
Effects of Postprandial Factors and Second Meal Intake Time on Bioequivalence Investigation of Tadalafil-Loaded Orodispersible Films in Human Volunteers

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preliminary Screening of Dispersion Agents
2.3. TDF Formulation Preparation
2.4. Physicochemical Characterization of TDF Formulations
2.4.1. Scanning Electron Microscopy (SEM)
2.4.2. Particle Size Distribution (PSD)
2.4.3. Dissolution Study
2.4.4. Disintegration Test
2.5. Analysis of TD Uniformity in TDF
2.6. The Simulation of Meal Viscosity by Water Intake Volumes
2.7. The Drug Diffusion Rate in the Postprandial Stomach Conditions
2.8. Pharmacokinetics and Bioequivalence Test in Healthy Human Volunteers
2.8.1. Design and Dosing Schedule
2.8.2. Fasting-State Study
2.8.3. Fed-State Study
2.8.4. Analysis of Drug Concentration in Blood
2.8.5. Pharmacokinetic Parameter Calculation and Analysis
3. Results and Discussion
3.1. Screening of Dispersion Agents and Formulation Design
3.2. The Disintegration and Dissolution Rate of TD-Loaded Formulations
3.3. The Drug Diffusion Rate in the Postprandial Stomach Conditions
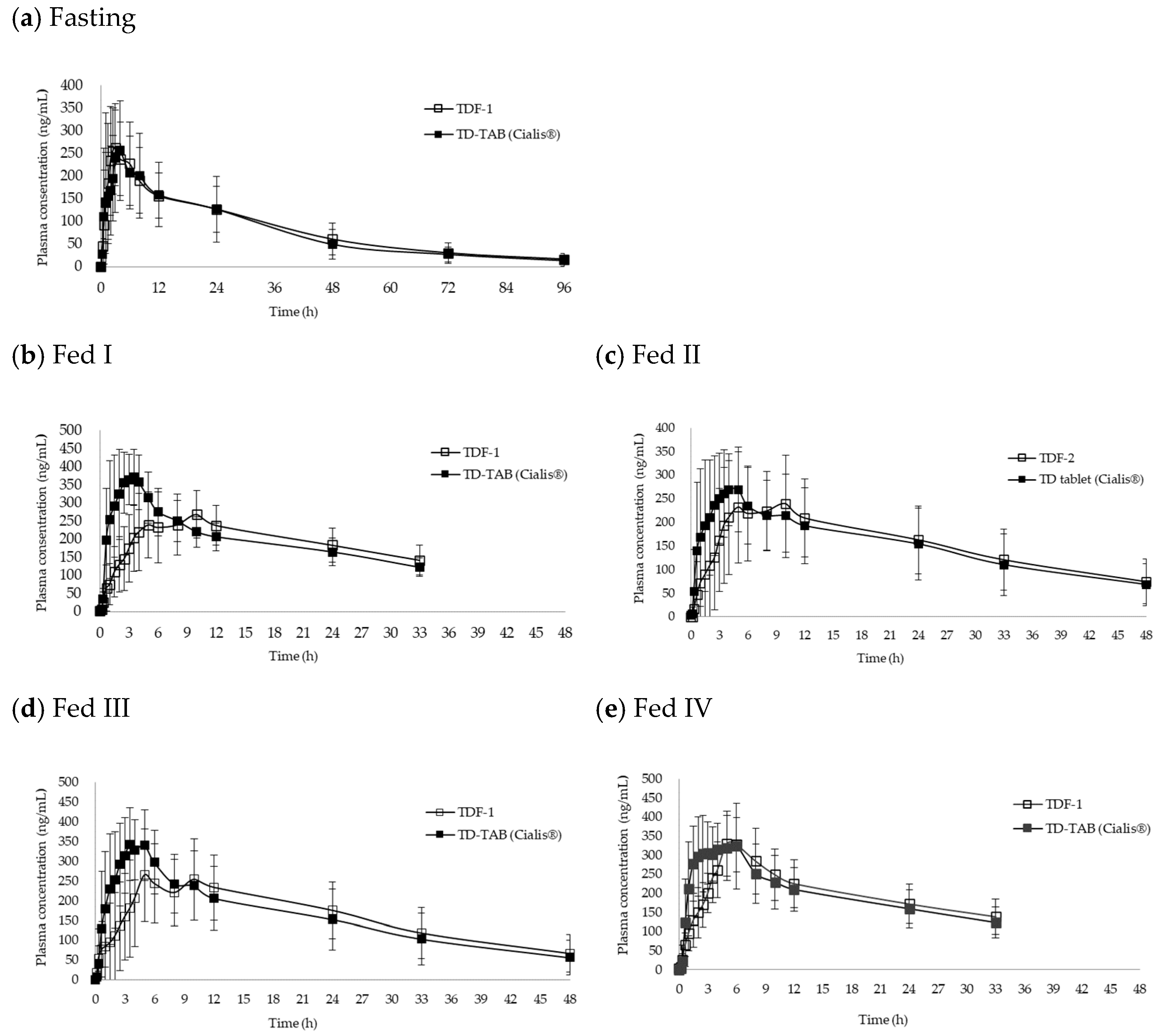
3.4. Comparative Bioequivalence Studies
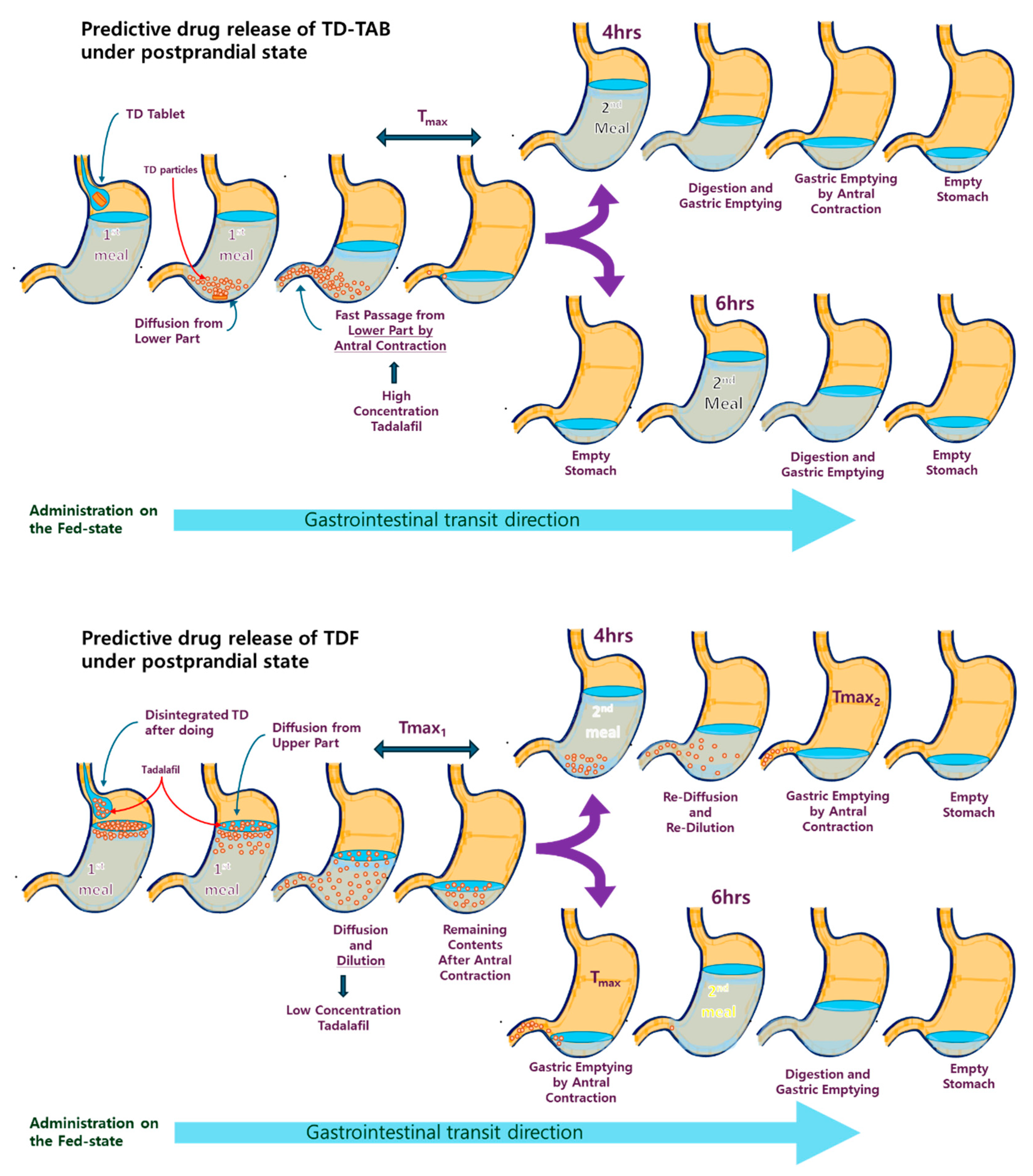
3.5. Mechanistic Understanding of the Importance of Second Meal Intake Time
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | antral contraction |
| ACW | antral contraction wave |
| AUC | area under the curve |
| BCS | biopharmaceutics classification |
| BE | bioequivalence |
| CI | confidence interval |
| CFD | computational fluid dynamics |
| GE | gastric emptying |
| GET | gastric emptying time |
| HPC-SL | hydroxypropyl cellulose SL |
| HPMC | hydroxypropylmethyl cellulose |
| IVIVC | in-vitro in-vivo correlation |
| LOD | loss on drying |
| ODF | orodispersible film |
| PET | polyethylene terephthalate |
| PSD | particle size distribution |
| RSD | relative standard deviation |
| SEM | scanning electron microscopy |
| TAC | terminal antrum contraction |
| TC | tonic contraction |
| TD | tadalafil |
| TDF | tadalafil-loaded ODF |
| TD-TAB | tadalafil-loaded tablet |
| T/R | test sample/reference sample ratio |
References
- Patel, A.A.; Patel, R.J.; Mishra, P. Nanosuspension for Oral Delivery of Tadalafil: Pharmacodynamic and Pharmacokinetic Studies. J. Drug Deliv. Sci. Technol. 2021, 61, 102203. [Google Scholar] [CrossRef]
- Anderson, N.R.; Hartauer, K.J.; Kral, M.A.; Stephenson, G.A. Beta-Carboline Drug Products. U.S. Patent 6,821,975 B1, 23 November 2004. [Google Scholar]
