
Enhancing Oral Bioavailability of Simvastatin Using Uncoated and Polymer-Coated Solid Lipid Nanoparticles


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Simvastatin-Loaded Solid Lipid Nanoparticles (SLNs)
2.2. Preparation of Solution of Na-Alginate/Chitosan Coating Layer at 1% and 0.5% (w/v)
2.3. Sizes, PDI, and Zeta Potential
2.4. Entrapment Efficiency of SVA (EE %)
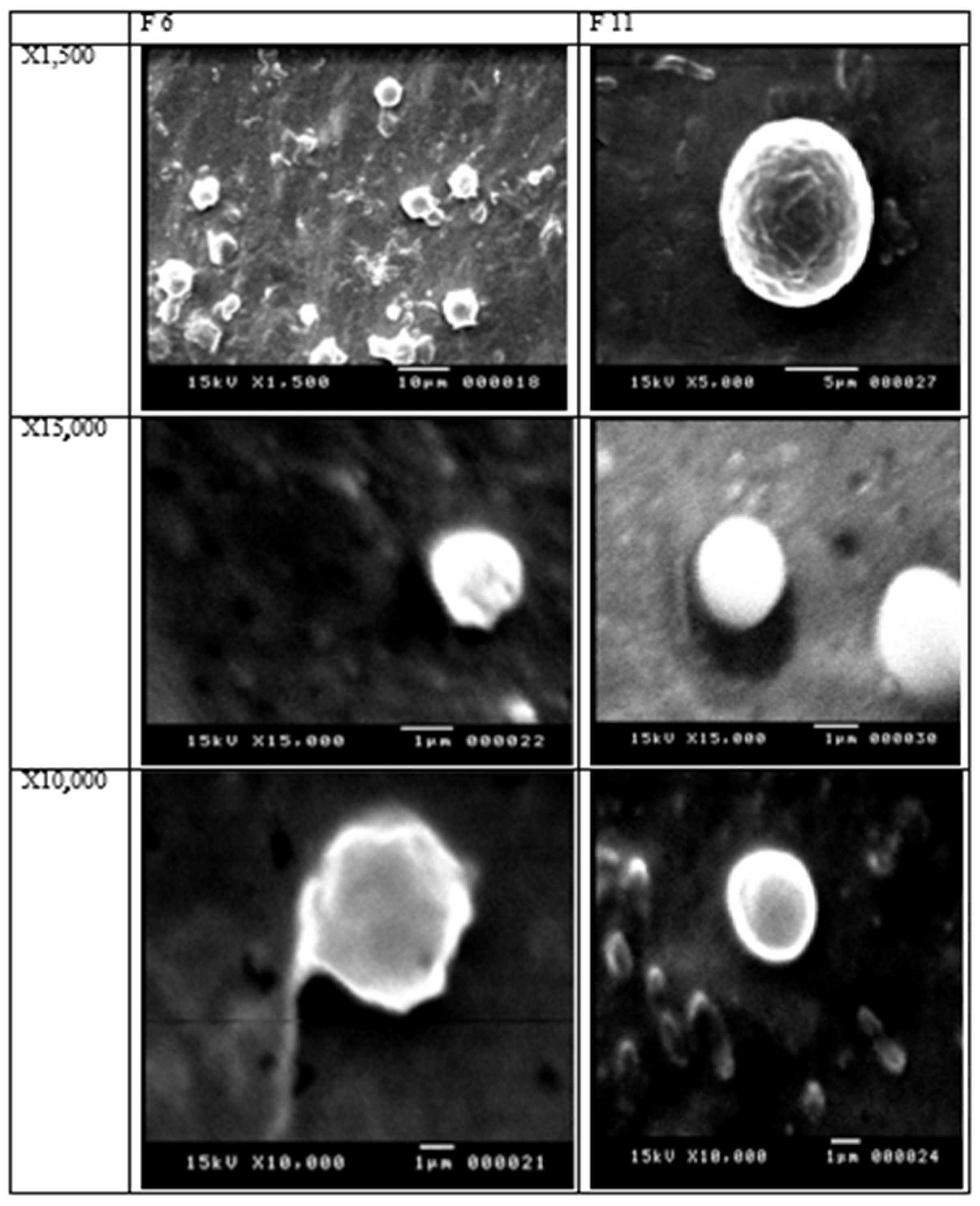
2.5. SEM
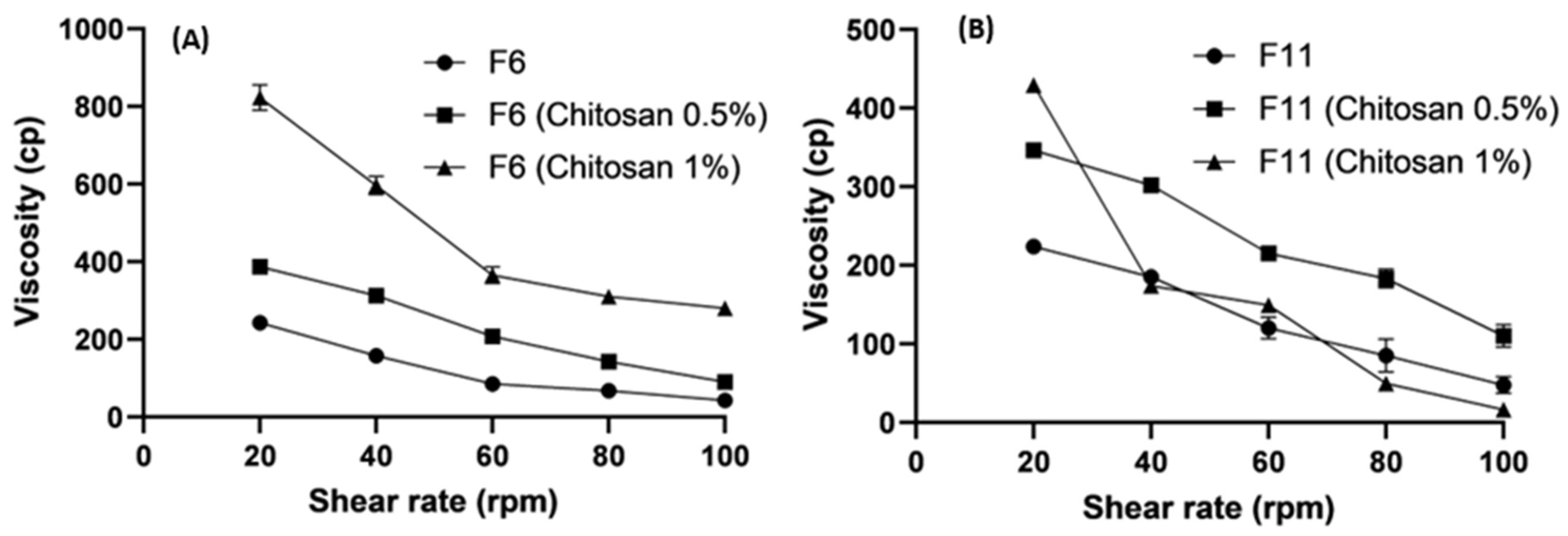
2.6. Viscosity
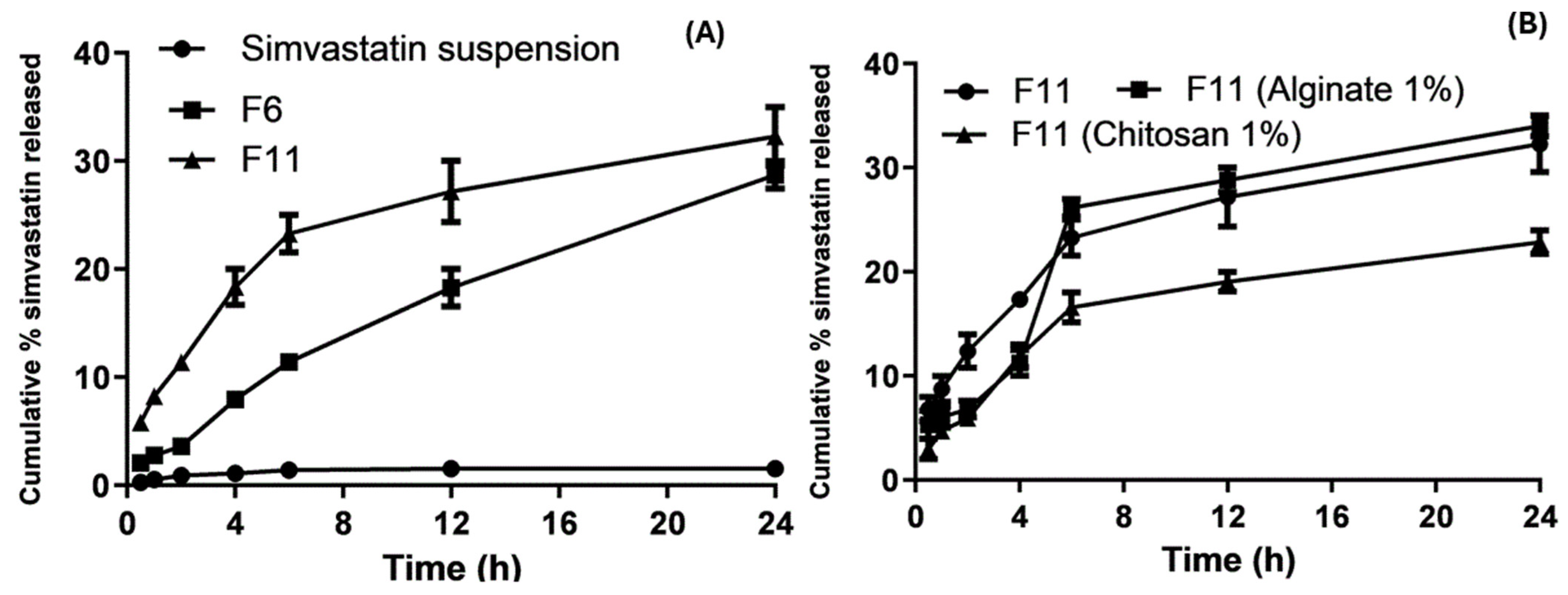
2.7. In Vitro Drug Release Studies
2.8. Release Kinetics
- Zero-order kinetics:
- 2.
