


Drug-Induced Reorganisation of Lipid Metabolism Limits the Therapeutic Efficacy of Ponatinib in Glioma Stem Cells
 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Neurosphere Formation and Self-Renewal Analysis
2.3. Subcellular Fractionation of Neurosphere Cells
2.4. Mass Spectrometry
2.4.1. MS/MS Spectral Library Generation
2.4.2. Quantitative Proteomics by SWATH Analysis
2.5. Western Blot Analysis
2.6. ASAH1 Overexpression
2.7. Data Analysis and Statistics
3. Results
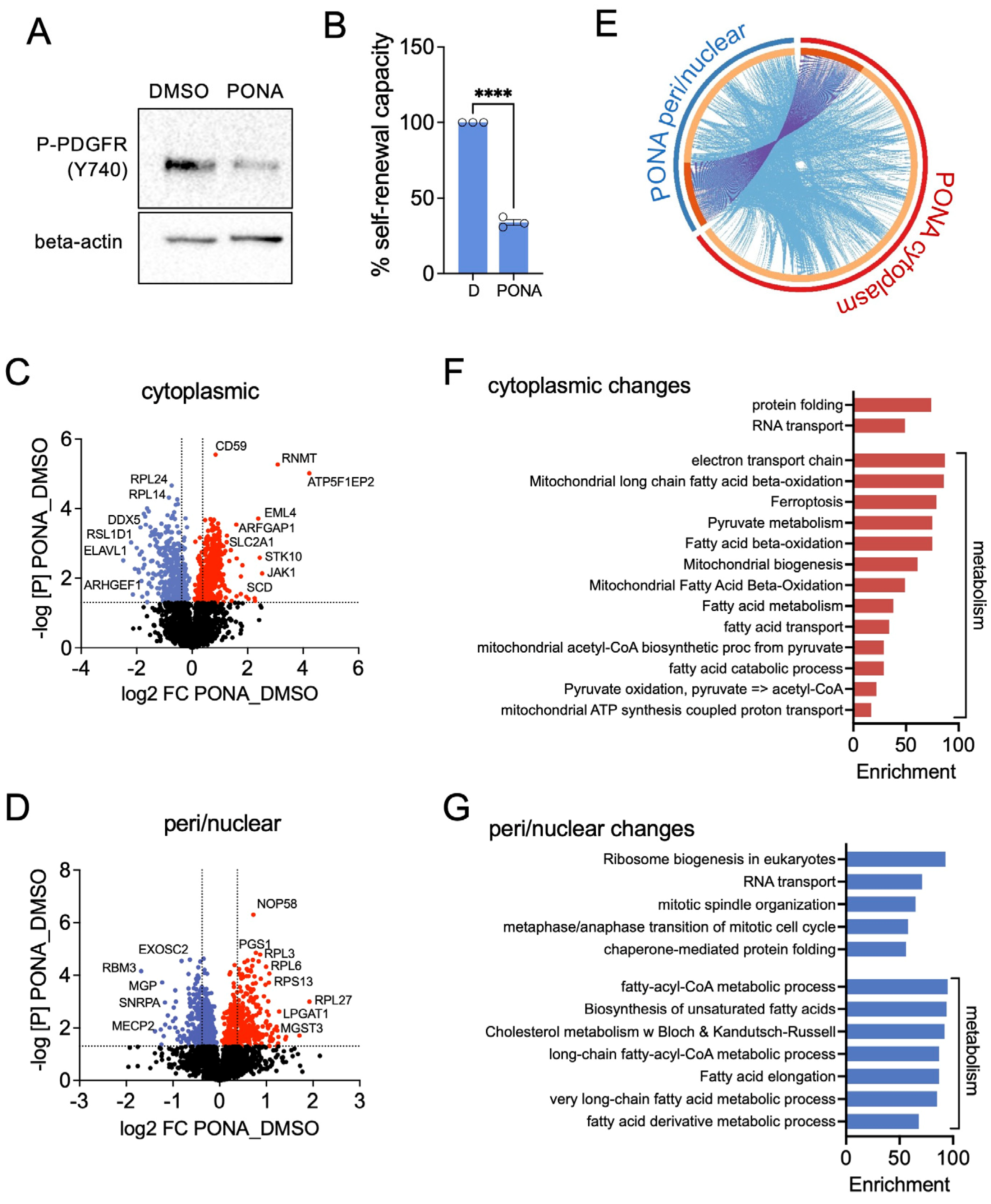
3.1. Ponatinib Induces Changes in Subcellular Protein Localisation in T98G Neurospheres
3.2. Ponatinib Reduces Glycolysis and Enhances Lipid Metabolism and OXPHOS in T98G Neurospheres
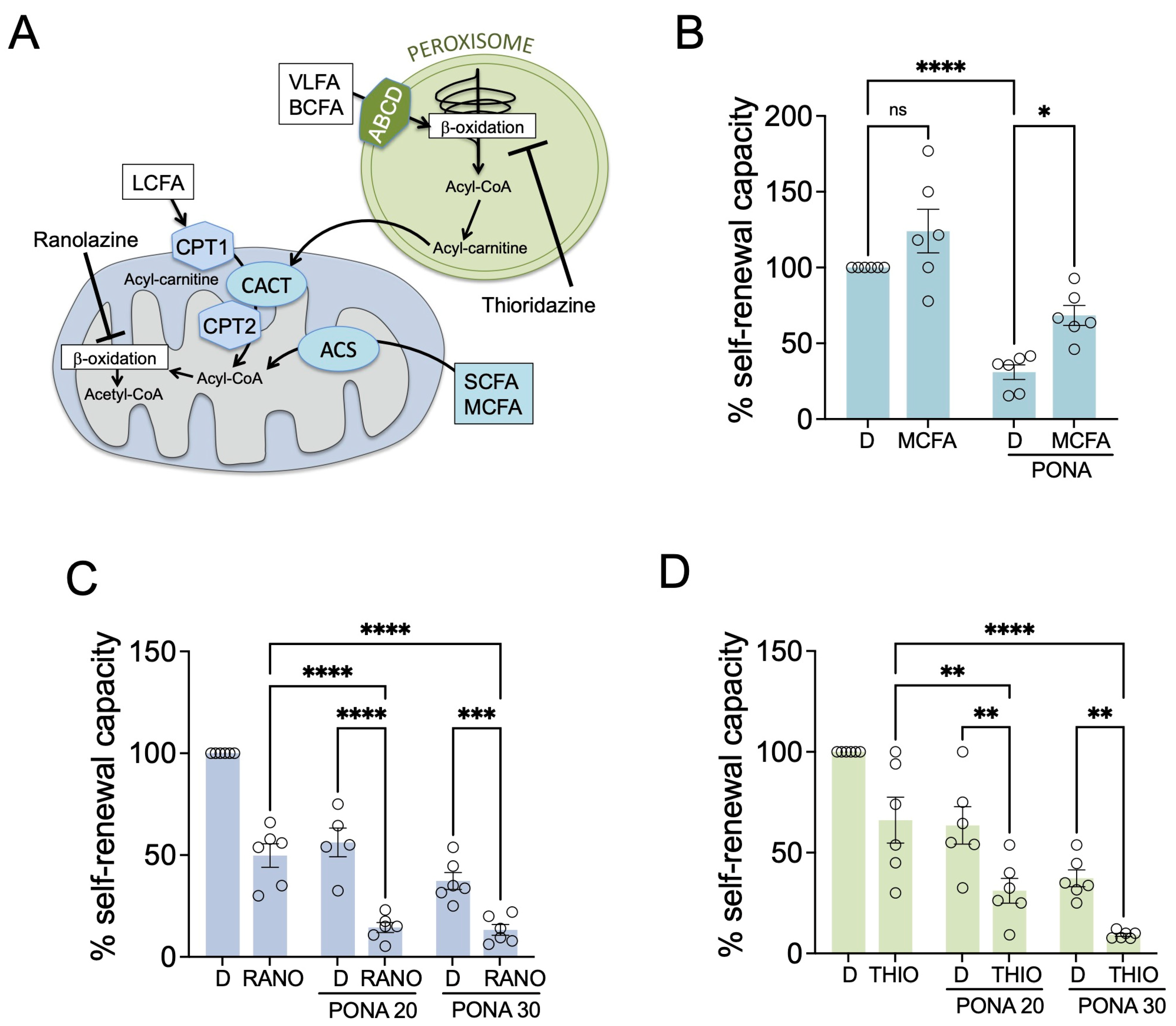
3.3. Inhibiting Fatty Acid Beta-Oxidation Potentiates the Response to Ponatinib in GSCs
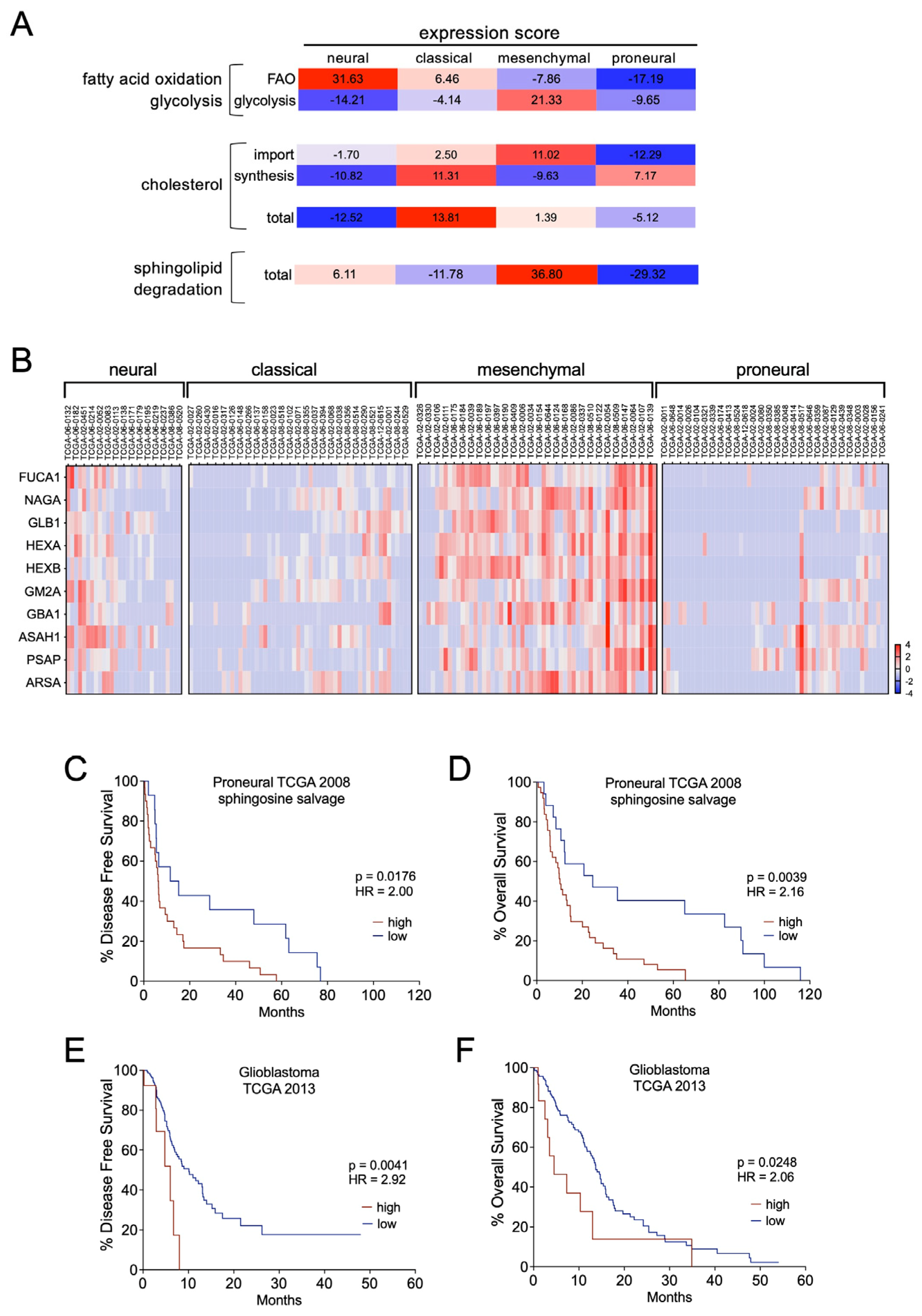
3.4. Cholesterol Synthesis Protects GSCs from Ponatinib-Mediated Inhibition
3.5. Ceramide Hydrolysis Counteracts Ponatinib Action in GSCs
3.6. Sphingolipid Degradation Is a Prognostic Factor in GBM
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dirks, P.B.; Gilbert, M.R.; Holland, E.C.; Maher, E.A.; Weiss, W.A. Translating Basic Science Discoveries into Improved Outcomes for Glioblastoma. Clin. Cancer Res. 2020, 26, 2457–2460. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849 e21. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef] [PubMed]
- Holland, E.C.; Hively, W.P.; DePinho, R.A.; Varmus, H.E. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998, 12, 3675–3685. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.J.; Appleman, V.A.; Wu, H.J.; Rose, C.M.; Pineda, J.J.; Yeo, A.T.; Delcuze, B.; Lee, C.; Gyuris, A.; Zhu, H.; et al. A PDGFRalpha-driven mouse model of glioblastoma reveals a stathmin1-mediated mechanism of sensitivity to vinblastine. Nat. Commun. 2018, 9, 3116. [Google Scholar] [CrossRef]