- Forgue, S.T.; Patterson, B.E.; Bedding, A.W.; Payne, C.D.; Phillips, D.L.; Wrishko, R.E.; Mitchell, M.I. Tadalafil Pharmacokinetics in Healthy Subjects. Br. J. Clin. Pharmacol. 2006, 61, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Patterson, B.; Bedding, A.; Jewell, H.; Payne, C.; Mitchell, M. Dose-Normalised Pharmacokinetics of Tadalafil Administered as a Single Dose to Healthy Volunteers. Eur. Urol. Suppl. Off. J. Eur. Assoc. Urol. 2002, 1, 152. [Google Scholar] [CrossRef]
- von Keitz, A.; Rajfer, J.; Segal, S.; Murphy, A.; Denne, J.; Costigan, T.; Lockhart, D.; Beasley, C.M.; Emmick, J.T. A Multicenter, Randomized, Double-Blind, Crossover Study to Evaluate Patient Preference between Tadalafil and Sildenafil. Eur. Urol. 2004, 45, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.; Zhang, R.L.; Cui, Y.; LaPointe, M.C.; Silver, B.; Chopp, M. Tadalafil, a Long-Acting Type 5 Phosphodiesterase Isoenzyme Inhibitor, Improves Neurological Functional Recovery in a Rat Model of Embolic Stroke. Brain Res. 2006, 1118, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Wrishko, R.; Sorsaburu, S.; Wong, D.; Strawbridge, A.; McGill, J. Safety, Efficacy, and Pharmacokinetic Overview of Low-Dose Daily Administration of Tadalafil. J. Sex. Med. 2009, 6, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.S.; Kumar, T.P.; Gowda, D.V. Orodispersible Thin Film: A New Patient-Centered Innovation. J. Drug Deliv. Sci. Technol. 2020, 59, 101843. [Google Scholar] [CrossRef]
- Sharma, R.; Kamboj, S.; Singh, G.; Rana, V. Development of Aprepitant Loaded Orally Disintegrating Films for Enhanced Pharmacokinetic Performance. Eur. J. Pharm. Sci. 2016, 84, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Morath, B.; Sauer, S.; Zaradzki, M.; Wagner, A.H. Orodispersible Films—Recent Developments and New Applications in Drug Delivery and Therapy. Biochem. Pharmacol. 2022, 200, 115036. [Google Scholar] [CrossRef]
- Musazzi, U.M.; Khalid, G.M.; Selmin, F.; Minghetti, P.; Cilurzo, F. Trends in the Production Methods of Orodispersible Films. Int. J. Pharm. 2020, 576, 118963. [Google Scholar] [CrossRef]
- Pacheco, M.S.; Barbieri, D.; da Silva, C.F.; de Moraes, M.A. A Review on Orally Disintegrating Films (ODFs) made from Natural Polymers such as Pullulan, Maltodextrin, Starch, and Others. Int. J. Biol. Macromol. 2021, 178, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.; Finke, J.H.; Kwade, A. Instant ODFs—Development of an Intermediate, Nanoparticle-Based Product Platform for Individualized Medication. Eur. J. Pharm. Biopharm. 2018, 126, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Turković, E.; Vasiljević, I.; Drašković, M.; Parojčić, J. Orodispersible Films—Pharmaceutical Development for Improved Performance: A Review. J. Drug Deliv. Sci. Technol. 2022, 75, 103708. [Google Scholar] [CrossRef]
- Ferlak, J.; Guzenda, W.; Osmałek, T. Orodispersible Films—Current State of the Art, Limitations, Advances and Future Perspectives. Pharmaceutics 2023, 15, 361. [Google Scholar] [CrossRef] [PubMed]
