
Cyclic Adenosine Monophosphate in Cardiac and Sympathoadrenal GLP-1 Receptor Signaling: Focus on Anti-Inflammatory Effects



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
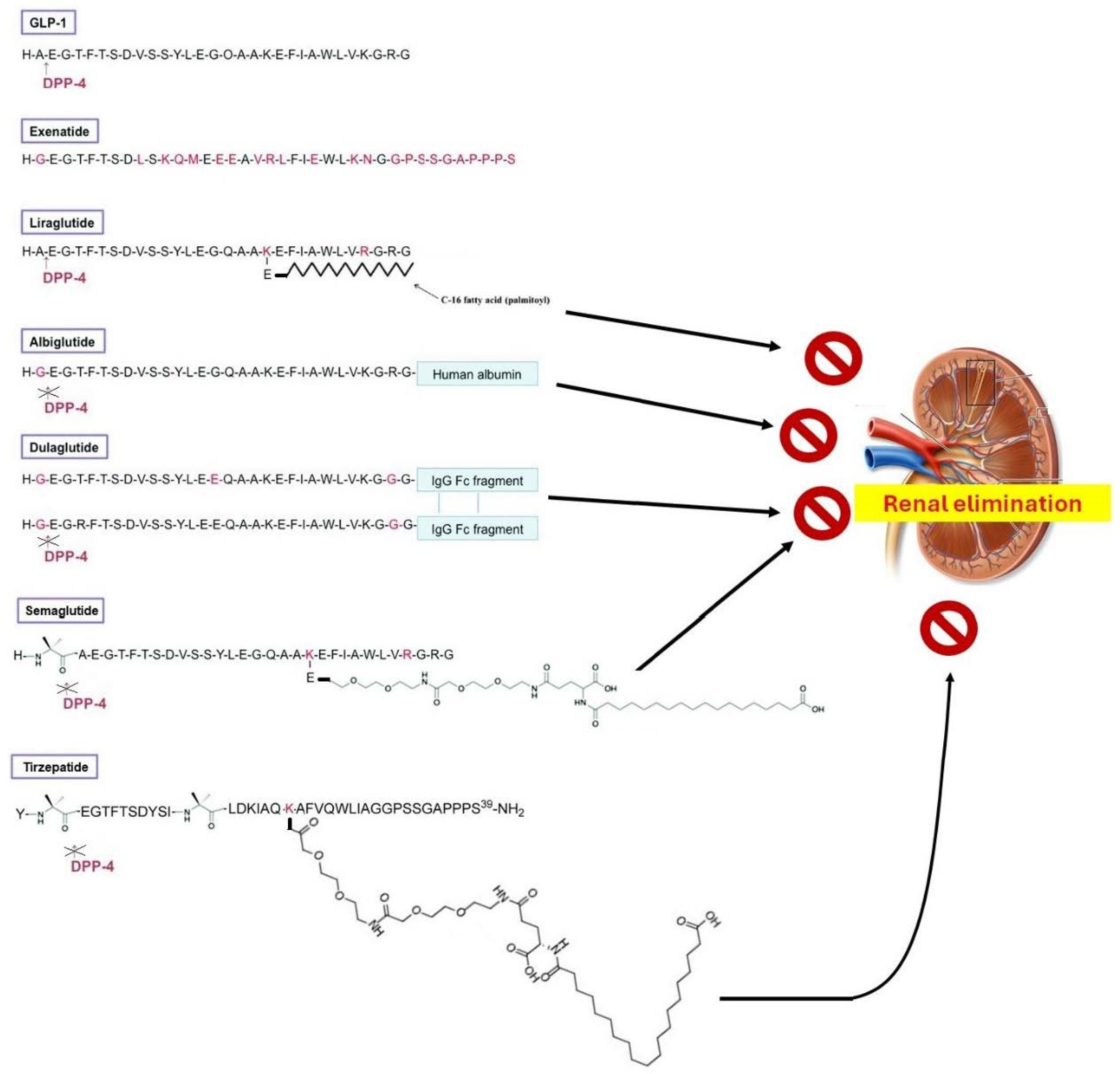
2. Chemistry of GLP-1R Agonists
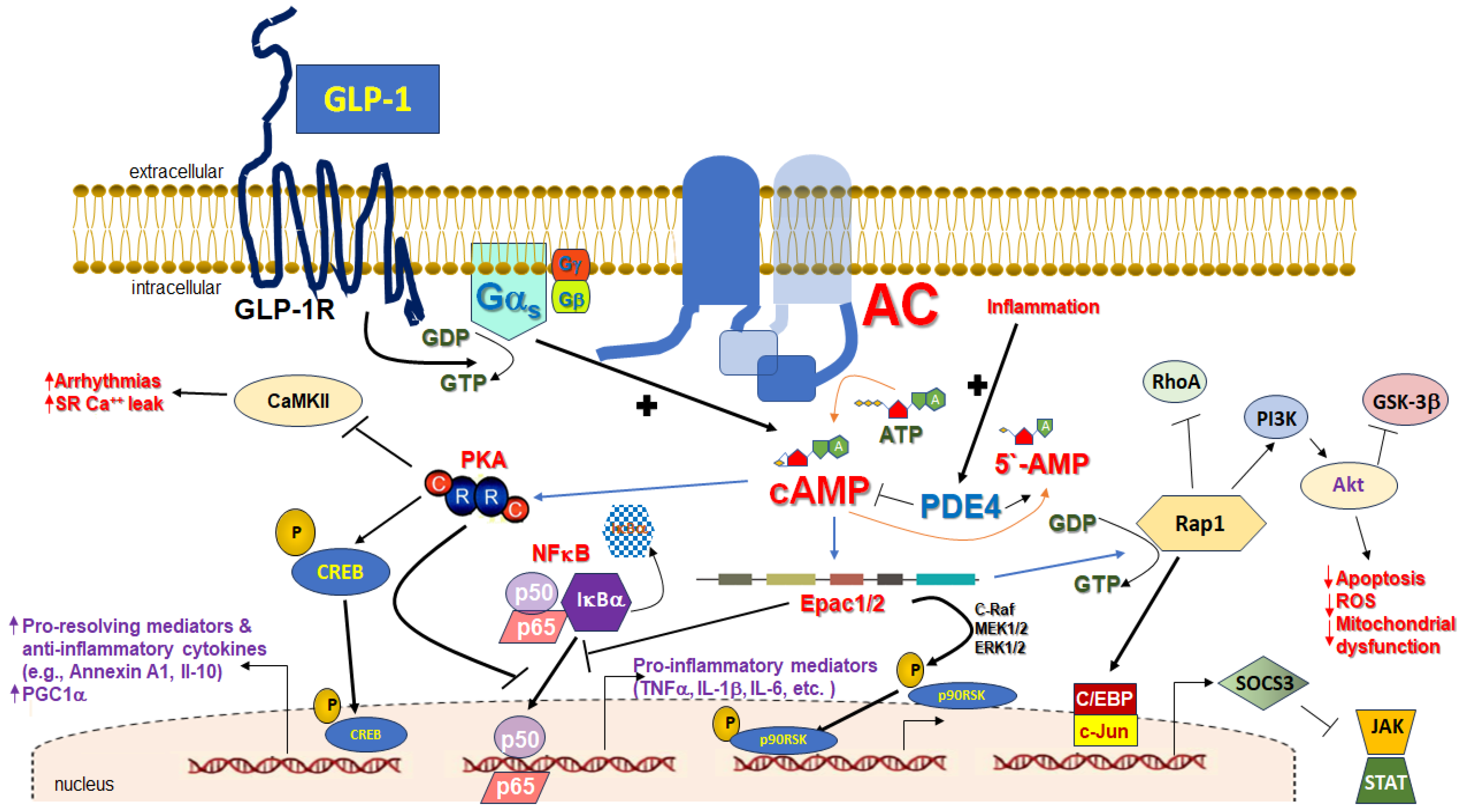
3. cAMP in Inflammation
4. Cardiac GLP-1R-Dependent, cAMP-Mediated Cardio-Protective Signaling
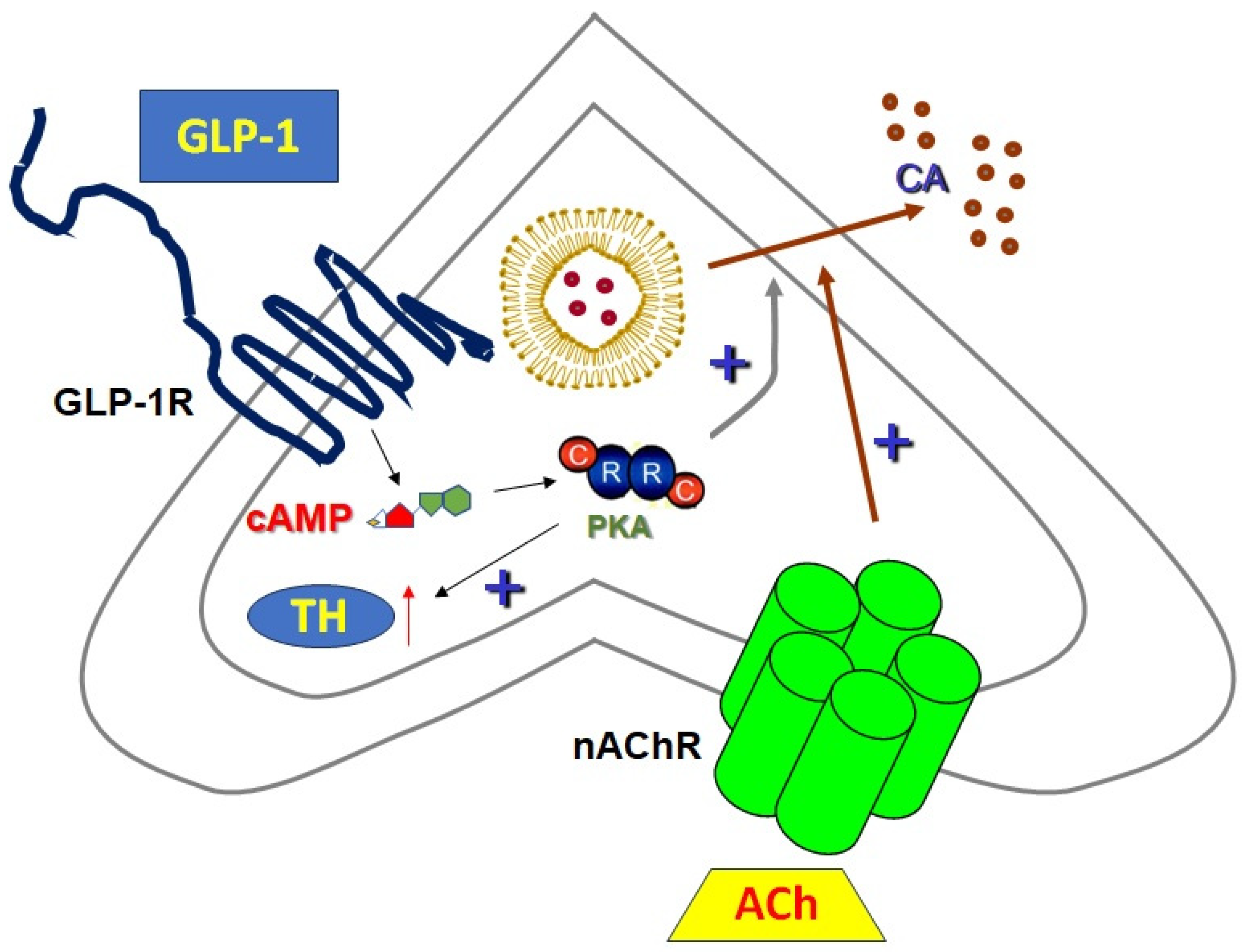
5. Sympathoadrenal GLP-1R-Dependent, cAMP-Mediated Signaling
6. Conclusions/Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Klen, J.; Dolžan, V. Glucagon-like Peptide-1 Receptor Agonists in the Management of Type 2 Diabetes Mellitus and Obesity: The Impact of Pharmacological Properties and Genetic Factors. Int. J. Mol. Sci. 2022, 23, 3451. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Meier, J.J. The incretin effect in healthy individuals and those with type 2 diabetes: Physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. 2016, 4, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Diz-Chaves, Y.; Mastoor, Z.; Spuch, C.; González-Matías, L.C.; Mallo, F. Anti-Inflammatory Effects of GLP-1 Receptor Activation in the Brain in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 9583. [Google Scholar] [CrossRef] [PubMed]
- Diz-Chaves, Y.; Herrera-Pérez, S.; González-Matías, L.C.; Lamas, J.A.; Mallo, F. Glucagon-Like Peptide-1 (GLP-1) in the Integration of Neural and Endocrine Responses to Stress. Nutrients 2020, 12, 3304. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D. The structure and function of the glucagon-like peptide-1 receptor and its ligands. Br. J. Pharmacol. 2012, 166, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Grieve, D.J.; Cassidy, R.S.; Green, B.D. Emerging cardiovascular actions of the incretin hormone glucagon-like peptide-1: Potential therapeutic benefits beyond glycaemic control? Br. J. Pharmacol. 2009, 157, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Graaf Cd Donnelly, D.; Wootten, D.; Lau, J.; Sexton, P.M.; Miller, L.J.; Ahn, J.M.; Liao, J.; Fletcher, M.M.; Yang, D.; Brown, A.J.; et al. Glucagon-Like Peptide-1 and Its Class B G Protein-Coupled Receptors: A Long March to Therapeutic Successes. Pharmacol. Rev. 2016, 68, 954–1013. [Google Scholar] [CrossRef] [PubMed]
- Willard, F.S.; Sloop, K.W. Physiology and emerging biochemistry of the glucagon-like peptide-1 receptor. Exp. Diabetes Res. 2012, 2012, 470851. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Lymperopoulos, A. Clinical pharmacology of cardiac cyclic AMP in human heart failure: Too much or too little? Expert Rev. Clin. Pharmacol. 2023, 16, 623–630. [Google Scholar] [CrossRef]
- Hsu, T.M.; Hahn, J.D.; Konanur, V.R.; Lam, A.; Kanoski, S.E. Hippocampal GLP-1 receptors influence food intake, meal size, and effort-based responding for food through volume transmission. Neuropsychopharmacology 2015, 40, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20 (Suppl. S1), 5–21. [Google Scholar] [CrossRef]
- Ussher, J.R.; Drucker, D.J. Glucagon-like peptide 1 receptor agonists: Cardiovascular benefits and mechanisms of action. Nat. Rev. Cardiol. 2023, 20, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The Cardiovascular Biology of Glucagon-like Peptide-1. Cell Metab. 2016, 24, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; McLean, B.A.; Baggio, L.L.; Koehler, J.A.; Hammoud, R.; Rittig, N.; Yabut, J.M.; Seeley, R.J.; Brown, T.J.; Drucker, D.J. Central glucagon-like peptide 1 receptor activation inhibits Toll-like receptor agonist-induced inflammation. Cell Metab. 2024, 36, 130–143.e5. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Z.; Ilyas, I.; Little, P.J.; Kamato, D.; Sahebka, A.; Chen, Z.; Luo, S.; Zheng, X.; Weng, J.; et al. GLP-1 receptor agonists (GLP-1RAs): Cardiovascular actions and therapeutic potential. Int. J. Biol. Sci. 2021, 17, 2050–2068. [Google Scholar] [CrossRef]
- Mehdi, S.F.; Pusapati, S.; Anwar, M.S.; Lohana, D.; Kumar, P.; Nandula, S.A.; Nawaz, F.K.; Tracey, K.; Yang, H.; LeRoith, D.; et al. Glucagon-like peptide-1: A multi-faceted anti-inflammatory agent. Front. Immunol. 2023, 14, 1148209. [Google Scholar] [CrossRef]
- Pandey, S.; Mangmool, S.; Parichatikanond, W. Multifaceted Roles of GLP-1 and Its Analogs: A Review on Molecular Mechanisms with a Cardiotherapeutic Perspective. Pharmaceuticals 2023, 16, 836. [Google Scholar] [CrossRef]