- Aldaz, P.; Arozarena, I. Tyrosine Kinase Inhibitors in Adult Glioblastoma: An (Un)Closed Chapter? Cancers 2021, 13, 5799. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Aldaz, P.; Auzmendi-Iriarte, J.; Durantez, M.; Lasheras-Otero, I.; Carrasco-Garcia, E.; Zelaya, M.V.; Bragado, L.; Olias-Arjona, A.; Egana, L.; Sampron, N.; et al. Identification of a Dexamethasone Mediated Radioprotection Mechanism Reveals New Therapeutic Vulnerabilities in Glioblastoma. Cancers 2021, 13, 361. [Google Scholar] [CrossRef]
- Ravi, K.; Franson, A.; Homan, M.J.; Roberts, H.; Pai, M.P.; Miklja, Z.; He, M.; Wen, B.; Benitez, L.L.; Perissinotti, A.J.; et al. Comparative pharmacokinetic analysis of the blood-brain barrier penetration of dasatinib and ponatinib in mice. Leuk. Lymphoma 2021, 62, 1990–1994. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhu, Y.; Miao, L.; Jia, T.; Mao, J.; Xue, L.; Wang, Y. Comparison of the Efficacy and Safety of Ponatinib and Dasatinib in Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia with Central Nervous System Relapse: A Retrospective Study. Technol. Cancer Res. Treat. 2023, 22, 15330338231165866. [Google Scholar] [CrossRef] [PubMed]
- Shilov, I.V.; Seymour, S.L.; Patel, A.A.; Loboda, A.; Tang, W.H.; Keating, S.P.; Hunter, C.L.; Nuwaysir, L.M.; Schaeffer, D.A. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteom. 2007, 6, 1638–1655. [Google Scholar] [CrossRef]
- Tang, W.H.; Shilov, I.V.; Seymour, S.L. Nonlinear fitting method for determining local false discovery rates from decoy database searches. J. Proteome Res. 2008, 7, 3661–3667. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Sanz, V.; Arozarena, I.; Crespo, P. Distinct carboxy-termini confer divergent characteristics to the mitogen-activated protein kinase p38alpha and its splice isoform Mxi2. FEBS Lett. 2000, 474, 169–174. [Google Scholar] [CrossRef]