- Islam, N.; Khan, N.U.; Razzaq, A.; Menaa, F.; Khan, Z.U.; Hussain, A.; Rehman, S.U.; Iqbal, H.; Ni, J. Loratadine Oral Bioavailability Enhancement via Solid Dispersion Loaded Oro-Dispersible Films: Formulation, Characterization and Pharmacokinetics. Colloids Surf. B Biointerfaces 2023, 230, 113526. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.L.; Fang, Y.; Han, H.; Li, Q.; Zhang, S.; Li, H.Y.; Chow, S.F.; Lam, T.N.; Lee, W.Y.T. Orally-Dissolving Film for Sublingual and Buccal Delivery of Ropinirole. Colloids Surf. B Biointerfaces 2018, 163, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Ravasi, E.; Melocchi, A.; Arrigoni, A.; Chiappa, A.; Gennari, C.G.M.; Uboldi, M.; Bertarelli, C.; Zema, L.; Briatico Vangosa, F. Electrospinning of Pullulan-Based Orodispersible Films Containing Sildenafil. Int. J. Pharm. 2023, 643, 123258. [Google Scholar] [CrossRef] [PubMed]
- Ṣen Karaman, D.; Patrignani, G.; Rosqvist, E.; Smått, J.; Orłowska, A.; Mustafa, R.; Preis, M.; Rosenholm, J.M. Mesoporous Silica Nanoparticles Facilitating the Dissolution of Poorly Soluble Drugs in Orodispersible Films. Eur. J. Pharm. Sci. 2018, 122, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Shen, C.; Yuan, X.; Bai, J.; Lv, Q.; Xu, H.; Dai, L.; Yu, C.; Han, J.; Yuan, H. Development and Characterization of an Orodispersible Film Containing Drug Nanoparticles. Eur. J. Pharm. Biopharm. 2013, 85, 1348–1356. [Google Scholar] [CrossRef]
- Zhao, Z.; Peng, D.; Liu, X.; Li, X.; Lan, L.; Wu, X.; Liu, X.; Cui, J.; Lee, B.; Shi, L.; et al. Formulation and Characterization of Tadalafil-Loaded Orodispersible Films with Enhanced Dissolution Rate and Membrane Permeability. J. Drug Deliv. Sci. Technol. 2023, 84, 104535. [Google Scholar] [CrossRef]
- Nguyen, V.K.; Dang, T.K.; Vu, L.T.D.; Ha, N.T.; Truong, H.D.; Tran, T.H. Orodispersible Film Incorporating Nanoparticulate Loratadine for an Enhanced Oral Bioavailability. J. Pharm. Investig. 2023, 53, 417. [Google Scholar] [CrossRef]
- Han, X.; Yan, J.; Ren, L.; Xue, M.; Yuan, Z.; Wang, T.; Yan, Z.; Yin, L.; Yang, L.; Qin, C. Preparation and Evaluation of Orally Disintegrating Film Containing Donepezil for Alzheimer Disease. J. Drug Deliv. Sci. Technol. 2019, 54, 101321. [Google Scholar] [CrossRef]
- Shaw, A.; Lawrence, T.E.; Yan, T.; Liu, M.; Summers, N.; Daggumati, V.; Austria, S.T.; Rondon, J.C.; Hackley, S.; Vignesh, S.O.; et al. Bioequivalence Studies of Sildenafil Citrate Orodispersible Film Administered with and without Water Vs ViagraⓇ Film-Coated Tablets in Healthy Male Volunteers. Curr. Ther. Res. 2023, 99, 100708. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, Y.; Lu, T.; Lee, C.; Chang, Y.; Chan, Y.; Mathew, P.; Lin, X.; Hsieh, W.; Huang, T.; et al. Design and Evaluation of Oral Formulation for Apixaban. Heliyon 2023, 9, e18422. [Google Scholar] [CrossRef]
