
pH-Triggered Hydrogel Nanoparticles for Efficient Anticancer Drug Delivery and Bioimaging Applications

 ,
,  ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
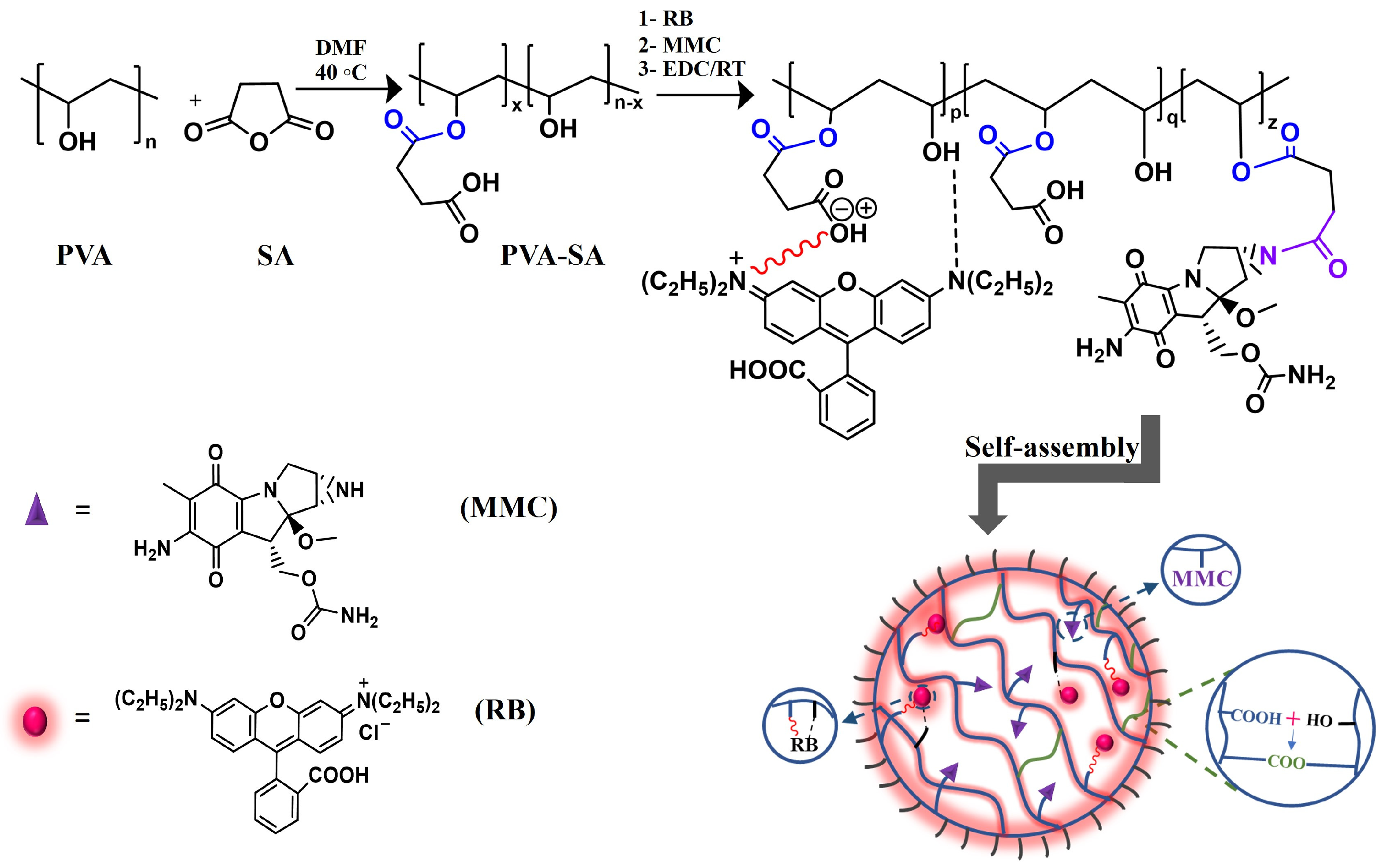
2.2. Synthesis of RB-PVA-SA and RB-PVA-SA-MMC Hydrogel NPs by a Simple One-Pot Reaction
2.3. Macromolecular Self-Assembly and Hydrogel NP Formation
2.4. Characterization of the Produced Hydrogel NPs
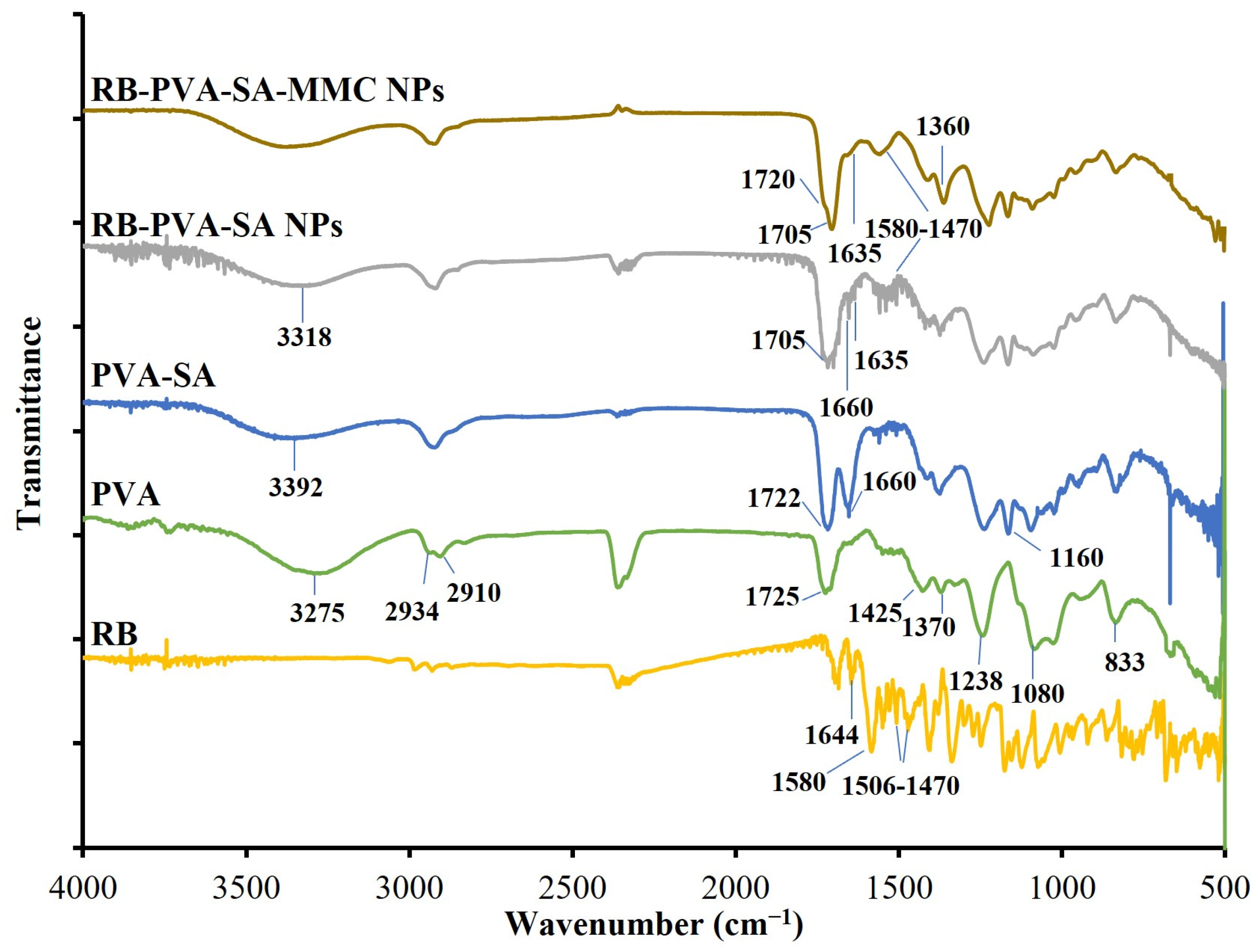
2.4.1. Structural Characterization and Compositional Analysis of the Synthesized Samples
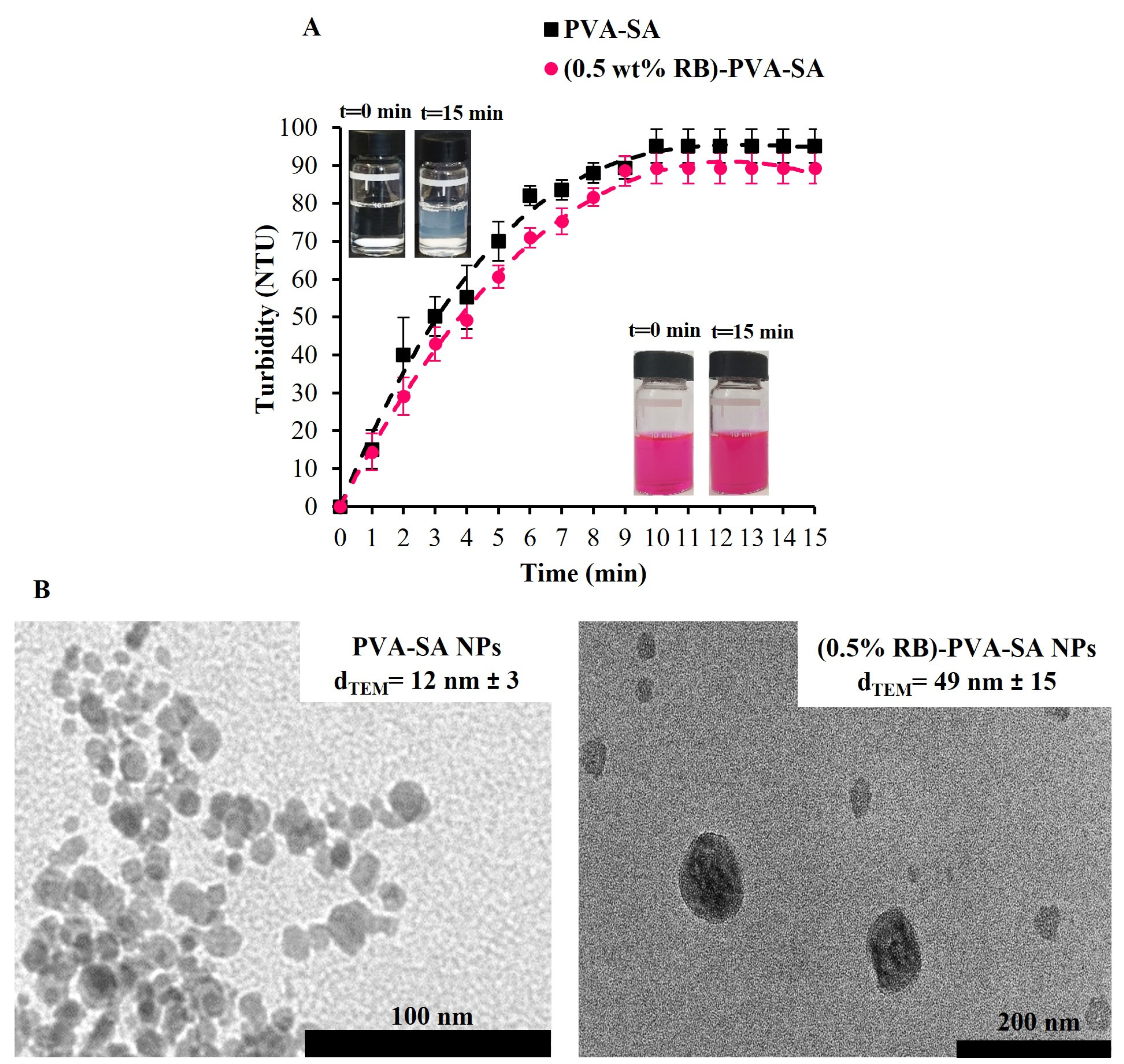
2.4.2. Particle Size, ζ-Potential, and Morphology Characterization
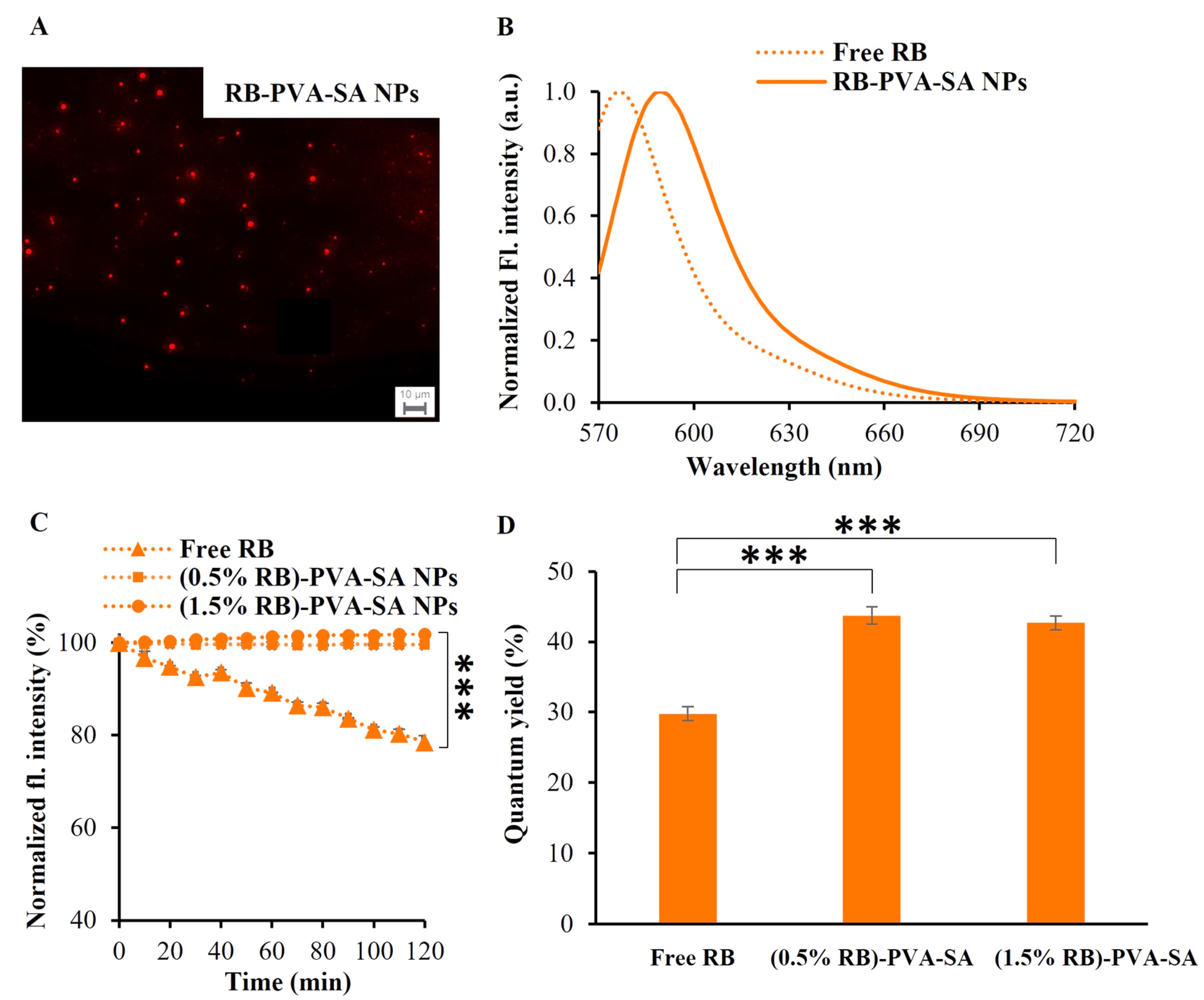
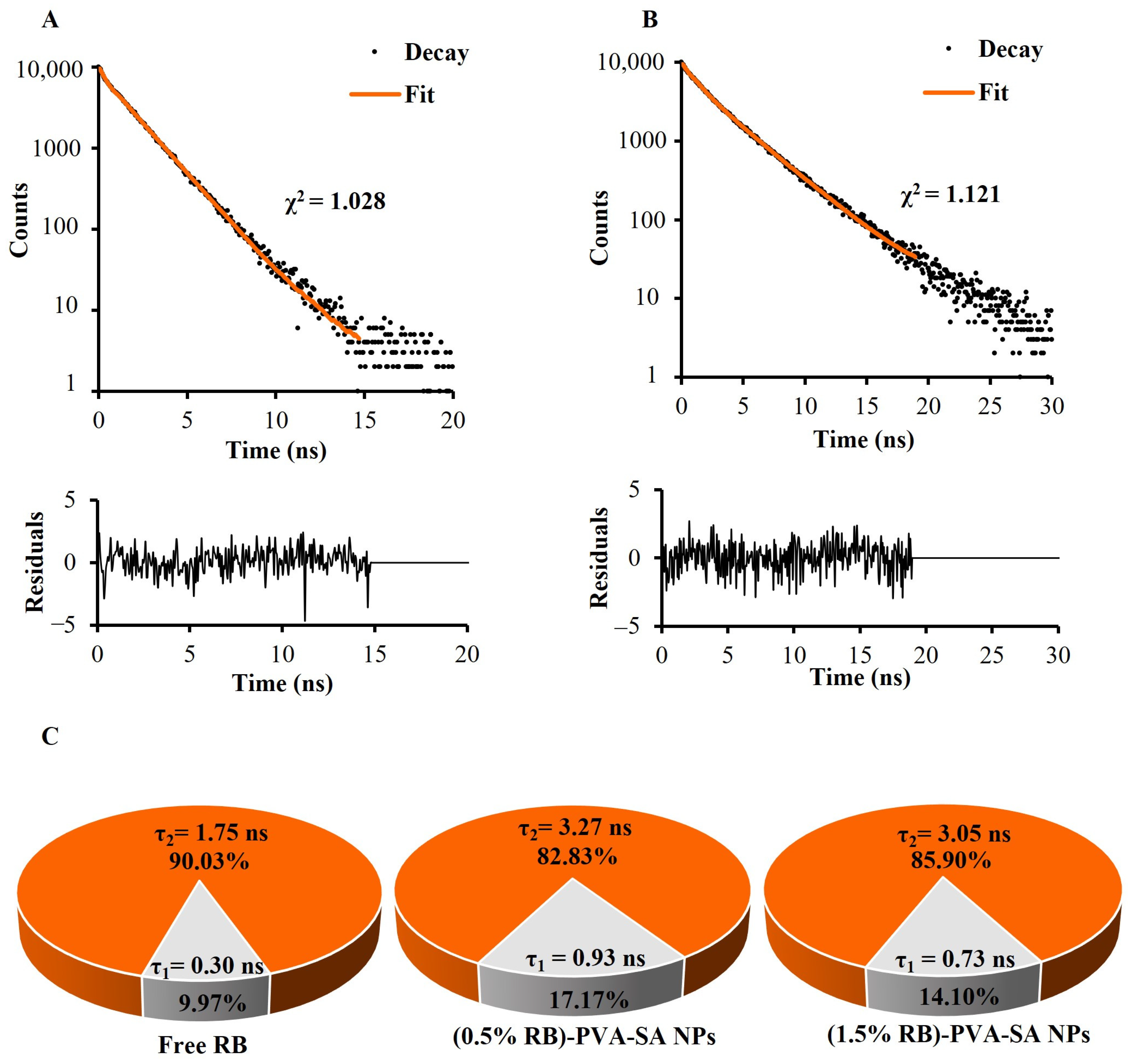
2.4.3. Fluorescence Emission and Decay Properties of RB-PVA-SA Hydrogel NPs
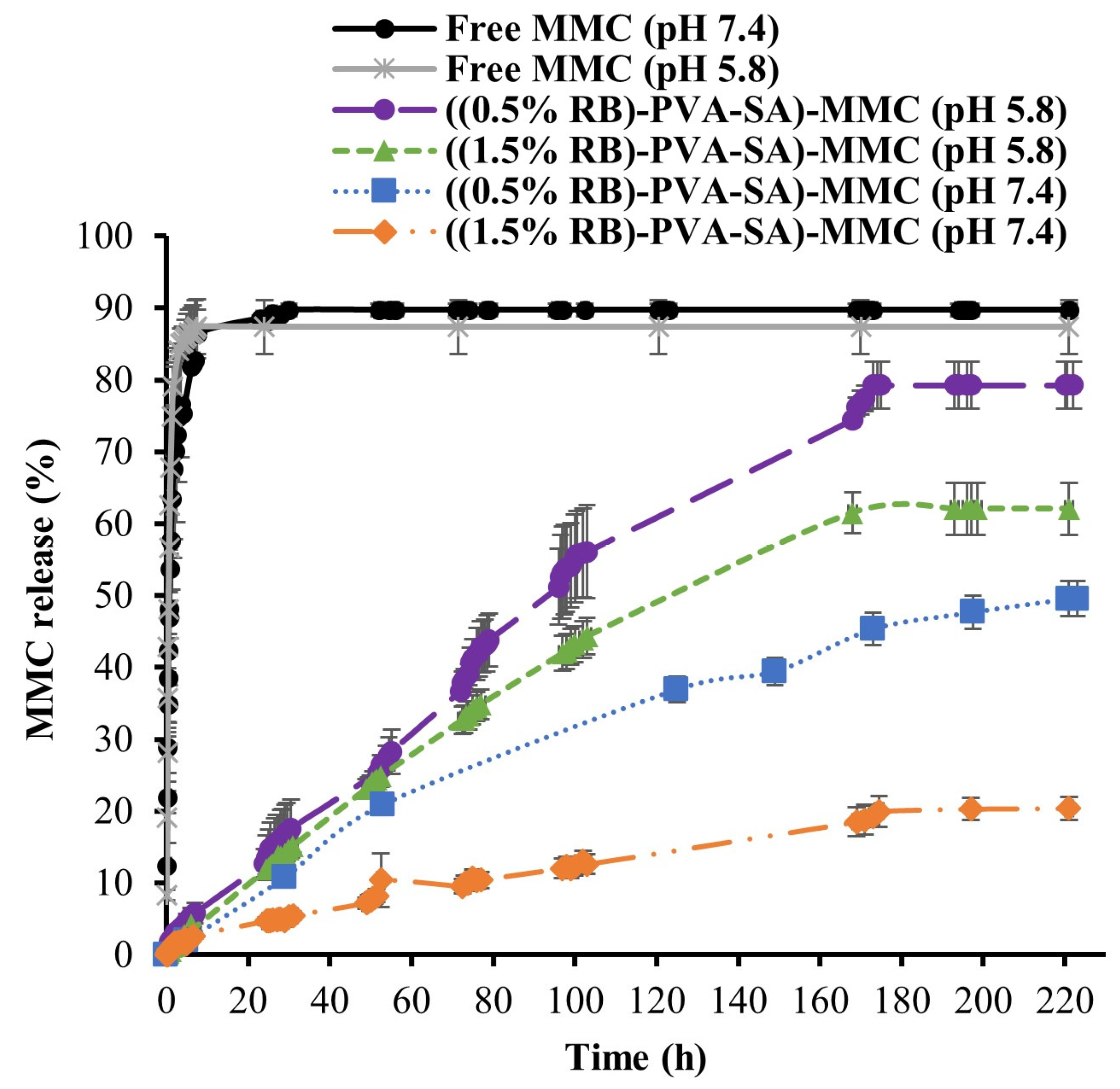
2.5. In Vitro Release of MMC from Resulting Hydrogel NPs
2.6. Cell Lines and Cell Culture
2.7. In Vitro Cytotoxicity Assays
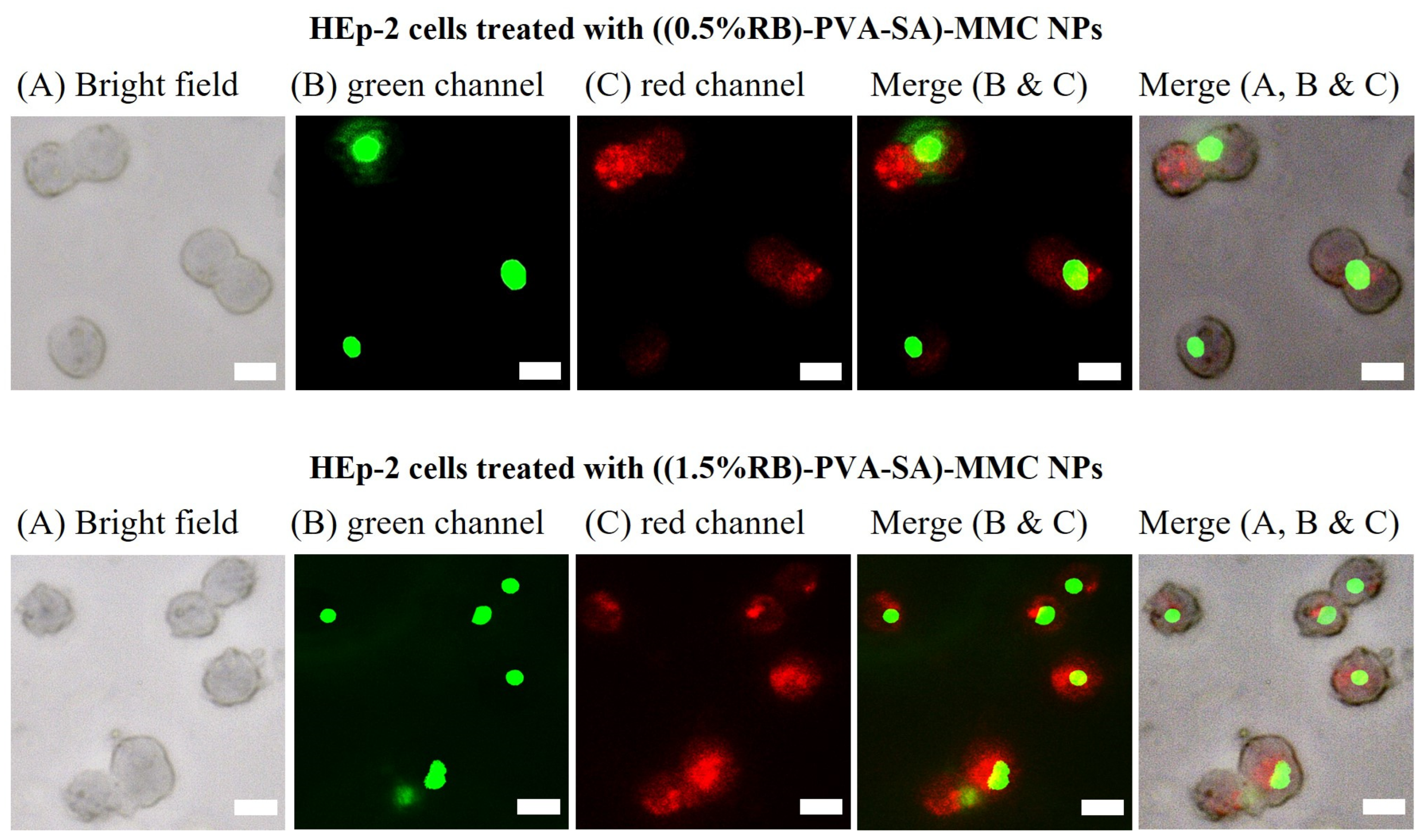