- Bedia, C.; Casas, J.; Andrieu-Abadie, N.; Fabrias, G.; Levade, T. Acid ceramidase expression modulates the sensitivity of A375 melanoma cells to dacarbazine. J. Biol. Chem. 2011, 286, 28200–28209. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Garcia, E.; Martinez-Lacaci, I.; Mayor-Lopez, L.; Tristante, E.; Carballo-Santana, M.; Garcia-Morales, P.; Ventero Martin, M.P.; Fuentes-Baile, M.; Rodriguez-Lescure, A.; Saceda, M. PDGFR and IGF-1R Inhibitors Induce a G2/M Arrest and Subsequent Cell Death in Human Glioblastoma Cell Lines. Cells 2018, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Shang, E.; Westhoff, M.A.; Karpel-Massler, G.; Siegelin, M.D. Therapeutic Drug-Induced Metabolic Reprogramming in Glioblastoma. Cells 2022, 11, 2956. [Google Scholar] [CrossRef] [PubMed]
- Redondo-Munoz, M.; Rodriguez-Baena, F.J.; Aldaz, P.; Caballe-Mestres, A.; Moncho-Amor, V.; Otaegi-Ugartemendia, M.; Carrasco-Garcia, E.; Olias-Arjona, A.; Lasheras-Otero, I.; Santamaria, E.; et al. Metabolic rewiring induced by ranolazine improves melanoma responses to targeted therapy and immunotherapy. Nat. Metab. 2023, 5, 1544–1562. [Google Scholar] [CrossRef] [PubMed]
- Duraj, T.; Garcia-Romero, N.; Carrion-Navarro, J.; Madurga, R.; Mendivil, A.O.; Prat-Acin, R.; Garcia-Canamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Schmoll Massari, M.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef]
- McCormack, J.G.; Stanley, W.C.; Wolff, A.A. Ranolazine: A novel metabolic modulator for the treatment of angina. Gen. Pharmacol. 1998, 30, 639–645. [Google Scholar] [CrossRef]
- Van den Branden, C.; Roels, F. Thioridazine: A selective inhibitor of peroxisomal beta-oxidation in vivo. FEBS Lett. 1985, 187, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhou, S.; Yang, Z.; Li, Z.A.; Hu, W.; Dai, L.; Liang, W.; Wang, X. Cholesterol metabolism and its implication in glioblastoma therapy. J. Cancer 2022, 13, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Heybrock, S.; Neculai, D.; Saftig, P. Cholesterol Handling in Lysosomes and Beyond. Trends Cell Biol. 2020, 30, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Maghe, C.; Trillet, K.; Andre-Gregoire, G.; Kerherve, M.; Merlet, L.; Jacobs, K.A.; Schauer, K.; Bidere, N.; Gavard, J. The paracaspase MALT1 controls cholesterol homeostasis in glioblastoma stem-like cells through lysosome proteome shaping. Cell Rep. 2024, 43, 113631. [Google Scholar] [CrossRef] [PubMed]