- Fredholt, F.; Di Meo, C.; Sloth, S.; Müllertz, A.; Berthelsen, R. Direct Visualizing of Paracetamol Immediate Release Tablet Disintegration In Vivo and In Vitro. Eur. J. Pharm. Biopharm. 2022, 180, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Parojčić, J.; Vasiljević, D.; Ibrić, S.; Djurić, Z. Tablet Disintegration and Drug Dissolution in Viscous Media: Paracetamol IR Tablets. Int. J. Pharm. 2008, 355, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Goodman, K.; Hodges, L.A.; Band, J.; Stevens, H.N.E.; Weitschies, W.; Wilson, C.G. Assessing Gastrointestinal Motility and Disintegration Profiles of Magnetic Tablets by a Novel Magnetic Imaging Device and Gamma Scintigraphy. Eur. J. Pharm. Biopharm. 2010, 74, 84–92. [Google Scholar] [CrossRef]
- Mulet-Cabero, A.; Rigby, N.M.; Brodkorb, A.; Mackie, A.R. Dairy Food Structures Influence the Rates of Nutrient Digestion through Different in Vitro Gastric Behaviour. Food Hydrocoll. 2017, 67, 63–73. [Google Scholar] [CrossRef]
- Cilurzo, F.; Musazzi, U.M.; Franzé, S.; Selmin, F.; Minghetti, P. Orodispersible Dosage Forms: Biopharmaceutical Improvements and Regulatory Requirements. Drug Discov. Today 2018, 23, 251–259. [Google Scholar] [CrossRef]
- Motawi, A.T.; Gamalel Din, S.F.; Meatmed, E.M.; Fahmy, I. Evaluation of Efficacy and Safety Profile of Tadalafil 5 mg Daily Dose in the Tablet Form Versus Oral Dispersible Film in Men with Mild-to-Moderate Erectile Dysfunction: A Comparative Placebo-Controlled Study. Int. Urol. Nephrol. 2024. [Google Scholar] [CrossRef]
- Park, S.; Heo, S.; Kim, G.; Chang, S.; Song, K.; Kim, M.; Jin, E.; Kim, J.; Lee, S.; Hong, J.H. Comparison of Tadalafil Pharmacokinetics after Administration of a New Orodispersible Film Versus a Film-Coated Tablet. Drug Des. Dev. Ther. 2018, 12, 935. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jin, Y. A CFD Model for Investigating the Dynamics of Liquid Gastric Contents in Human-Stomach Induced by Gastric Motility. J. Food Eng. 2021, 296, 110461. [Google Scholar] [CrossRef]
- Li, C.; Jin, Y. Digestion of Meat Proteins in a Human-Stomach: A CFD Simulation Study. Innov. Food Sci. Emerg. Technol. 2023, 83, 103252. [Google Scholar] [CrossRef]
- Zhong, C.; Langrish, T. A Comparison of Different Physical Stomach Models and an Analysis of Shear Stresses and Strains in these System. Food Res. Int. 2020, 135, 109296. [Google Scholar] [CrossRef] [PubMed]
- Donis-Rabanales, F.; López-Ruiz, T.; Ruiz-Huerta, L.; Ascanio, G.; Brito-de la Fuente, E.; Caballero-Ruiz, A. Development of an in Vitro Distal Gastric Simulator to Mimic the Mechanical Action of the Human Stomach. Food Res. Int. 2022, 161, 111902. [Google Scholar] [CrossRef] [PubMed]