- First-order kinetics:
- 3.
- Hixson–Crowell model:
- 4.
- Korsmeyer-Peppas kinetics model:
- 5.
- Baker and Lonsdale kinetic model:
- 6.
- Higuchi kinetic model:
2.9. In Vivo Study
2.10. Statistical Analysis
3. Results and Discussion
3.1. Particle Size and Polydispersity Index
3.2. Zeta Potential (ζ) Analysis
3.3. Entrapment Efficiency (EE%)
3.4. Scanning Electron Microscope (SEM)
3.5. Rheological Studies
3.5.1. Effect of Lipids on Viscosity
3.5.2. Effect of Polymer Concentrations on Viscosity
3.6. In Vitro Drug Release
Release Kinetics Design for Optimized SLNs and Coated Nanoparticles
3.7. In Vivo Pharmacokinetic Evaluation
- Enzymatic lipid breakdown in the small intestine results in the formation of monoglycerides and diglycerides, which eventually separate from the surface of nanoparticles and form micelles. These micelles combine with surface-active bile salts to generate mixed micelles, which are then absorbed via a transcellular pathway.
- Chylomicrons created from micelles eventually reach the lymphatic system. These micellar products could improve the in vivo drug dissolution rates and the ability of enterocytes to absorb them.
- Lipid nanoparticles (SLNs) can penetrate M cells via transcytosis, travel to the lymphatic system, and exit the systemic circulation through the thoracic duct. They have particular benefits in lymphatic transport due to chylomicron production, rapid absorption by M cells, and improved bioavailability by extending their time at the absorption site and preserving the medication from hepatic breakdown and the potential intestinal wall.
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mc Namara, K.; Alzubaidi, H.; Jackson, J.K. Cardiovascular disease as a leading cause of death: How are pharmacists getting involved? Integr. Pharm. Res. Pract. 2019, 8, 1–11. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927. [Google Scholar]
- O’Mahoney, P.R.; Wong, D.T.; Ray, J.G. Retinal vein occlusion and traditional risk factors for atherosclerosis. Arch. Ophthalmol. 2008, 126, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Rudnicka, A.R. Statin safety: A systematic review. Am. J. Cardiol. 2006, 97, S52–S60. [Google Scholar] [CrossRef]
- O’Sullivan, S. Statins—A review of benefits and risks. Trinity Stud. Med. J. 2007, 8, 52–56. [Google Scholar]
- Vanderbist, F.; Sereno, A.; Baudier, P.; Galephar, M.F. Oral Pharmaceutical Composition Containing a Statin Derivative. U.S. Patent US20040235935A1, 25 November 2004. [Google Scholar]
- Tahamtan, S.; Shirban, F.; Bagherniya, M.; Johnston, T.P.; Sahebkar, A. The effects of statins on dental and oral health: A review of preclinical and clinical studies. J. Transl. Med. 2020, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Neto, R.N.; Barros Gomes, E.D.; Weba-Soares, L.; Dias, L.R.; da Silva, L.C.; de Miranda, R.D. Biotechnological Production of Statins: Metabolic Aspects and Genetic Approaches. Curr. Pharm. Biotechnol. 2019, 20, 1244–1259. [Google Scholar] [CrossRef]
- Pathak, D.; Dahiya, S.; Pathak, K. Solid dispersion of meloxicam: Factorially designed dosage form for geriatric population. Acta Pharm. 2008, 58, 99–110. [Google Scholar] [CrossRef]
- Tiwari, R.; Pathak, K. Statins therapy: A review on conventional and novel formulation approaches. J. Pharm. Pharmacol. 2011, 63, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Ghadi, R.; Dand, N. BCS class IV drugs: Highly notorious candidates for formulation development. J. Control. Release 2017, 248, 71–95. [Google Scholar] [CrossRef]