- Allende, M.L.; Zhu, H.; Kono, M.; Hoachlander-Hobby, L.E.; Huso, V.L.; Proia, R.L. Genetic defects in the sphingolipid degradation pathway and their effects on microglia in neurodegenerative disease. Cell Signal 2021, 78, 109879. [Google Scholar] [CrossRef]
- Hawkins, C.C.; Ali, T.; Ramanadham, S.; Hjelmeland, A.B. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules 2020, 10, 1357. [Google Scholar] [CrossRef] [PubMed]
- Mahajan-Thakur, S.; Bien-Moller, S.; Marx, S.; Schroeder, H.; Rauch, B.H. Sphingosine 1-phosphate (S1P) signaling in glioblastoma multiforme—A systematic review. Int. J. Mol. Sci. 2017, 18, 2448. [Google Scholar] [CrossRef] [PubMed]
- Zaibaq, F.; Dowdy, T.; Larion, M. Targeting the Sphingolipid Rheostat in Gliomas. Int. J. Mol. Sci. 2022, 23, 9255. [Google Scholar] [CrossRef]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef]
- Aloia, A.; Mullhaupt, D.; Chabbert, C.D.; Eberhart, T.; Fluckiger-Mangual, S.; Vukolic, A.; Eichhoff, O.; Irmisch, A.; Alexander, L.T.; Scibona, E.; et al. A Fatty Acid Oxidation-dependent Metabolic Shift Regulates the Adaptation of BRAF-mutated Melanoma to MAPK Inhibitors. Clin. Cancer Res. 2019, 25, 6852–6867. [Google Scholar] [CrossRef]
- Shen, S.; Faouzi, S.; Souquere, S.; Roy, S.; Routier, E.; Libenciuc, C.; Andre, F.; Pierron, G.; Scoazec, J.Y.; Robert, C. Melanoma Persister Cells Are Tolerant to BRAF/MEK Inhibitors via ACOX1-Mediated Fatty Acid Oxidation. Cell Rep. 2020, 33, 108421. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Jahangiri, A.; Chen, W.; Nguyen, A.T.; Yagnik, G.; Pereira, M.P.; Jain, S.; Garcia, J.H.; Shah, S.S.; Wadhwa, H.; et al. Clonal ZEB1-Driven Mesenchymal Transition Promotes Targetable Oncologic Antiangiogenic Therapy Resistance. Cancer Res. 2020, 80, 1498–1511. [Google Scholar] [CrossRef]
- Longo, J.; van Leeuwen, J.E.; Elbaz, M.; Branchard, E.; Penn, L.Z. Statins as Anticancer Agents in the Era of Precision Medicine. Clin. Cancer Res. 2020, 26, 5791–5800. [Google Scholar] [CrossRef] [PubMed]