- Lee, Y.S.; Jun, H.S. Anti-Inflammatory Effects of GLP-1-Based Therapies beyond Glucose Control. Mediat. Inflamm. 2016, 2016, 3094642. [Google Scholar] [CrossRef]
- Bendotti, G.; Montefusco, L.; Lunati, M.E.; Usuelli, V.; Pastore, I.; Lazzaroni, E.; Assi, E.; Seelam, A.J.; El Essawy, B.; Jang, J.; et al. The anti-inflammatory and immunological properties of GLP-1 Receptor Agonists. Pharmacol. Res. 2022, 182, 106320. [Google Scholar] [CrossRef]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef] [PubMed]
- Diz-Chaves, Y.; Herrera-Pérez, S.; González-Matías, L.C.; Mallo, F. Effects of Glucagon-like peptide 1 (GLP-1) analogs in the hippocampus. Vitam. Horm. 2022, 118, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Akindehin, S.E.; Orsso, C.E.; Waldner, R.C.; DiMarchi, R.D.; Müller, T.D.; Haqq, A.M. Recent Advances in Incretin-Based Pharmacotherapies for the Treatment of Obesity and Diabetes. Front. Endocrinol. 2022, 13, 838410. [Google Scholar] [CrossRef] [PubMed]
- Niman, S.; Hardy, J.; Goldfaden, R.F.; Reid, J.; Sheikh-Ali, M.; Sutton, D.; Choksi, R. A Review on the Efficacy and Safety of Oral Semaglutide. Drugs R&D 2021, 21, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Misra, S. A review of an investigational drug retatrutide, a novel triple agonist agent for the treatment of obesity. Eur. J. Clin. Pharmacol. 2024, 80, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Tavares, L.P.; Negreiros-Lima, G.L.; Lima, K.M.; ESilva, P.M.R.; Pinho, V.; Teixeira, M.M.; Sousa, L.P. Blame the signaling: Role of cAMP for the resolution of inflammation. Pharmacol. Res. 2020, 159, 105030. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, M.B.; Insel, P.A. Inhaled β2 Adrenergic Agonists and Other cAMP-Elevating Agents: Therapeutics for Alveolar Injury and Acute Respiratory Disease Syndrome? Pharmacol. Rev. 2021, 73, 488–526. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular Organization of the cAMP Signaling Pathway. Pharmacol. Rev. 2021, 73, 278–309. [Google Scholar] [CrossRef] [PubMed]
- Gruscheski, L.; Brand, T. The Role of POPDC Proteins in Cardiac Pacemaking and Conduction. J. Cardiovasc. Dev. Dis. 2021, 8, 160. [Google Scholar] [CrossRef]
- Page, C.P.; Spina, D. Phosphodiesterase inhibitors in the treatment of inflammatory diseases. Handb. Exp. Pharmacol. 2011, 204, 391–414. [Google Scholar] [CrossRef]
- Ghosh, M.; Garcia-Castillo, D.; Aguirre, V.; Golshani, R.; Atkins, C.M.; Bramlett, H.M.; Dietrich, W.D.; Pearse, D.D. Proinflammatory cytokine regulation of cyclic AMP-phosphodiesterase 4 signaling in microglia in vitro and following CNS injury. Glia 2012, 60, 1839–1859. [Google Scholar] [CrossRef] [PubMed]
- Lima, K.M.; Vago, J.P.; Caux, T.R.; Negreiros-Lima, G.L.; Sugimoto, M.A.; Tavares, L.P.; Arribada, R.G.; Carmo, A.A.F.; Galvão, I.; Costa, B.R.C.; et al. The resolution of acute inflammation induced by cyclic AMP is dependent on annexin A1. J. Biol. Chem. 2017, 292, 13758–13773. [Google Scholar] [CrossRef] [PubMed]
- Antonicelli, F.; De Coupade, C.; Russo-Marie, F.; Le Garrec, Y. CREB is involved in mouse annexin A1 regulation by cAMP and glucocorticoids. Eur. J. Biochem. 2001, 268, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Lin, Y.; Perez-Polo, J.R.; Uretsky, B.F.; Ye, Z.; Tieu, B.C.; Birnbaum, Y. Phosphorylation of 5-lipoxygenase at ser523 by protein kinase A determines whether pioglitazone and atorvastatin induce proinflammatory leukotriene B4 or anti-inflammatory 15-epi-lipoxin a4 production. J. Immunol. 2008, 181, 3515–3523. [Google Scholar] [CrossRef] [PubMed]
- Warne, J.P.; John, C.D.; Christian, H.C.; Morris, J.F.; Flower, R.J.; Sugden, D.; Solito, E.; Gillies, G.E.; Buckingham, J.C. Gene deletion reveals roles for annexin A1 in the regulation of lipolysis and IL-6 release in epididymal adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1264–E1273. [Google Scholar] [CrossRef] [PubMed]
- Aronoff, D.M.; Carstens, J.K.; Chen, G.H.; Toews, G.B.; Peters-Golden, M. Short communication: Differences between macrophages and dendritic cells in the cyclic AMP-dependent regulation of lipopolysaccharide-induced cytokine and chemokine synthesis. J. Interferon Cytokine Res. 2006, 26, 827–833. [Google Scholar] [CrossRef] [PubMed]
