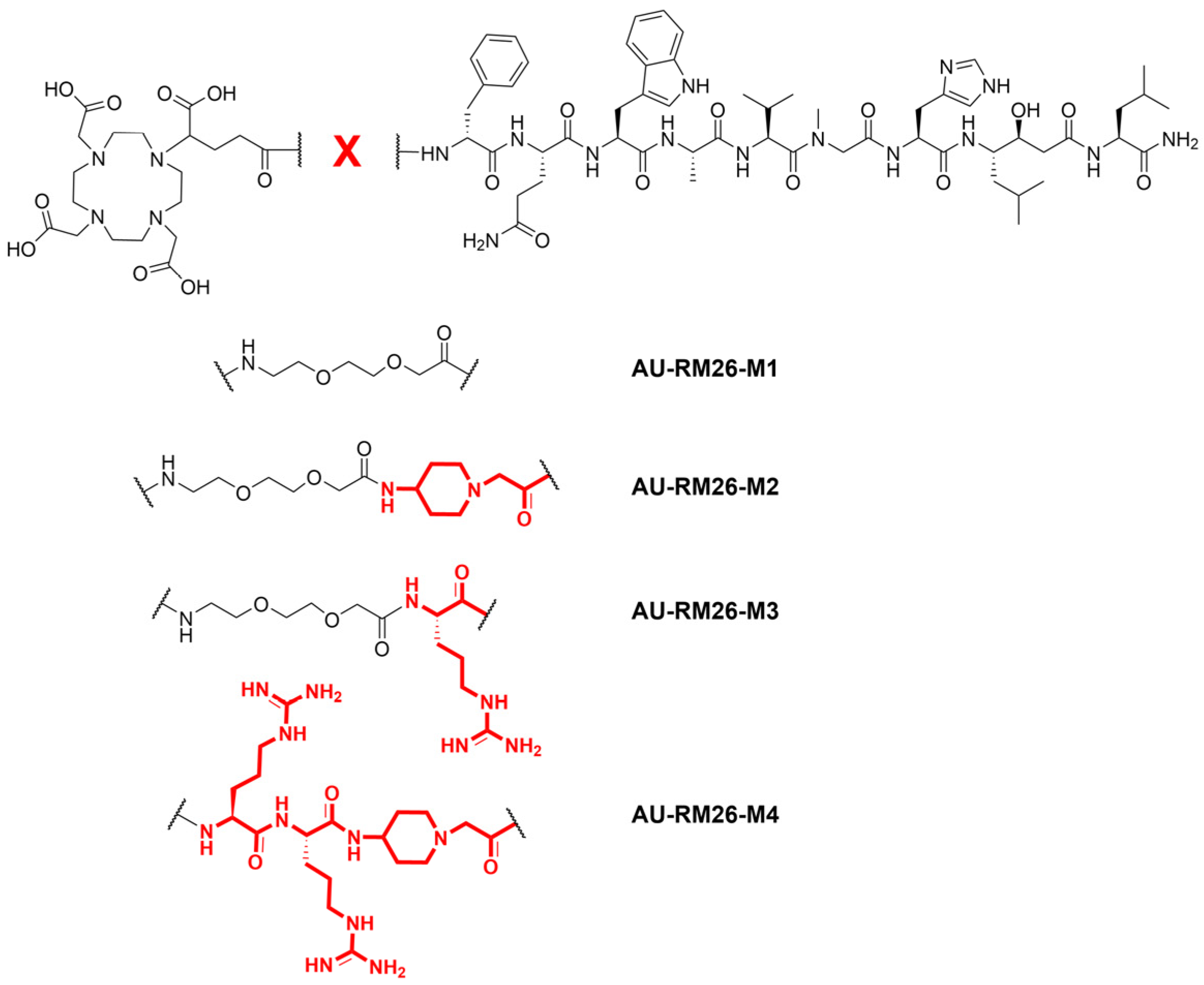








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | iTLC * RCY (%) (n) | ×1000 EDTA (1 h) (n) | PBS (n) | HPLC ** RCP (%) |
|---|---|---|---|---|
| [111In]In-AU-RM26-M2 | 97 ± 3 (7) | 5.7 ± 0.7% (3) | 5.7 ± 0.2% (3) | 93 ± 3 |
| [111In]In-AU-RM26-M3 | 98 ± 2 (5) | 3.9 ± 0.3% (3) | 3.7 ± 0.7% (3) | 96.1 ± 0.8 |
| [111In]In-AU-RM26-M4 | 99 ± 1 (5) | 3.2 ± 0.1% (3) | 1.9 ± 0.1% (3) | 94 ± 2 |
| Interaction Constants | [111In]In-AU-RM26-M2 | [111In]In-AU-RM26-M3 | [111In]In-AU-RM26-M4 |
|---|---|---|---|
| ka1 (M−1s−1) | 7.93 × 104 | 1.02 × 105 | 3.44 × 105 |
| kd1 (s−1) | 2.50 × 10−5 | 2.50 × 10−5 | 2.00 × 10−6 |
| KD1 (M) | 3.15 × 10−10 | 2.45 × 10−10 | 5.80 × 10−12 |
| ka2 (M−1s−1) | 1.39 × 105 | 2.82 × 105 | 3.46 × 105 |
| kd2 (s−1) | 2.33 × 104 | 3.94 × 10−4 | 2.08 × 10−6 |
| KD2 (M) | 1.68 × 10−9 | 1.40 × 10−9 | 6.0 × 10−12 |
| Compound | Control | Entresto® Treated | Control/ Entresto® |
|---|---|---|---|
| [111In]In-AU-RM26-M1 (ref.) | 88 ± 8 | 82 ± 1 NS | NS |
| [111In]In-AU-RM26-M2 | 78 ± 2 * | 91 ± 2 NS | p < 0.001 |
| [111In]In-AU-RM26-M3 | 25 ± 5 *** | 94 ± 0.8 NS | p < 0.0001 |