- Gu, F.; Ning, J.; Fan, H.; Wu, C.; Wang, Y. Preparation and characterization of simvastatin/DMβCD complex and its pharmacokinetics in rats. Acta Pharm. 2018, 68, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Doktorovová, S.; Kovačević, A.B.; Garcia, M.L.; Souto, E.B. Preclinical safety of solid lipid nanoparticles and nanostructured lipid carriers: Current evidence from in vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2016, 108, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Tekade, R.K.; Maheshwari, R.; Tekade, M.; Chougule, M.B. Solid lipid nanoparticles for targeting and delivery of drugs and genes. In Nanotechnology-Based Approaches for Targeting and Delivery of Drugs and Genes; Elsevier: Amsterdam, The Netherlands, 2017; pp. 256–286. [Google Scholar]
- Shahraeini, S.; Akbari, J.; Saeedi, M.; Morteza-Semnani, K.; Abootorabi, S.; Dehghanpoor, M.; Rostamkalaei, S.S.; Nokhodchi, A. Atorvastatin solid lipid nanoparticles as a promising approach for dermal delivery and an anti-inflammatory agent. AAPS PharmSciTech 2020, 21, 263. [Google Scholar] [CrossRef] [PubMed]
- Dawoud, M. Chitosan coated solid lipid nanoparticles as promising carriers for docetaxel. J. Drug Deliv. Sci. Technol. 2021, 62, 102409. [Google Scholar] [CrossRef]
- Wang, F.; Yang, S.; Yuan, J.; Gao, Q.; Huang, C. Effective method of chitosan-coated alginate nanoparticles for target drug delivery applications. J. Biomater. Appl. 2016, 31, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Sastri, K.T.; Radha, G.V.; Pidikiti, S.; Vajjhala, P. Solid lipid nanoparticles: Preparation techniques, their characterization, and an update on recent studies. J. Appl. Pharm. Sci. 2020, 10, 126–141. [Google Scholar]
- Khairnar, S.V.; Pagare, P.; Thakre, A.; Nambiar, A.R.; Junnuthula, V.; Abraham, M.C.; Kolimi, P.; Nyavanandi, D.; Dyawanapelly, S. Review on the scale-up methods for the preparation of solid lipid nanoparticles. Pharmaceutics 2022, 14, 1886. [Google Scholar] [CrossRef] [PubMed]
- El-hafian, E.A.; Elgannoudi, E.S.; Mainal, A.; Yahaya, A.H. Characterization of chitosan in acetic acid: Rheological and thermal studies. Turk. J. Chem. 2010, 34, 47–56. [Google Scholar] [CrossRef]
- Yallapu, M.M.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C. Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells. J. Colloid Interface Sci. 2010, 351, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tan, T.; Gao, L. Preparation of oridonin-loaded solid lipid nanoparticles and studies on them in vitro and in vivo. Nanotechnology 2006, 17, 5821. [Google Scholar] [CrossRef]
- Khan, M.F.; Ur Rehman, A.; Howari, H.; Alhodaib, A.; Ullah, F.; Mustafa, Z.U.; Elaissari, A.; Ahmed, N. Hydrogel containing solid lipid nanoparticles loaded with argan oil and simvastatin: Preparation, in vitro and ex vivo assessment. Gels 2022, 8, 277. [Google Scholar] [CrossRef] [PubMed]
- Lokhandwala, H.; Deshpande, A.; Deshpande, S. Kinetic modeling and dissolution profiles comparison: An overview. Int. J. Pharma Bio Sci. 2013, 4, 728–773. [Google Scholar]
- Havlík, T. (Ed.) Woodhead Publishing Series in Metals and Surface Engineering, Hydrometallurgy; Woodhead Publishing: Cambridge, UK, 2008. [Google Scholar]
- Mircioiu, C.; Voicu, V.; Anuta, V.; Tudose, A.; Celia, C.; Paolino, D.; Fresta, M.; Sandulovici, R.; Mircioiu, I. Mathematical modeling of release kinetics from supramolecular drug delivery systems. Pharmaceutics 2019, 11, 140. [Google Scholar] [CrossRef]
- Rizvi, S.Z.; Shah, F.A.; Khan, N.; Muhammad, I.; Ali, K.H.; Ansari, M.M.; ud Din, F.; Qureshi, O.S.; Kim, K.W.; Choe, Y.H.; et al. Simvastatin-loaded solid lipid nanoparticles for enhanced anti-hyperlipidemic activity in hyperlipidemia animal model. Int. J. Pharm. 2019, 560, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Aburahma, M.H.; Badr-Eldin, S.M. Compritol 888 ATO: A multifunctional lipid excipient in drug delivery systems and nanopharmaceuticals. Expert Opin. Drug Deliv. 2014, 11, 1865–1883. [Google Scholar] [CrossRef]