- Nadia, J.; Olenskyj, A.G.; Stroebinger, N.; Hodgkinson, S.M.; Estevez, T.G.; Subramanian, P.; Singh, H.; Singh, R.P.; Bornhorst, G.M. Carbohydrate Digestion: The Importance of the Proximal and Distal Stomach during Digestion in Growing Pigs. Anim.-Sci. Proc. 2022, 13, 127–132. [Google Scholar] [CrossRef]
- Guerra, A.; Etienne-Mesmin, L.; Livrelli, V.; Denis, S.; Blanquet-Diot, S.; Alric, M. Relevance and Challenges in Modeling Human Gastric and Small Intestinal Digestion. Trends Biotechnol. 2012, 30, 591–600. [Google Scholar] [CrossRef]
- Martens, B.M.J.; Bruininx, E.M.A.M.; Gerrits, W.J.J.; Schols, H.A. The Importance of Amylase Action in the Porcine Stomach to Starch Digestion Kinetics. Anim. Feed Sci. Technol. 2020, 267, 114546. [Google Scholar] [CrossRef]
- Mennah-Govela, Y.A.; Swackhamer, C.; Bornhorst, G.M. Gastric Secretion Rate and Protein Concentration Impact Intragastric pH and Protein Hydrolysis during Dynamic in Vitro Gastric Digestion. Food Hydrocoll. Health 2021, 1, 100027. [Google Scholar] [CrossRef]
- Papenkort, S.; Borsdorf, M.; Böl, M.; Siebert, T. A Geometry Model of the Porcine Stomach Featuring Mucosa and Muscle Layer Thicknesses. J. Mech. Behav. Biomed. Mater. 2023, 142, 105801. [Google Scholar] [CrossRef]
- Camilleri, M. Integrated Upper Gastrointestinal Response to Food Intake. Gastroenterology 2006, 131, 640–658. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yu, W.; Wu, P.; Chen, X.D. Current in Vitro Digestion Systems for Understanding Food Digestion in Human Upper Gastrointestinal Tract. Trends Food Sci. Technol. 2020, 96, 114–126. [Google Scholar] [CrossRef]
- Liu, W.; Fu, D.; Zhang, X.; Chai, J.; Tian, S.; Han, J. Development and Validation of a New Artificial Gastric Digestive System. Food Res. Int. 2019, 122, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Guidance for Industry on Food-Effect Bioavailability and Fed Bioequivalence Studies: Availability. J. Pharm. Investig. 2004, 34, 223–228. [Google Scholar] [CrossRef]
- Wlodarski, K.; Sawicki, W.; Paluch, K.J.; Tajber, L.; Grembecka, M.; Hawelek, L.; Wojnarowska, Z.; Grzybowska, K.; Talik, E.; Paluch, M. The Influence of Amorphization Methods on the Apparent Solubility and Dissolution Rate of Tadalafil. Eur. J. Pharm. Sci. 2014, 62, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, K.; Sawicki, W.; Haber, K.; Knapik, J.; Wojnarowska, Z.; Paluch, M.; Lepek, P.; Hawelek, L.; Tajber, L. Physicochemical Properties of Tadalafil Solid Dispersions—Impact of Polymer on the Apparent Solubility and Dissolution Rate of Tadalafil. Eur. J. Pharm. Biopharm. 2015, 94, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs. 2021. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioequivalence-studies-pharmacokinetic-endpoints-drugs-submitted-under-abbreviated-new-drug (accessed on 15 November 2023).