2.8. In Vitro Cellular Uptake of RB-PVA-SA-MMC Hydrogel NPs
2.9. Statistical Analysis of Results
3. Results
3.1. Structural Characterization and Compositional Analysis of the Synthesized Samples
3.2. Self-Assembly of Polymeric Chains and Formation of RB-PVA-SA Hydrogel NPs
3.3. Maximization of RB Content in the Hydrogel Composite Particles
3.4. Fluorescence Emission and Decay Properties of RB-PVA-SA Hydrogel NPs
3.5. One-Pot Synthesis of Fluorescent MMC-Loaded Hydrogel NPs
3.6. In Vitro MMC Release Study
3.7. In Vitro Cytotoxicity
3.7.1. Cytotoxicity of Labeled Polymeric NPs (RB-PVA-SA NPs)
3.7.2. Cytotoxicity of MMC-Loaded Hydrogel NPs
3.8. In Vitro Cellular Uptake of RB-PVA-SA-MMC Hydrogel NPs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feng, X.; Lv, F.; Liu, L.; Tang, H.; Xing, C.; Yang, Q.; Wang, S. Conjugated Polymer Nanoparticles for Drug Delivery and Imaging. ACS Appl. Mater. Interfaces 2010, 2, 2429–2435. [Google Scholar] [CrossRef]