- van der Pouw Kraan, T.C.; Boeije, L.C.; Smeenk, R.J.; Wijdenes, J.; Aarden, L.A. Prostaglandin-E2 is a potent inhibitor of human interleukin 12 production. J. Exp. Med. 1995, 181, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Jones, S.M.; Flamand, N.; Aronoff, D.M.; Peters-Golden, M.; Brock, T.G. Phosphorylation by protein kinase a inhibits nuclear import of 5-lipoxygenase. J. Biol. Chem. 2005, 280, 40609–40616. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, W.A.; Vanaudenaerde, B.M.; Dupont, L.J.; Demedts, M.G.; Verleden, G.M. Modulation by cAMP of IL-1beta-induced eotaxin and MCP-1 expression and release in human airway smooth muscle cells. Eur. Respir. J. 2003, 22, 220–226. [Google Scholar] [CrossRef]
- Kawashita, E.; Tsuji, D.; Toyoshima, M.; Kanno, Y.; Matsuno, H.; Itoh, K. Prostaglandin E2 reverses aberrant production of an inflammatory chemokine by microglia from Sandhoff disease model mice through the cAMP-PKA pathway. PLoS ONE 2011, 6, e16269. [Google Scholar] [CrossRef]
- Lea, S.; Metryka, A.; Li, J.; Higham, A.; Bridgewood, C.; Villetti, G.; Civelli, M.; Facchinetti, F.; Singh, D. The modulatory effects of the PDE4 inhibitors CHF6001 and roflumilast in alveolar macrophages and lung tissue from COPD patients. Cytokine 2019, 123, 154739. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.M.; Alves, A.C.; Serra, M.F.; Pires, A.L.; Silva, J.P.; Barreto, E.O.; Cordeiro, R.S.; Jose, P.J.; Teixeira, M.M.; Lagente, V.; et al. Modulation of eotaxin formation and eosinophil migration by selective inhibitors of phosphodiesterase type 4 isoenzyme. Br. J. Pharmacol. 2001, 134, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Vang, T.; Torgersen, K.M.; Sundvold, V.; Saxena, M.; Levy, F.O.; Skålhegg, B.S.; Hansson, V.; Mustelin, T.; Taskén, K. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J. Exp. Med. 2001, 193, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Welch, E.J.; Gao, X.P.; Malik, A.B.; Ye, R.D. Lysophosphatidylcholine modulates neutrophil oxidant production through elevation of cyclic AMP. J. Immunol. 2005, 174, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Laudanna, C.; Campbell, J.J.; Butcher, E.C. Elevation of intracellular cAMP inhibits RhoA activation and integrin-dependent leukocyte adhesion induced by chemoattractants. J. Biol. Chem. 1997, 272, 24141–24144. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Tetsuka, T.; Uranishi, H.; Okamoto, T. Inhibition of the NF-kappaB transcriptional activity by protein kinase A. Eur. J. Biochem. 2002, 269, 4559–4565. [Google Scholar] [CrossRef] [PubMed]
- Procópio, D.O.; Teixeira, M.M.; Camargo, M.M.; Travassos, L.R.; Ferguson, M.A.; Almeida, I.C.; Gazzinelli, R.T. Differential inhibitory mechanism of cyclic AMP on TNF-alpha and IL-12 synthesis by macrophages exposed to microbial stimuli. Br. J. Pharmacol. 1999, 127, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Grieshammer, T.; Chuvpilo, S.; Kneitz, B.; Lohoff, M.; Schimpl, A.; Franza, B.R., Jr.; Serfling, E. RelA/p65 is a molecular target for the immunosuppressive action of protein kinase A. EMBO J. 1995, 14, 1991–2004. [Google Scholar] [CrossRef]
- Kamthong, P.J.; Wu, M. Inhibitor of nuclear factor-kappaB induction by cAMP antagonizes interleukin-1-induced human macrophage-colony-stimulating-factor expression. Biochem. J. 2001, 356 Pt 2, 525–530. [Google Scholar] [CrossRef]
- Sousa, L.P.; Carmo, A.F.; Rezende, B.M.; Lopes, F.; Silva, D.M.; Alessandri, A.L.; Bonjardim, C.A.; Rossi, A.G.; Teixeira, M.M.; Pinho, V. Cyclic AMP enhances resolution of allergic pleurisy by promoting inflammatory cell apoptosis via inhibition of PI3K/Akt and NF-kappaB. Biochem. Pharmacol. 2009, 78, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.A.; Arora, M.; Bohlson, S.S.; Lozano, E.; Tenner, A.J. Generation of inhibitory NFkappaB complexes and phosphorylated cAMP response element-binding protein correlates with the anti-inflammatory activity of complement protein C1q in human monocytes. J. Biol. Chem. 2007, 282, 7360–7367. [Google Scholar] [CrossRef] [PubMed]
- Wall, E.A.; Zavzavadjian, J.R.; Chang, M.S.; Randhawa, B.; Zhu, X.; Hsueh, R.C.; Liu, J.; Driver, A.; Bao, X.R.; Sternweis, P.C.; et al. Suppression of LPS-induced TNF-alpha production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci. Signal. 2009, 2, ra28. [Google Scholar] [CrossRef] [PubMed]