- Roh, H.; Son, H.; Lee, D.; Yeon, K.J.; Kim, H.S.; Kim, H.; Park, K. Pharmacokinetic Comparison of an Orally Disintegrating Film Formulation with a Film-Coated Tablet Formulation of Sildenafil in Healthy Korean Subjects: A Randomized, Open-Label, Single-Dose, 2-Period Crossover Study. Clin. Ther. 2013, 35, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Coward, R.M.; Carson, C.C. Tadalafil in the treatment of erectile dysfunction. Ther. Clin. Risk Manag. 2008, 4, 1315–1329. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.; Pham, C.V.; Myung, C.; Cho, C. Tadalafil-Loaded Nanostructured Lipid Carriers using Permeation Enhancers. Int. J. Pharm. 2015, 495, 701–709. [Google Scholar] [CrossRef]
- Deconinck, E.; Andriessens, S.; Bothy, J.L.; Courselle, P.; De Beer, J.O. Comparative Dissolution Study on Counterfeit Medicines of PDE-5 Inhibitors. J. Pharm. Anal. 2014, 4, 250. [Google Scholar] [CrossRef]
- Scheff, J.D.; Almon, R.R.; Dubois, D.C.; Jusko, W.J.; Androulakis, I.P. Assessment of Pharmacologic Area under the Curve When Baselines Are Variable. Pharm. Res. 2012, 28, 1081. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.S.R.; de Oliveira Cruvinel, K.; Oliveira-Nascimento, L. A Mini-Review on Drug Delivery through Wafer Technology: Formulation and Manufacturing of Buccal and Oral Lyophilizates. J. Adv. Res. 2019, 20, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, F.A.; Aamir, M.N.; Haseeb, M.T.; Abbas Bukhari, S.N.; Farid ul Haq, M.; Akhtar, N. Design, Physico-Chemical Assessment and Pharmacokinetics of a Non-Toxic Orodispersible Film for Potential Application in Musculo-Skeletal Disorder. J. Drug Deliv. Sci. Technol. 2021, 65, 102726. [Google Scholar] [CrossRef]
- Rao, Q.; Qiu, Z.; Huang, D.; Lu, T.; Zhang, Z.J.; Luo, D.; Pan, P.; Zhang, L.; Liu, Y.; Guan, S.; et al. Enhancement of the Apparent Solubility and Bioavailability of Tadalafil Nanoparticles Via Antisolvent Precipitation. Eur. J. Pharm. Sci. 2019, 128, 222–231. [Google Scholar] [CrossRef]
- Kozu, H.; Kobayashi, I.; Nakajima, M.; Uemura, K.; Sato, S.; Ichikawa, S. Simulation of Motion of Gastric Fluid Induced by Peristalsis using CFD. J. Biosci. Bioeng. 2009, 108, S136. [Google Scholar] [CrossRef]
- Grimm, M.; Koziolek, M.; Kühn, J.; Weitschies, W. Interindividual and Intraindividual Variability of Fasted State Gastric Fluid Volume and Gastric Emptying of Water. Eur. J. Pharm. Biopharm. 2018, 127, 309–317. [Google Scholar] [CrossRef]