- Khaled, A.; Abdel-Hamid, S.; Nasr, M.; Sammour, O.A. Fabrication of extended-dissolution divalproex tablets: A green solvent-free granulation technique. Drug Dev. Ind. Pharm. 2020, 46, 975–987. [Google Scholar] [CrossRef]
- Kotmakçı, M.; Akbaba, H.; Erel, G.; Ertan, G.; Kantarcı, G. Improved method for solid lipid nanoparticle preparation based on hot microemulsions: Preparation, characterization, cytotoxicity, and hemocompatibility evaluation. AAPS PharmSciTech 2017, 18, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Singhal, G.B.; Patel, R.P.; Prajapati, B.G.; Patel, N.A. Solid lipid nanoparticles and nano lipid carriers: As novel solid lipid based drug carrier. Int. Res. J. Pharm. 2011, 2, 20–52. [Google Scholar]
- Siddiqui, A.; Alayoubi, A.; El-Malah, Y.; Nazzal, S. Modeling the effect of sonication parameters on size and dispersion temperature of solid lipid nanoparticles (SLNs) by response surface methodology (RSM). Pharm. Dev. Technol. 2014, 19, 342–346. [Google Scholar] [CrossRef]
- Shlear, H.; Nabeel, S.O.; Kafia, M.S. Solid lipid nanoparticles for topical delivery of meloxicam: Development and in vitro characterization. Eur. Sci. J. ESJ 2013, 3, 779–798. [Google Scholar]
- Rowe, R.C.; Sheskey, P.; Quinn, M. Handbook of Pharmaceutical Excipients; Libros Digitales-Pharmaceutical Press: London, UK, 2009. [Google Scholar]
- Housaindokht, M.R.; Pour, A.N. Study the effect of HLB of surfactant on particle size distribution of hematite nanoparticles prepared via the reverse microemulsion. Solid State Sci. 2012, 14, 622–625. [Google Scholar] [CrossRef]
- Mura, P.; Maestrelli, F.; D’Ambrosio, M.; Luceri, C.; Cirri, M. Evaluation and comparison of solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) as vectors to develop hydrochlorothiazide effective and safe pediatric oral liquid formulations. Pharmaceutics 2021, 13, 437. [Google Scholar] [CrossRef] [PubMed]
- Matricardi, P.; Meo, C.D.; Coviello, T.; Alhaique, F. Recent advances and perspectives on coated alginate microspheres for modified drug delivery. Expert Opin. Drug Deliv. 2008, 5, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Fangueiro, J.F.; Andreani, T.; Egea, M.A.; Garcia, M.L.; Souto, S.B.; Souto, E.B. Experimental factorial design applied to mucoadhesive lipid nanoparticles via multiple emulsion process. Colloids Surf. B Biointerfaces 2012, 100, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Takka, S.; Gürel, A. Evaluation of chitosan/alginate beads using experimental design: Formulation and in vitro characterization. AAPS PharmSciTech 2010, 11, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Stainmesse, S.; Fessi, H.; Devissaguet, J.-P.; Puisieux, F.; Theis, C. Process for the Preparation of Dispersible Colloidal Systems of a Substance in the Form of Nanoparticles. U.S. Patent US5133908A, 28 July 1992. [Google Scholar]
- Dwivedi, P.; Karumbaiah, K.M.; Das, R. Nano-size polymers via precipitation of polymer solutions. In Nano-Size Polymers; Springer: Berlin/Heidelberg, Germany, 2016; pp. 251–282. [Google Scholar]
- Mishra, V.; Bansal, K.K.; Verma, A.; Yadav, N.; Thakur, S.; Sudhakar, K.; Rosenholm, J.M. Solid lipid nanoparticles: Emerging colloidal nano drug delivery systems. Pharmaceutics 2018, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- Hassas, B.V.; Karakaş, F.; Çelik, M.S. Ultrafine coal dewatering: Relationship between hydrophilic lipophilic balance (HLB) of surfactants and coal rank. Int. J. Miner. Process. 2014, 133, 97–104. [Google Scholar] [CrossRef]
- Zimmermann, E.; Müller, R.H. Electrolyte-and pH-stabilities of aqueous solid lipid nanoparticle (SLN™) dispersions in artificial gastrointestinal media. Eur. J. Pharm. Biopharm. 2001, 52, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Graciaa, A.; Creux, P.; Dicharry, C.; Lachaise, J. Measurement of the zeta potential of oil drops with the spinning tube zetameter. J. Dispers. Sci. Technol. 2002, 23, 301–307. [Google Scholar] [CrossRef]