- Scheibner, K.A.; Boodoo, S.; Collins, S.; Black, K.E.; Chan-Li, Y.; Zarek, P.; Powell, J.D.; Horton, M.R. The adenosine a2a receptor inhibits matrix-induced inflammation in a novel fashion. Am. J. Respir. Cell Mol. Biol. 2009, 40, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Sands, W.A.; Woolson, H.D.; Milne, G.R.; Rutherford, C.; Palmer, T.M. Exchange protein activated by cyclic AMP (Epac)-mediated induction of suppressor of cytokine signaling 3 (SOCS-3) in vascular endothelial cells. Mol. Cell Biol. 2006, 26, 6333–6346. [Google Scholar] [CrossRef] [PubMed]
- Woolson, H.D.; Thomson, V.S.; Rutherford, C.; Yarwood, S.J.; Palmer, T.M. Selective inhibition of cytokine-activated extracellular signal-regulated kinase by cyclic AMP via Epac1-dependent induction of suppressor of cytokine signalling-3. Cell. Signal. 2009, 21, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Fujita, T.; Jin, M.; Kurotani, R.; Hidaka, Y.; Cai, W.; Suita, K.; Prajapati, R.; Liang, C.; Ohnuki, Y.; et al. Epac activation inhibits IL-6-induced cardiac myocyte dysfunction. J. Physiol. Sci. 2018, 68, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Varghese, L.N.; Nicola, N.A. Inhibition of IL-6 family cytokines by SOCS3. Semin. Immunol. 2014, 26, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Yen, J.H.; Ganea, D. A novel signaling pathway mediates the inhibition of CCL3/4 expression by prostaglandin E2. J. Biol. Chem. 2004, 279, 55176–55186. [Google Scholar] [CrossRef]
- Hertz, A.L.; Bender, A.T.; Smith, K.C.; Gilchrist, M.; Amieux, P.S.; Aderem, A.; Beavo, J.A. Elevated cyclic AMP and PDE4 inhibition induce chemokine expression in human monocyte-derived macrophages. Proc. Natl. Acad. Sci. USA 2009, 106, 21978–21983. [Google Scholar] [CrossRef]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Dao, K.K.; Teigen, K.; Kopperud, R.; Hodneland, E.; Schwede, F.; Christensen, A.E.; Martinez, A.; Døskeland, S.O. Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition. J. Biol. Chem. 2006, 281, 21500–21511. [Google Scholar] [CrossRef] [PubMed]
- Gros, R.; You, X.; Baggio, L.L.; Kabir, M.G.; Sadi, A.M.; Mungrue, I.N.; Parker, T.G.; Huang, Q.; Drucker, D.J.; Husain, M. Cardiac function in mice lacking the glucagon-like peptide-1 receptor. Endocrinology 2003, 144, 2242–2252. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, S.M.; March, J.E.; Kemp, P.A.; Bennett, T. Autonomic nervous system-dependent and -independent cardiovascular effects of exendin-4 infusion in conscious rats. Br. J. Pharmacol. 2008, 154, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Ang, R.; Mastitskaya, S.; Hosford, P.S.; Basalay, M.; Specterman, M.; Aziz, Q.; Li, Y.; Orini, M.; Taggart, P.; Lambiase, P.D.; et al. Modulation of Cardiac Ventricular Excitability by GLP-1 (Glucagon-Like Peptide-1). Circ. Arrhythm. Electrophysiol. 2018, 11, e006740. [Google Scholar] [CrossRef] [PubMed]
- Vila Petroff, M.G.; Egan, J.M.; Wang, X.; Sollott, S.J. Glucagon-like peptide-1 increases cAMP but fails to augment contraction in adult rat cardiac myocytes. Circ. Res. 2001, 89, 445–452. [Google Scholar] [CrossRef]
- Zhao, T.; Parikh, P.; Bhashyam, S.; Bolukoglu, H.; Poornima, I.; Shen, Y.T.; Shannon, R.P. Direct effects of glucagon-like peptide-1 on myocardial contractility and glucose uptake in normal and postischemic isolated rat hearts. J. Pharmacol. Exp. Ther. 2006, 317, 1106–1113. [Google Scholar] [CrossRef]
- Ban, K.; Noyan-Ashraf, M.H.; Hoefer, J.; Bolz, S.S.; Drucker, D.J.; Husain, M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 2008, 117, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Kanagasundram, A.N.; Richardson, T.D.; Stevenson, W.G. The Heart Rate of Ventricular Tachycardia. Circulation 2021, 143, 227–229. [Google Scholar] [CrossRef]
- Soderling, S.H.; Beavo, J.A. Regulation of cAMP and cGMP signaling: New phosphodiesterases and new functions. Curr. Opin. Cell Biol. 2000, 12, 174–179. [Google Scholar] [CrossRef]