- Hur, S.J.; Lim, B.O.; Decker, E.A.; McClements, D.J. In Vitro Human Digestion Models for Food Applications. Food Chem. 2011, 125, 1–12. [Google Scholar] [CrossRef]
- Humphrey, S.P.; Williamson, R.T. A Review of Saliva: Normal Composition, Flow, and Function. J. Prosthet. Dent. 2001, 85, 162–169. [Google Scholar] [CrossRef]
- Neyraud, E.; Palicki, O.; Schwartz, C.; Nicklaus, S.; Feron, G. Variability of Human Saliva Composition: Possible Relationships with Fat Perception and Liking. Arch. Oral Biol. 2012, 57, 556–566. [Google Scholar] [CrossRef]
- Liu, J.; Duan, Y. Saliva: A Potential Media for Disease Diagnostics and Monitoring. Oral Oncol. 2012, 48, 569–577. [Google Scholar] [CrossRef]
- Fadda, H.M.; Hellström, P.M.; Webb, D. Intra- and Inter-Subject Variability in Gastric pH Following a Low-Fat, Low-Calorie Meal. Int. J. Pharm. 2022, 625, 122069. [Google Scholar] [CrossRef] [PubMed]
- Ulleberg, E.K.; Comi, I.; Holm, H.; Herud, E.B.; Jacobsen, M.; Vegarud, G.E. Human Gastrointestinal Juices Intended for use in in Vitro Digestion Models. Food Dig. 2011, 2, 52. [Google Scholar] [CrossRef] [PubMed]
- Zullo, A.; Germanà, B.; Galliani, E.; Iori, A.; de Pretis, G.; Manfredi, G.; Buscarini, E.; Ciuffi, M.; Ignomirelli, O.; Farinati, F.; et al. Real-Time Determination of Gastric Juice pH with EndoFaster® for Atrophic Gastritis Assessment. Dig. Liver Dis. 2022, 54, 1646–1648. [Google Scholar] [CrossRef] [PubMed]
- McQuilken, S.A. The Mouth, Stomach and Intestines. Anaesth. Intensive Care Med. 2021, 22, 330–335. [Google Scholar] [CrossRef]
- Longo-Silva, G.; Bezerra de Oliveira, P.M.; Pedrosa, A.K.P.; Ribeiro da Silva, J.; Bernardes, R.S.; Egito de Menezes, R.C.; Marinho, P.d.M. Breakfast Skipping and Timing of Lunch and Dinner: Relationship with BMI and Obesity. Obes. Res. Clin. Pract. 2022, 16, 507–513. [Google Scholar] [CrossRef]
- Lopez-Minguez, J.; Gómez-Abellán, P.; Garaulet, M. Timing of Breakfast, Lunch, and Dinner. Effects on Obesity and Metabolic Risk. Nutrients 2019, 11, 2624. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | TDF Formulation | Water Intake Volume (mL) | First Meal Intake Time (Breakfast) | Second Meal Intake Time (h) after Administration |
|---|---|---|---|---|
| Fasting | TDF-1 | 20 | No | 4 |
| Fed I | TDF-1 | 20 | Yes | 4 |
| Fed II | TDF-2 | 20 | Yes | 4 |
| Fed III | TDF-1 | 240 | Yes | 4 |
| Fed IV | TDF-1 | 20 | Yes | 6 |
| Polymer | Dispersion Agent | % RSD | Visual Appearance |
|---|---|---|---|
| HPMC (5 cp) | Polyethylene glycol 400 | 0.44 | Surface crack, Surface roughness |
| Polyoxyl 35 castor oil | 2.79 | Surface roughness, TD aggregation | |
| Polyethylene glycol 40 hydrogenated castor oil | 3.08 | TD aggregation | |
| Triethyl citrate | 2.84 | Non-homogenous layer | |
| Polyethylene oxide | 3.83 | Surface crack | |