- Liu, M.; Wang, F.; Pu, C.; Tang, W.; Sun, Q. Nanoencapsulation of lutein within lipid-based delivery systems: Characterization and comparison of zein peptide stabilized nano-emulsion, solid lipid nanoparticle, and nano-structured lipid carrier. Food Chem. 2021, 358, 129840. [Google Scholar] [CrossRef]
- Szabó, L.; Gerber-Lemaire, S.; Wandrey, C. Strategies to functionalize the anionic biopolymer Na-alginate without restricting its polyelectrolyte properties. Polymers 2020, 12, 919. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, Z.; Peng, Y.; Feng, L.; Li, X.; Zhao, C.; Sarfaraz, K. Novel cationic polymer modified magnetic chitosan beads for efficient adsorption of heavy metals and dyes over a wide pH range. Int. J. Biol. Macromol. 2020, 156, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Motwani, S.K.; Chopra, S.; Talegaonkar, S.; Kohli, K.; Ahmad, F.J.; Khar, R.K. Chitosan–sodium alginate nanoparticles as submicroscopic reservoirs for ocular delivery: Formulation, optimisation and in vitro characterisation. Eur. J. Pharm. Biopharm. 2008, 68, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Vigani, B.; Valentino, C.; Sandri, G.; Listro, R.; Fagiani, F.; Collina, S.; Lanni, C.; Bonferoni, M.C.; Caramella, C.M.; Rossi, S.; et al. A composite nanosystem as a potential tool for the local treatment of glioblastoma: Chitosan-coated solid lipid nanoparticles embedded in electrospun nanofibers. Polymers 2021, 13, 1371. [Google Scholar] [CrossRef] [PubMed]
- Pyo, Y.-C.; Tran, P.; Kim, D.-H.; Park, J.-S. Chitosan-coated nanostructured lipid carriers of fenofibrate with enhanced oral bioavailability and efficacy. Colloids Surf. B Biointerfaces 2020, 196, 111331. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.J. An overview of techniques for multifold enhancement in solubility of poorly soluble drugs. Curr. Issues Pharm. Med. Sci. 2019, 32, 203–209. [Google Scholar] [CrossRef]
- Krishnam Raju, K.; Sudhakar, B.; Murthy, K.V. Factorial design studies and biopharmaceutical evaluation of simvastatin loaded solid lipid nanoparticles for improving the oral bioavailability. Int. Sch. Res. Not. 2014, 2014, 951016. [Google Scholar] [CrossRef]
- Luo, Y.; Teng, Z.; Li, Y.; Wang, Q. Solid lipid nanoparticles for oral drug delivery: Chitosan coating improves stability, controlled delivery, mucoadhesion and cellular uptake. Carbohydr. Polym. 2015, 122, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Piazzini, V.; Cinci, L.; D’Ambrosio, M.; Luceri, C.; Bilia, A.R.; Bergonzi, M.C. Solid lipid nanoparticles and chitosan-coated solid lipid nanoparticles as promising tool for silybin delivery: Formulation, characterization, and in vitro evaluation. Curr. Drug Deliv. 2019, 16, 142–152. [Google Scholar] [CrossRef]
- Bodnár, T.; Sequeira, A. Analysis of the Shear-Thinning Viscosity Behavior of the Johnson–Segalman Viscoelastic Fluids. Fluids 2022, 7, 36. [Google Scholar] [CrossRef]
- Cetin, F.N. Effect of the Sodium Alginate Concentration and the Ionic Strength on the Rheological Properties of Sodium Alginate-Based Hydrogels. Ph.D. Thesis, UNITESI, Auckland, New Zealand, 2022. [Google Scholar]
- Samimi Gharaie, S.; Dabiri, S.M.H.; Akbari, M. Smart shear-thinning hydrogels as injectable drug delivery systems. Polymers 2018, 10, 1317. [Google Scholar] [CrossRef] [PubMed]
- Isaac, V.L.B.; Chiari-Andréo, B.G.; Marto, J.M.; Moraes, J.D.D.; Leone, B.A.; Corrêa, M.A.; Ribeiro, H.M. Rheology as a tool to predict the release of alpha-lipoic acid from emulsions used for the prevention of skin aging. BioMed Res. Int. 2015, 2015, 818656. [Google Scholar] [CrossRef] [PubMed]