- Koshibu, M.; Mori, Y.; Saito, T.; Kushima, H.; Hiromura, M.; Terasaki, M.; Takada, M.; Fukui, T.; Hirano, T. Antiatherogenic effects of liraglutide in hyperglycemic apolipoprotein E-null mice via AMP-activated protein kinase-independent mechanisms. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E895–E907. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Kurobe, H.; Nishio, C.; Tanaka, K.; Fukuda, D.; Uematsu, E.; Nishimoto, S.; Soeki, T.; Harada, N.; Sakaue, H.; et al. Exendin-4, a glucagon-like peptide-1 receptor agonist, attenuates neointimal hyperplasia after vascular injury. Eur. J. Pharmacol. 2013, 699, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Bjørnholm, K.D.; Skovsted, G.F.; Mitgaard-Thomsen, A.; Rakipovski, G.; Tveden-Nyborg, P.; Lykkesfeldt, J.; Povlsen, G.K. Liraglutide treatment improves endothelial function in the Ldlr-/- mouse model of atherosclerosis and affects genes involved in vascular remodelling and inflammation. Basic Clin. Pharmacol. Toxicol. 2021, 128, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, X.; Fang, Q.; Ding, M.; Li, C. Liraglutide attenuates atherosclerosis via inhibiting ER-induced macrophage derived microvesicles production in T2DM rats. Diabetol. Metab. Syndr. 2017, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Yusta, B.; Mulvihill, E.E.; Cao, X.; Streutker, C.J.; Butany, J.; Cappola, T.P.; Margulies, K.B.; Drucker, D.J. GLP-1 Receptor Expression Within the Human Heart. Endocrinology 2018, 159, 1570–1584. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, Q.; Yang, X.; Chen, X. Protective effect of glucagon-like peptide-1 agents on reperfusion injury for acute myocardial infarction: A meta-analysis of randomized controlled trials. Ann. Med. 2017, 49, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Cora, N.; Maning, J.; Brill, A.R.; Sizova, A. Signaling and function of cardiac autonomic nervous system receptors: Insights from the GPCR signalling universe. FEBS J. 2021, 288, 2645–2659. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, D.; Wang, F.; Shi, S.; Chen, Y.; Yang, B.; Tang, Y.; Huang, C. Exendin-4 inhibits structural remodeling and improves Ca2+ homeostasis in rats with heart failure via the GLP-1 receptor through the eNOS/cGMP/PKG pathway. Peptides 2017, 90, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Darwesh, A.M.; El-Azab, M.F.; Abo-Gresha, N.M.; El-Sayed, N.M.; Moustafa, Y.M. Cardioprotective Mechanisms of Exenatide in Isoprenaline-induced Myocardial Infarction: Novel Effects on Myocardial α-Estrogen Receptor Expression and IGF-1/IGF-2 System. J. Cardiovasc. Pharmacol. 2018, 71, 160–173. [Google Scholar] [CrossRef]
- Robinson, E.; Cassidy, R.S.; Tate, M.; Zhao, Y.; Lockhart, S.; Calderwood, D.; Church, R.; McGahon, M.K.; Brazil, D.P.; McDermott, B.J.; et al. Exendin-4 protects against post-myocardial infarction remodelling via specific actions on inflammation and the extracellular matrix. Basic Res. Cardiol. 2015, 110, 20. [Google Scholar] [CrossRef]
- Winiarska, A.; Knysak, M.; Nabrdalik, K.; Gumprecht, J.; Stompór, T. Inflammation and Oxidative Stress in Diabetic Kidney Disease: The Targets for SGLT2 Inhibitors and GLP-1 Receptor Agonists. Int. J. Mol. Sci. 2021, 22, 10822. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Inoguchi, T.; Sonoda, N.; Hendarto, H.; Makimura, H.; Sasaki, S.; Yokomizo, H.; Fujimura, Y.; Miura, D.; Takayanagi, R. GLP-1 analog liraglutide protects against cardiac steatosis, oxidative stress and apoptosis in streptozotocin-induced diabetic rats. Atherosclerosis 2015, 240, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, C.; Zhu, Q.; Li, N.; Zhou, H. Liraglutide, a glucagon-like peptide-1 analog, inhibits high glucose-induced oxidative stress and apoptosis in neonatal rat cardiomyocytes. Exp. Ther. Med. 2019, 17, 3734–3740. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.Y.; Chen, Z.W.; Gao, Y.H.; Wang, X.X.; Ma, J.Y.; Chang, S.F.; Qian, J.Y.; Ge, J.B. Exenatide Reduces Tumor Necrosis Factor-α-induced Apoptosis in Cardiomyocytes by Alleviating Mitochondrial Dysfunction. Chin. Med. J. 2015, 128, 3211–3218. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Liu, J.; Qin, S.; Jiang, Y.; Zhang, P.; Yu, H.; Lu, K.; Zhang, N.; Cao, L.; Wang, Y.; et al. Cardioprotection by exenatide: A novel mechanism via improving mitochondrial function involving the GLP-1 receptor/cAMP/PKA pathway. Int. J. Mol. Med. 2018, 41, 1693–1703. [Google Scholar] [CrossRef]