- Salari, N.; Faraji, F.; Torghabeh, F.M.; Faraji, F.; Mansouri, K.; Abam, F.; Shohaimi, S.; Akbari, H.; Mohammadi, M. Polymer-Based Drug Delivery Systems for Anticancer Drugs: A Systematic Review. Cancer Treat. Res. Commun. 2022, 32, 100605. [Google Scholar] [CrossRef] [PubMed]
- Abdelghafour, M.M.; Deák, Á.; Szabó, D.; Dékány, I.; Rovó, L.; Janovák, L. Use of Self-Assembled Colloidal Prodrug Nanoparticles for Controlled Drug Delivery of Anticancer, Antifibrotic and Antibacterial Mitomycin. Int. J. Mol. Sci. 2022, 23, 6807. [Google Scholar] [CrossRef]
- Rivera-Hernández, G.; Antunes-Ricardo, M.; Martínez-Morales, P.; Sánchez, M.L. Polyvinyl Alcohol Based-Drug Delivery Systems for Cancer Treatment. Int. J. Pharm. 2021, 600, 120478. [Google Scholar] [CrossRef]
- Yang, C.; Huang, S.; Wang, X.; Wang, M. Theranostic Unimolecular Micelles of Highly Fluorescent Conjugated Polymer Bottlebrushes for Far Red/near Infrared Bioimaging and Efficient Anticancer Drug Delivery. Polym. Chem. 2016, 7, 7455–7468. [Google Scholar] [CrossRef]