| Polyvinylalcohol–Polyethylene glycol copolymer | 3.82 | Non-homogenous layer | |
| HPC-SL | Polyethylene glycol 400 | 0.43 | Very good |
| Polyoxyl 35 castor oil | 2.46 | TD aggregation, Surface crack | |
| Polyethylene glycol 40 hydrogenated castor oil | 2.52 | Surface crack | |
| Triethyl citrate | 3.11 | Non-homogenous layer | |
| Polyethylene oxide | 2.65 | Surface roughness | |
| Polyvinylalcohol–Polyethylene glycol copolymer | 2.72 | Non-homogenous layer | |
| Pullulan | Polyethylene glycol 400 | 0.31 | Surface roughness |
| Polyoxyl 35 castor oil | 2.68 | TD aggregation | |
| Polyethylene glycol 40 hydrogenated castor oil | 2.54 | Surface roughness | |
| Triethyl citrate | 3.02 | Non-homogenous layer | |
| Polyethylene oxide | 3.04 | TD aggregation, Surface roughness | |
| Polyvinylalcohol–Polyethylene glycol copolymer | 2.99 | Non-homogenous layer |
| Condition | Items | Variable | ||||
|---|---|---|---|---|---|---|
| Cmax (ng/mL) | T/R, 90% CI | AUCt (h*ng/mL) | T/R, 90% CI | Tmax (h) | ||
| Fasting (n = 37) | TD-TAB | 268.2 ± 77.7 | 0.936–1.105 | 6399.8 ± 2447.9 | 1.012–1.153 | 2.6 ± 1.7 |
| TDF-1 | 276.3 ± 83.9 | 6871.8 ± 2234.5 | 3.2 ± 2.2 | |||
| Fed I (n = 10) | TD-TAB | 416.4 ± 81.0 | 0.610–0.798 | 6562.4 ± 1269.4 | 0.884–1.022 | 2.3 ± 1.0 |
| TDF-1 | 290.4 ± 81.0 | 6237.3 ± 1548.0 | 7.9 ± 3.2 | |||
| Fed II (n = 12) | TD-TAB | 326.8 ± 77.4 | 0.735–0.972 | 6549.6 ± 3034.0 | 0.894–1.137 | 2.6 ± 2.6 |
| TDF-2 | 276.2 ± 100.3 | 6602.3 ± 3028.6 | 5.7 ± 6.1 | |||
| Fed III (n = 10) | TD-TAB | 408.6 ± 153.6 | 0.635–0.847 | 7097.6 ± 2631.7 | 0.929–1.074 | 3.2 ± 1.5 |
| TDF-1 | 299.6 ± 102.3 | 7089.9 ± 2525.1 | 6.8 ± 3.2 | |||
| Fed IV (n = 8) | TD-TAB | 400.8 ± 61.2 | 0.851–0.998 | 6354.9 ± 1542.9 | 0.958–1.045 | 2.4 ± 2.1 |
| TDF-1 | 367.3 ± 59.2 | 6382.5 ± 1501.7 | 4.8 ± 1.7 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-J.; Gil, M.-C.; Lee, B.-S.; Jung, M.; Lee, B.-J. Effects of Postprandial Factors and Second Meal Intake Time on Bioequivalence Investigation of Tadalafil-Loaded Orodispersible Films in Human Volunteers. Pharmaceutics 2024, 16, 915. https://doi.org/10.3390/pharmaceutics16070915
Park S-J, Gil M-C, Lee B-S, Jung M, Lee B-J. Effects of Postprandial Factors and Second Meal Intake Time on Bioequivalence Investigation of Tadalafil-Loaded Orodispersible Films in Human Volunteers. Pharmaceutics. 2024; 16(7):915. https://doi.org/10.3390/pharmaceutics16070915
Chicago/Turabian StylePark, Su-Jun, Myung-Chul Gil, Bong-Sang Lee, Minji Jung, and Beom-Jin Lee. 2024. "Effects of Postprandial Factors and Second Meal Intake Time on Bioequivalence Investigation of Tadalafil-Loaded Orodispersible Films in Human Volunteers" Pharmaceutics 16, no. 7: 915. https://doi.org/10.3390/pharmaceutics16070915