- Wu, J.; Xie, F.; Qin, Y.; Liu, J.; Yang, Z. Notch signaling is involved in the antiapoptotic effects of liraglutide on rat H9c2 cardiomyocytes exposed to hypoxia followed by reoxygenation. J. Int. Med. Res. 2020, 48, 300060520948394. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, L.; Feng, B.; He, N.; Zhang, Y.; Ye, H. Protective effects of glucagon-like peptide-1 on cardiac remodeling by inhibiting oxidative stress through mammalian target of rapamycin complex 1/p70 ribosomal protein S6 kinase pathway in diabetes mellitus. J. Diabetes Investig. 2020, 11, 39–51. [Google Scholar] [CrossRef] [PubMed]
- XiaoTian, L.; QiNan, W.; XiaGuang, G.; WuQuan, D.; Bing, C.; ZiWen, L. Exenatide Activates the APPL1-AMPK-PPARα Axis to Prevent Diabetic Cardiomyocyte Apoptosis. J. Diabetes Res. 2016, 2016, 4219735. [Google Scholar] [CrossRef]
- Hu, S.Y.; Zhang, Y.; Zhu, P.J.; Zhou, H.; Chen, Y.D. Liraglutide directly protects cardiomyocytes against reperfusion injury possibly via modulation of intracellular calcium homeostasis. J. Geriatr. Cardiol. 2017, 14, 57–66. [Google Scholar] [CrossRef]
- Chen, J.; Xu, S.; Zhou, W.; Wu, L.; Wang, L.; Li, W. Exendin-4 Reduces Ventricular Arrhythmia Activity and Calcium Sparks-Mediated Sarcoplasmic Reticulum Ca Leak in Rats with Heart Failure. Int. Heart J. 2020, 61, 145–152. [Google Scholar] [CrossRef]
- Brayden, J.E. Functional roles of KATP channels in vascular smooth muscle. Clin. Exp. Pharmacol. Physiol. 2002, 29, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.J.; Hao, J.T.; Zheng, R.H.; Yan, C.P.; Wang, J.; Yang, C.H.; Zhang, W.F.; Zhao, Z.Q. Glucagon-Like Peptide-1 Analog Liraglutide Attenuates Pressure-Overload Induced Cardiac Hypertrophy and Apoptosis through Activating ATP Sensitive Potassium Channels. Cardiovasc. Drugs Ther. 2021, 35, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Ambrosy, A.P.; DeVore, A.D.; Margulies, K.B.; McNulty, S.E.; Mentz, R.J.; Hernandez, A.F.; Michael Felker, G.; Cooper, L.B.; Lala, A.; et al. Liraglutide and weight loss among patients with advanced heart failure and a reduced ejection fraction: Insights from the FIGHT trial. ESC Heart Fail. 2018, 5, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Jorsal, A.; Kistorp, C.; Holmager, P.; Tougaard, R.S.; Nielsen, R.; Hänselmann, A.; Nilsson, B.; Møller, J.E.; Hjort, J.; Rasmussen, J.; et al. Effect of liraglutide, a glucagon-like peptide-1 analogue, on left ventricular function in stable chronic heart failure patients with and without diabetes (LIVE)—A multicentre, double-blind, randomised, placebo-controlled trial. Eur. J. Heart Fail. 2017, 19, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Mui, D.; Zhu, H.; Zhang, Y. Exenatide inhibits NF-κB and attenuates ER stress in diabetic cardiomyocyte models. Aging 2020, 12, 8640–8651. [Google Scholar] [CrossRef] [PubMed]
- Okabe, K.; Matsushima, S.; Ikeda, S.; Ikeda, M.; Ishikita, A.; Tadokoro, T.; Enzan, N.; Yamamoto, T.; Sada, M.; Deguchi, H.; et al. DPP (Dipeptidyl Peptidase)-4 Inhibitor Attenuates Ang II (Angiotensin II)-Induced Cardiac Hypertrophy via GLP (Glucagon-Like Peptide)-1-Dependent Suppression of Nox (Nicotinamide Adenine Dinucleotide Phosphate Oxidase) 4-HDAC (Histone Deacetylase) 4 Pathway. Hypertension 2020, 75, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Chen, H.; Cheng, C.; Zhang, J.; Yan, Z.; Kuang, J.; Kong, F.; Li, C.; Lu, Q. Liraglutide protects high-glucose-stimulated fibroblasts by activating the CD36-JNK-AP1 pathway to downregulate P4HA1. Biomed. Pharmacother. 2019, 118, 109224. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Yang, F.; Wang, W.; Li, X.; Liu, D.; Zhang, Y.; Yin, G.; Lv, F.; Guo, Z.; Mehta, J.L.; et al. Liraglutide Attenuates Myocardial Fibrosis via Inhibition of AT1R-Mediated ROS Production in Hypertensive Mice. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Zhang, F.X.; Yang, F.; Ding, Z.F.; Agarwal, N.; Guo, Z.K.; Mehta, J.L. Effects of linagliptin and liraglutide on glucose- and angiotensin II-induced collagen formation and cytoskeleton degradation in cardiac fibroblasts in vitro. Acta Pharmacol. Sin. 2016, 37, 1349–1358. [Google Scholar] [CrossRef]
- Zheng, R.H.; Bai, X.J.; Zhang, W.W.; Wang, J.; Bai, F.; Yan, C.P.; James, E.A.; Bose, H.S.; Wang, N.P.; Zhao, Z.Q. Liraglutide attenuates cardiac remodeling and improves heart function after abdominal aortic constriction through blocking angiotensin II type 1 receptor in rats. Drug Des. Devel. Ther. 2019, 13, 2745–2757. [Google Scholar] [CrossRef]