- Xue, Y.; Lee, J.; Kim, H.-J.; Cho, H.-J.; Zhou, X.; Liu, Y.; Tebon, P.; Hoffman, T.; Qu, M.; Ling, H.; et al. Rhodamine Conjugated Gelatin Methacryloyl Nanoparticles for Stable Cell Imaging. ACS Appl. Bio Mater. 2020, 3, 6908–6918. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, D.; Denis, S.; Mougin, J.; Jacobs, M.; Guillaneuf, Y.; Mura, S.; Couvreur, P.; Nicolas, J. A Facile Route to Heterotelechelic Polymer Prodrug Nanoparticles for Imaging, Drug Delivery and Combination Therapy. J. Control. Release 2018, 286, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.W.K.; Abdelghafour, M.M.; Hornok, V.; Kiss, T.; Szabó, D.; Rovó, L.; Janovák, L. Mitomycin Loaded Self-Assembled Colloidal Prodrug Nanoparticles for Magnetic Drug Targeting. J. Drug Deliv. Sci. Technol. 2023, 88, 104948. [Google Scholar] [CrossRef]
- Dalabehera, N.R.; Meher, S.; Bhusana Palai, B.; Sharma, N.K. Instability of Amide Bond with Trifluoroacetic Acid (20%): Synthesis, Conformational Analysis, and Mechanistic Insights into Cleavable Amide Bond Comprising β-Troponylhydrazino Acid. ACS Omega 2020, 5, 26141–26152. [Google Scholar] [CrossRef]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic Microenvironment in Cancer: Molecular Mechanisms and Therapeutic Interventions. Signal Transduct. Target. Ther. 2023, 8, 70. [Google Scholar] [CrossRef]
- Sutoo, S.; Maeda, T.; Suzuki, A.; Kato, Y. Adaptation to Chronic Acidic Extracellular pH Elicits a Sustained Increase in Lung Cancer Cell Invasion and Metastasis. Clin. Exp. Metastasis 2020, 37, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Cong, V.T.; Tilley, R.D.; Sharbeen, G.; Phillips, P.A.; Gaus, K.; Gooding, J.J. How to Exploit Different Endocytosis Pathways to Allow Selective Delivery of Anticancer Drugs to Cancer Cells over Healthy Cells. Chem. Sci. 2021, 12, 15407–15417. [Google Scholar] [CrossRef] [PubMed]