- Simões, A.; Miranda, M.; Cardoso, C.; Veiga, F.; Vitorino, C. Rheology by design: A regulatory tutorial for analytical method validation. Pharmaceutics 2020, 12, 820. [Google Scholar] [CrossRef] [PubMed]
- Abo-Zalam, H.B.; El-Denshary, E.S.; Abdelsalam, R.M.; Khalil, I.A.; Khattab, M.M.; Hamzawy, M.A. Therapeutic advancement of simvastatin-loaded solid lipid nanoparticles (SV-SLNs) in treatment of hyperlipidemia and attenuating hepatotoxicity, myopathy and apoptosis: Comprehensive study. Biomed. Pharmacother. 2021, 139, 111494. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, F.; Faghfouri, L.; Hamidi, M. Preparation, optimization, and in-vitro characterization of α-tocopherol-loaded solid lipid nanoparticles (SLNs). Drug Dev. Ind. Pharm. 2020, 46, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Sarathchandiran, I.; Koumaravelou, K.; Selvasudha, N. Interaction pattern and in vitro, in vivo release behavior of simvastatin-loaded chitosan nanoformulation. Drug Dev. Ind. Pharm. 2019, 45, 1725–1739. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Munoz, N.; Urbán-Morlán, Z.; Leyva-Gómez, G.; de la Luz Zambrano-Zaragoza, M.; Quintanar-Guerrero, D. Solid lipid nanoparticles: An approach to improve oral drug delivery. J. Pharm. Pharm. Sci. 2021, 24, 509–532. [Google Scholar] [CrossRef]
- Rahamathulla, M.; H.V, G.; Veerapu, G.; Hani, U.; Alhamhoom, Y.; Alqahtani, A.; Moin, A. Characterization, optimization, in vitro and in vivo evaluation of simvastatin proliposomes, as a drug delivery. AAPS PharmSciTech 2020, 21, 129. [Google Scholar] [CrossRef]
- Jana, S.; Maiti, S. Chitosan-based nanoparticulate systems for oral drug delivery. Nanostructures for Oral Medicine. In Nanostructures for Oral Medicine; Jana, S., Maiti, S., Eds.; Elsevier: Philadelphia, PA, USA, 2017; pp. 607–638. [Google Scholar]
- Klinkesorn, U. The role of chitosan in emulsion formation and stabilization. Food Rev. Int. 2013, 29, 371–393. [Google Scholar] [CrossRef]
- Laye, C.; McClements, D.J.; Weiss, J. Formation of biopolymer-coated liposomes by electrostatic deposition of chitosan. J. Food Sci. 2008, 73, N7–N15. [Google Scholar] [CrossRef]
- Selvasudha, N.; Koumaravelou, K. The multifunctional synergistic effect of chitosan on simvastatin loaded nanoparticulate drug delivery system. Carbohydr. Polym. 2017, 163, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Dolatabadi, S.; Karimi, M.; Nasirizadeh, S.; Hatamipour, M.; Golmohammadzadeh, S.; Jaafari, M.R. Preparation, characterization and in vivo pharmacokinetic evaluation of curcuminoids-loaded solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs). J. Drug Deliv. Sci. Technol. 2021, 62, 102352. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 | F11 | F12 | F13 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compritol 888 ATO (mg) | 200 | 400 | 200 | 200 | 100 | 100 | |||||||
| Precirol (mg) | 200 | 400 | 200 | 100 | 100 | 100 | |||||||
| Geleol (mg) | 200 | 400 | 200 | 100 | 100 | 100 | |||||||
| Gelucire43/01 (mg) | 200 | 400 | 100 | 100 | |||||||||
| Poloxamer407 (mg) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Tween 80 (%) | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Formulation Code | P.S (nm) | PDI | ζ (mV) | % EE |
|---|---|---|---|---|
| F1 | 663.9 ± 90.92 | 0.612 ± 0.084 | −4.92 ± 0.22 ** | 100 ± 2.85 ** |
| F2 | 338.57± 52.86 ** | 0.438 ± 0.051 | −5.53 ± 0.49 ** | 99.75 ± 0.52 |
| F3 | 332.6 ± 7.71 ** | 0.907 ± 0.03 | −21.86 ± 1.52 * | 97.97 ± 0.39 |
| F4 | 1167.67 ± 73.33 * | 0.611 ± 0.09 | −4.79 ± 0.13 | 98.96 ± 2.66 |
| F5 | 396.7 ± 8.63 | 0.832 ± 0.073 | −3.21 ± 0.15 | 101.01 ± 2.85 |
| F6 * | 231.87 ± 108.22 | 0.4926 ± 0.014 | −1.8 ± 0.15 | 100.1 ± 2.55 |
| F7 | 387.3 ± 1.7 ** | 0.518 ± 0.013 | −6.75 ± 1.7 | 99.26± 1.56 |
| F8 | 871.83 ± 26.25 | 0.65 ± 0.034 | −2.57 ± 0.23 | 99.26 ± 0.28 |
| F9 | 765.17 ± 29.18 | 0.619 ± 0.002 | −8.17 ± 0.63 | 99.72 ± 0.41 |