- Maja, M.; Tyteca, D. Alteration of cholesterol distribution at the plasma membrane of cancer cells: From evidence to pathophysiological implication and promising therapy strategy. Front. Physiol. 2022, 13, 999883. [Google Scholar] [CrossRef] [PubMed]
- Wahlen, E.; Olsson, F.; Soderberg, O.; Lennartsson, J.; Heldin, J. Differential impact of lipid raft depletion on platelet-derived growth factor (PDGF)-induced ERK1/2 MAP-kinase, SRC and AKT signaling. Cell. Signal. 2022, 96, 110356. [Google Scholar] [CrossRef]
- Happold, C.; Gorlia, T.; Nabors, L.B.; Erridge, S.C.; Reardon, D.A.; Hicking, C.; Picard, M.; Stupp, R.; Weller, M.; Group, E.B.T.; et al. Do statins, ACE inhibitors or sartans improve outcome in primary glioblastoma? J. Neurooncol. 2018, 138, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Seliger, C.; Schaertl, J.; Gerken, M.; Luber, C.; Proescholdt, M.; Riemenschneider, M.J.; Leitzmann, M.F.; Hau, P.; Klinkhammer-Schalke, M. Use of statins or NSAIDs and survival of patients with high-grade glioma. PLoS ONE 2018, 13, e0207858. [Google Scholar] [CrossRef]
- Marfia, G.; Campanella, R.; Navone, S.E.; Di Vito, C.; Riccitelli, E.; Hadi, L.A.; Bornati, A.; de Rezende, G.; Giussani, P.; Tringali, C.; et al. Autocrine/paracrine sphingosine-1-phosphate fuels proliferative and stemness qualities of glioblastoma stem cells. Glia 2014, 62, 1968–1981. [Google Scholar] [CrossRef]
- Janneh, A.H.; Ogretmen, B. Targeting Sphingolipid Metabolism as a Therapeutic Strategy in Cancer Treatment. Cancers 2022, 14, 2183. [Google Scholar] [CrossRef]
- Doan, N.B.; Nguyen, H.S.; Al-Gizawiy, M.M.; Mueller, W.M.; Sabbadini, R.A.; Rand, S.D.; Connelly, J.M.; Chitambar, C.R.; Schmainda, K.M.; Mirza, S.P. Acid ceramidase confers radioresistance to glioblastoma cells. Oncol. Rep. 2017, 38, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.B.; Alhajala, H.; Al-Gizawiy, M.M.; Mueller, W.M.; Rand, S.D.; Connelly, J.M.; Cochran, E.J.; Chitambar, C.R.; Clark, P.; Kuo, J.; et al. Acid ceramidase and its inhibitors: A de novo drug target and a new class of drugs for killing glioblastoma cancer stem cells with high efficiency. Oncotarget 2017, 8, 112662–112674. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.C.; Jones, A.B.; Gordon, E.R.; Harsh, Y.; Ziebro, J.K.; Willey, C.D.; Griguer, C.; Crossman, D.K.; Cooper, S.J.; Ramanadham, S.; et al. Carmofur prevents cell cycle progression by reducing E2F8 transcription in temozolomide-resistant glioblastoma cells. Cell Death Discov. 2023, 9, 451. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldaz, P.; Olias-Arjona, A.; Lasheras-Otero, I.; Ausin, K.; Redondo-Muñoz, M.; Wellbrock, C.; Santamaria, E.; Fernandez-Irigoyen, J.; Arozarena, I. Drug-Induced Reorganisation of Lipid Metabolism Limits the Therapeutic Efficacy of Ponatinib in Glioma Stem Cells. Pharmaceutics 2024, 16, 728. https://doi.org/10.3390/pharmaceutics16060728
Aldaz P, Olias-Arjona A, Lasheras-Otero I, Ausin K, Redondo-Muñoz M, Wellbrock C, Santamaria E, Fernandez-Irigoyen J, Arozarena I. Drug-Induced Reorganisation of Lipid Metabolism Limits the Therapeutic Efficacy of Ponatinib in Glioma Stem Cells. Pharmaceutics. 2024; 16(6):728. https://doi.org/10.3390/pharmaceutics16060728
Chicago/Turabian StyleAldaz, Paula, Ana Olias-Arjona, Irene Lasheras-Otero, Karina Ausin, Marta Redondo-Muñoz, Claudia Wellbrock, Enrique Santamaria, Joaquin Fernandez-Irigoyen, and Imanol Arozarena. 2024. "Drug-Induced Reorganisation of Lipid Metabolism Limits the Therapeutic Efficacy of Ponatinib in Glioma Stem Cells" Pharmaceutics 16, no. 6: 728. https://doi.org/10.3390/pharmaceutics16060728
APA StyleAldaz, P., Olias-Arjona, A., Lasheras-Otero, I., Ausin, K., Redondo-Muñoz, M., Wellbrock, C., Santamaria, E., Fernandez-Irigoyen, J., & Arozarena, I. (2024). Drug-Induced Reorganisation of Lipid Metabolism Limits the Therapeutic Efficacy of Ponatinib in Glioma Stem Cells. Pharmaceutics, 16(6), 728. https://doi.org/10.3390/pharmaceutics16060728