- Pollard, C.M.; Desimine, V.L.; Wertz, S.L.; Perez, A.; Parker, B.M.; Maning, J.; McCrink, K.A.; Shehadeh, L.A.; Lymperopoulos, A. Deletion of Osteopontin Enhances β₂-Adrenergic Receptor-Dependent Anti-Fibrotic Signaling in Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 1396. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Murray, F.; Yokoyama, U.; Romano, S.; Yun, H.; Brown, L.; Snead, A.; Lu, D.; Aroonsakool, N. cAMP and Epac in the regulation of tissue fibrosis. Br. J. Pharmacol. 2012, 166, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Surinkaew, S.; Aflaki, M.; Takawale, A.; Chen, Y.; Qi, X.Y.; Gillis, M.A.; Shi, Y.F.; Tardif, J.C.; Chattipakorn, N.; Nattel, S. Exchange protein activated by cyclic-adenosine monophosphate (Epac) regulates atrial fibroblast function and controls cardiac remodelling. Cardiovasc. Res. 2019, 115, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Bu, R.; Yang, Q.; Jia, J.; Li, T.; Wang, Q.; Chen, Y. Exendin-4 Protects against Hyperglycemia-Induced Cardiomyocyte Pyroptosis via the AMPK-TXNIP Pathway. J. Diabetes Res. 2019, 2019, 8905917. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Chen, Z.; Xia, Y.; Lu, D.; Yang, X.; Sun, A.; Zou, Y.; Qian, J.; Ge, J. Liraglutide attenuates NLRP3 inflammasome-dependent pyroptosis via regulating SIRT1/NOX4/ROS pathway in H9c2 cells. Biochem. Biophys. Res. Commun. 2018, 499, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Gong, T.; Jiang, W.; Zhou, R. GPCRs in NLRP3 Inflammasome Activation, Regulation, and Therapeutics. Trends Pharmacol. Sci. 2018, 39, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Pharmacological Inhibition of the NLRP3 Inflammasome: Structure, Molecular Activation, and Inhibitor-NLRP3 Interaction. Pharmacol. Rev. 2023, 75, 487–520. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Tuo, X.; Li, B.; Deng, Z.; Qiu, Y.; Xie, H. Semaglutide attenuates excessive exercise-induced myocardial injury through inhibiting oxidative stress and inflammation in rats. Life Sci. 2020, 250, 117531. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, N.; Han, Y.; Xu, J.; Xu, Z. Dulaglutide Alleviates LPS-Induced Injury in Cardiomyocytes. ACS Omega 2021, 6, 8271–8278. [Google Scholar] [CrossRef]
- Nuamnaichati, N.; Mangmool, S.; Chattipakorn, N.; Parichatikanond, W. Stimulation of GLP-1 Receptor Inhibits Methylglyoxal-Induced Mitochondrial Dysfunctions in H9c2 Cardiomyoblasts: Potential Role of Epac/PI3K/Akt Pathway. Front. Pharmacol. 2020, 11, 805. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Wang, R.C.; Cheng, K.; Ring, B.Z.; Su, L. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer Biol. Med. 2017, 14, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Winzeler, B.; da Conceição, I.; Refardt, J.; Sailer, C.O.; Dutilh, G.; Christ-Crain, M. Effects of Glucagon-Like Peptide-1 Receptor Agonists on Hypothalamic-Pituitary-Adrenal Axis in Healthy Volunteers. J. Clin. Endocrinol. Metab. 2019, 104, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Kishi, T.; Lee, C.E.; Choi, B.J.; Fang, H.; Hollenberg, A.N.; Drucker, D.J.; Elmquist, J.K. Glucagon-like peptide-1-responsive catecholamine neurons in the area postrema link peripheral glucagon-like peptide-1 with central autonomic control sites. J. Neurosci. 2003, 23, 2939–2946. [Google Scholar] [CrossRef] [PubMed]
- Barragán, J.M.; Eng, J.; Rodríguez, R.; Blázquez, E. Neural contribution to the effect of glucagon-like peptide-1-(7-36) amide on arterial blood pressure in rats. Am. J. Physiol. 1999, 277, E784–E791. [Google Scholar] [CrossRef]
- Yamamoto, H.; Lee, C.E.; Marcus, J.N.; Williams, T.D.; Overton, J.M.; Lopez, M.E.; Hollenberg, A.N.; Baggio, L.; Saper, C.B.; Drucker, D.J.; et al. Glucagon-like peptide-1 receptor stimulation increases blood pressure and heart rate and activates autonomic regulatory neurons. J. Clin. Investg. 2002, 110, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Heuvelman, V.D.; Van Raalte, D.H.; Smits, M.M. Cardiovascular effects of glucagon-like peptide 1 receptor agonists: From mechanistic studies in humans to clinical outcomes. Cardiovasc. Res. 2020, 116, 916–930. [Google Scholar] [CrossRef] [PubMed]