- Landarani-Isfahani, A.; Moghadam, M.; Mohammadi, S.; Royvaran, M.; Moshtael-Arani, N.; Rezaei, S.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I. Elegant pH-Responsive Nanovehicle for Drug Delivery Based on Triazine Dendrimer Modified Magnetic Nanoparticles. Langmuir 2017, 33, 8503–8515. [Google Scholar] [CrossRef] [PubMed]
- Oyekanmi, A.A.; Ahmad, A.; Hossain, K.; Rafatullah, M. Adsorption of Rhodamine B Dye from Aqueous Solution onto Acid Treated Banana Peel: Response Surface Methodology, Kinetics and Isotherm Studies. PLoS ONE 2019, 14, e0216878. [Google Scholar] [CrossRef] [PubMed]
- Tsakos, M.; Schaffert, E.S.; Clement, L.L.; Villadsen, N.L.; Poulsen, T.B. Ester Coupling Reactions—An Enduring Challenge in the Chemical Synthesis of Bioactive Natural Products. Nat. Prod. Rep. 2015, 32, 605–632. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, L.; Atif, R.; Eldeen, T.S.; Yahya, I.; Omara, A.; Eltayeb, M. Study the Using of Nanoparticles as Drug Delivery System Based on Mathematical Models for Controlled Release. IJLTEMAS 2019, 8, 52–56. [Google Scholar]