| F10 | 528.27 ± 20.88 | 0.772 ± 0.12 | −2.5 ± 0.39 | 99.20 ± 0.32 |
| F11 * | 260.1 ± 3.72 | 0.409 ± 0.005 | −4.26 ± 0.26 | 99.36 ± 0.68 |
| F12 | 894.3 ± 56.14 | 0.611 ± 0.034 | −7.97 ± 0.71 | 99.44 ± 1.21 |
| F13 | 488.4 ± 19.24 | 0.738 ± 0.11 | −7.98 ± 0.81 | 99.53 ± 0.28 |
| F6 (Chitosan 0.5%) | 487.5 ± 13.09 * | 0.127 ± 0.028 | 44.7 ± 1.63 | 98.999 ± 0.02 |
| F6 (Chitosan 1%) | 844.4 ± 117.9 * | 0.504 ± 0.057 | 52.9 ± 2.24 | 98 ± 0.28 |
| F6 (Alginate 0.5%) | 1181 ± 129.7 ** | 0.831 ± 0.072 | −22.0 ± 0.781 | 97.9866 ± 0.23 |
| F6 (Alginate 1%) | 906.0 ± 42.12 ** | 0.621 ± 0.09 | −27 ± 0.321 | 97.99 ± 0.06 |
| F11 (Chitosan 0.5%) | 477.3 ± 21.4 | 0.504 ± 0.057 | 33.4 ± 1.85 | 99.4793 ± 0.08 |
| F11 (Chitosan 1%) | 524.3 ± 80.31 | 0.249 ± 0.016 | 42.6 ± 1.75 | 99.756 ± 0.15 |
| F11 (alginate 0.5%) | 157 ± 16.35 | 0.067 ± 0.019 | −9.48 ± 0.174 | 98.798 ± 0.10 |
| F11 (Alginate 1%) | 1071 ± 45.63 | 0.852 ± 0.084 | −24.8 ± 0.346 | 99.314 ± 0.31 |
| Code Formula | Zero | First | Higuchi | Hixon–Crowell | Baker & Lonsdale | Korysmeyer-Peppas | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R2 * | K ** | R2 | K | R2 | KH | R2 | KHC | R2 | K3 | R2 | N *** | |
| SVA | 0.58 | 0.72 | 0.49 | −0.16 | 0.76 | 0.44 | 0.52 | 0.01 | 0.63 | 0.001 | 0.88 | 0.33 |
| F6 | 0.98 | 2.29 | 0.79 | 0.50 | 0.98 | −4.17 | 0.87 | 0.07 | 0.97 | −0.001 | 0.97 | 0.72 |
| F11 | 0.89 | 9.8 | 0.64 | 2.23 | 0.85 | 2.02 | 0.69 | 2.1 | 0.92 | 0.001 | 0.97 | 0.45 |
| F6 (Alginate 0.5%) | 0.81 | 7.81 | 0.79 | 0.879 | 0.92 | −0.21 | 0.74 | 0.006 | 0.91 | 0.0006 | 0.92 | 0.53 |
| F6 (Alginate 1%) | 0.738 | 15.81 | 0.665 | 1.187 | 0.88 | 8.27 | 0.69 | 0.04 | 0.81 | 0.005 | 0.93 | 0.33 |
| F6 (Chitosan 0.5%) | 0.69 | 25.12 | 0.54 | 1.3 | 0.86 | 1.33 | 0.69 | 0.09 | 0.96 | −0.006 | 0.87 | 0.61 |
| F6 (Chitosan 1%) | 0.775 | 3.22 | 0.58 | 0.397 | 0.92 | −1.11 | 0.65 | 0.058 | 0.91 | 0.0002 | 0.94 | 0.79 |
| F11 (Alginate 0.5%) | 0.75 | 10.79 | 0.67 | 1.008 | 0.86 | 0.55 | 0.7 | 0.065 | 0.83 | 0.002 | 0.89 | 0.51 |
| F11 (Alginate 1%) | 0.79 | 8.77 | 0.7 | 0.93 | 0.89 | 0.896 | 0.73 | 0.06 | 0.88 | 0.001 | 0.89 | 0.48 |
| F11 (Chitosan 0.5%) | 0.68 | 32.13 | 0.51 | 1.4 | 0.85 | 9.97 | 0.57 | 0.075 | 0.90 | 0.02 | 0.92 | 0.53 |
| F11 (Chitosan 1%) | 0.83 | 6.30 | 0.68 | 0.78 | 0.95 | 1.27 | 0.73 | 0.05 | 0.93 | 0.005 | 0.95 | 0.47 |
| Parameters | SVA Suspension | F11 | F11 (Chitosan 1%) |
|---|---|---|---|
| AUC0→24 (ng·h/mL) * | 272 ± 197.89 | 1880.4 ± 222.08 | 3562.18 ± 1256.03 |
| Cmax (ng/mL) ** | 57.1.6 ± 22.93 | 251.88 ± 17.50 | 228.3 ± 21.76 |
| Tmax (h) *** | 1.41 ± 0.32 | 1.74 ± 0.36 | 5.15 ± 1.13 |
| t1/2 (h) **** | 0.54 ± 1.12 | 3.97 ± 0.99 | 11.43 ± 5.85 |
| MRT (h) ***** | 3.7 ± 0.52 | 6.29 ± 0.84 | 19.1 ± 1.03 |
| Ka (h−1) | 0.604 ± 0.06 | 1.32 ± 0.21 | 0.40 ± 0.03 |
| Kel (h−1) | 0.160 ± 0.02 | 0.030 ± 0.02 | 0.011 ± 0.05 |
| t1/2Ka (h) | 1.15 ± 0.12 | 0.52 ± 0.04 | 1.52 ± 0.20 |
| F% | 691.28 ± 0.61 | 1309.66 ± 0.92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abd-Elghany, A.E.; El-Garhy, O.; Fatease, A.A.; Alamri, A.H.; Abdelkader, H. Enhancing Oral Bioavailability of Simvastatin Using Uncoated and Polymer-Coated Solid Lipid Nanoparticles. Pharmaceutics 2024, 16, 763. https://doi.org/10.3390/pharmaceutics16060763
Abd-Elghany AE, El-Garhy O, Fatease AA, Alamri AH, Abdelkader H. Enhancing Oral Bioavailability of Simvastatin Using Uncoated and Polymer-Coated Solid Lipid Nanoparticles. Pharmaceutics. 2024; 16(6):763. https://doi.org/10.3390/pharmaceutics16060763
Chicago/Turabian StyleAbd-Elghany, Amira E., Omar El-Garhy, Adel Al Fatease, Ali H. Alamri, and Hamdy Abdelkader. 2024. "Enhancing Oral Bioavailability of Simvastatin Using Uncoated and Polymer-Coated Solid Lipid Nanoparticles" Pharmaceutics 16, no. 6: 763. https://doi.org/10.3390/pharmaceutics16060763