| [111In]In-AU-RM26-M4 | 83 ± 2 NS | 92 ± 1 NS | p < 0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obeid, K.; Kanellopoulos, P.; Abouzayed, A.; Mattsson, A.; Tolmachev, V.; Nock, B.A.; Maina, T.; Orlova, A. GRPR-Antagonists Carrying DOTAGA-Chelator via Positively Charged Linkers: Perspectives for Prostate Cancer Theranostics. Pharmaceutics 2024, 16, 513. https://doi.org/10.3390/pharmaceutics16040513
Obeid K, Kanellopoulos P, Abouzayed A, Mattsson A, Tolmachev V, Nock BA, Maina T, Orlova A. GRPR-Antagonists Carrying DOTAGA-Chelator via Positively Charged Linkers: Perspectives for Prostate Cancer Theranostics. Pharmaceutics. 2024; 16(4):513. https://doi.org/10.3390/pharmaceutics16040513
Chicago/Turabian StyleObeid, Karim, Panagiotis Kanellopoulos, Ayman Abouzayed, Adam Mattsson, Vladimir Tolmachev, Berthold A. Nock, Theodosia Maina, and Anna Orlova. 2024. "GRPR-Antagonists Carrying DOTAGA-Chelator via Positively Charged Linkers: Perspectives for Prostate Cancer Theranostics" Pharmaceutics 16, no. 4: 513. https://doi.org/10.3390/pharmaceutics16040513
APA StyleObeid, K., Kanellopoulos, P., Abouzayed, A., Mattsson, A., Tolmachev, V., Nock, B. A., Maina, T., & Orlova, A. (2024). GRPR-Antagonists Carrying DOTAGA-Chelator via Positively Charged Linkers: Perspectives for Prostate Cancer Theranostics. Pharmaceutics, 16(4), 513. https://doi.org/10.3390/pharmaceutics16040513