- Smits, M.M.; Muskiet, M.H.; Tonneijck, L.; Hoekstra, T.; Kramer, M.H.; Diamant, M.; van Raalte, D.H. Exenatide acutely increases heart rate in parallel with augmented sympathetic nervous system activation in healthy overweight males. Br. J. Clin. Pharmacol. 2016, 81, 613–620. [Google Scholar] [CrossRef]
- Baggio, L.L.; Ussher, J.R.; McLean, B.A.; Cao, X.; Kabir, M.G.; Mulvihill, E.E.; Mighiu, A.S.; Zhang, H.; Ludwig, A.; Seeley, R.J.; et al. The autonomic nervous system and cardiac GLP-1 receptors control heart rate in mice. Mol. Metab. 2017, 6, 1339–1349. [Google Scholar] [CrossRef]
- Smits, M.M.; Tonneijck, L.; Muskiet, M.H.; Hoekstra, T.; Kramer, M.H.; Diamant, M.; van Raalte, D.H. Heart rate acceleration with GLP-1 receptor agonists in type 2 diabetes patients: An acute and 12-week randomised, double-blind, placebo-controlled trial. Eur. J. Endocrinol. 2017, 176, 77–86. [Google Scholar] [CrossRef]
- Baraboi, E.D.; St-Pierre, D.H.; Shooner, J.; Timofeeva, E.; Richard, D. Brain activation following peripheral administration of the GLP-1 receptor agonist exendin-4. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1011–R1024. [Google Scholar] [CrossRef]
- Wong, C.K.; Yusta, B.; Koehler, J.A.; Baggio, L.L.; McLean, B.A.; Matthews, D.; Seeley, R.J.; Drucker, D.J. Divergent roles for the gut intraepithelial lymphocyte GLP-1R in control of metabolism, microbiota, and T cell-induced inflammation. Cell Metab. 2022, 34, 1514–1531.e7. [Google Scholar] [CrossRef] [PubMed]
- González-Santana, A.; Estévez-Herrera, J.; Seward, E.P.; Borges, R.; Machado, J.D. Glucagon-like peptide-1 receptor controls exocytosis in chromaffin cells by increasing full-fusion events. Cell Rep. 2021, 36, 109609. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S. Adrenal responses to stress. Cell. Mol. Neurobiol. 2010, 30, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Brill, A.; McCrink, K.A. GPCRs of adrenal chromaffin cells & catecholamines: The plot thickens. Int. J. Biochem. Cell Biol. 2016, 77 Pt B, 213–219. [Google Scholar] [CrossRef]
- Jafferjee, M.; Reyes Valero, T.; Marrero, C.; McCrink, K.A.; Brill, A.; Lymperopoulos, A. GRK2 Up-Regulation Creates a Positive Feedback Loop for Catecholamine Production in Chromaffin Cells. Mol. Endocrinol. 2016, 30, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Funakoshi, H.; Eckhart, A.D.; Koch, W.J. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 2007, 13, 315–323. [Google Scholar] [CrossRef]
- Perrais, D.; Kleppe, I.C.; Taraska, J.W.; Almers, W. Recapture after exocytosis causes differential retention of protein in granules of bovine chromaffin cells. J. Physiol. 2004, 560 Pt 2, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Yim, Y.Y.; Zurawski, Z.; Hamm, H. GPCR regulation of secretion. Pharmacol. Ther. 2018, 192, 124–140. [Google Scholar] [CrossRef]
- Holt, M.K.; Trapp, S. The physiological role of the brain GLP-1 system in stress. Cogent Biol. 2016, 2, 1229086. [Google Scholar] [CrossRef]
- Ceasrine, A.M.; Lin, E.E.; Lumelsky, D.N.; Iyer, R.; Kuruvilla, R. Adrb2 controls glucose homeostasis by developmental regulation of pancreatic islet vasculature. Elife 2018, 7, e39689. [Google Scholar] [CrossRef]
- Xu, X.; Kaindl, J.; Clark, M.J.; Hübner, H.; Hirata, K.; Sunahara, R.K.; Gmeiner, P.; Kobilka, B.K.; Liu, X. Binding pathway determines norepinephrine selectivity for the human β1AR over β2AR. Cell Res. 2021, 31, 569–579. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lymperopoulos, A.; Borges, J.I.; Stoicovy, R.A. Cyclic Adenosine Monophosphate in Cardiac and Sympathoadrenal GLP-1 Receptor Signaling: Focus on Anti-Inflammatory Effects. Pharmaceutics 2024, 16, 693. https://doi.org/10.3390/pharmaceutics16060693
Lymperopoulos A, Borges JI, Stoicovy RA. Cyclic Adenosine Monophosphate in Cardiac and Sympathoadrenal GLP-1 Receptor Signaling: Focus on Anti-Inflammatory Effects. Pharmaceutics. 2024; 16(6):693. https://doi.org/10.3390/pharmaceutics16060693
Chicago/Turabian StyleLymperopoulos, Anastasios, Jordana I. Borges, and Renee A. Stoicovy. 2024. "Cyclic Adenosine Monophosphate in Cardiac and Sympathoadrenal GLP-1 Receptor Signaling: Focus on Anti-Inflammatory Effects" Pharmaceutics 16, no. 6: 693. https://doi.org/10.3390/pharmaceutics16060693