- Stipniece, L.; Salma-Ancane, K.; Rjabovs, V.; Juhnevica, I.; Turks, M.; Narkevica, I.; Berzina-Cimdina, L. Development of Functionalized Hydroxyapatite/Poly(Vinyl Alcohol) Composites. J. Cryst. Growth 2016, 444, 14–20. [Google Scholar] [CrossRef]
- Zhang, B.; Huang, Q.; Luo, F.; Fu, X.; Jiang, H.; Jane, J. Effects of Octenylsuccinylation on the Structure and Properties of High-Amylose Maize Starch. Carbohydr. Polym. 2011, 84, 1276–1281. [Google Scholar] [CrossRef]
- Guan, H.-M.; Chung, T.-S.; Huang, Z.; Chng, M.L.; Kulprathipanja, S. Poly(Vinyl Alcohol) Multilayer Mixed Matrix Membranes for the Dehydration of Ethanol–Water Mixture. J. Membr. Sci. 2006, 268, 113–122. [Google Scholar] [CrossRef]
- Hansen, P.E.; Spanget-Larsen, J. NMR and IR Investigations of Strong Intramolecular Hydrogen Bonds. Molecules 2017, 22, 552. [Google Scholar] [CrossRef]
- Padmakumari, R.; Ravindrachary, V.; Mahantesha, B.K.; Sagar, R.N.; Sahanakumari, R.; Bhajantri, R.F. Modification of Fluorescence and Optical Properties of Rhodamine B Dye Doped PVA/Chitosan Polymer Blend Films. In Proceedings of the 2nd International Conference on Condensed Matter and Applied Physics (ICC 2017), Bikaner, India, 24–25 November 2017; 2018; p. 030248. [Google Scholar]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; Wiley: New York, NY, USA, 2000; ISBN 978-0-471-97670-7. [Google Scholar]
- Haigh, J.M.; Van Dam, M.A.; Thornton, D.A. Studies on Complexes and Ligands: Part II Complexes of Mercury(II) and Zinc(II) with Primary Aromatic Amines: Infra-Red Spectra and Bonding. S. Afr. J. Chem. 1967, 20, 113–122. [Google Scholar]
- Sadat, A.; Joye, I.J. Peak Fitting Applied to Fourier Transform Infrared and Raman Spectroscopic Analysis of Proteins. Appl. Sci. 2020, 10, 5918. [Google Scholar] [CrossRef]
- Ji, Y.; Yang, X.; Ji, Z.; Zhu, L.; Ma, N.; Chen, D.; Jia, X.; Tang, J.; Cao, Y. DFT-Calculated IR Spectrum Amide I, II, and III Band Contributions of N -Methylacetamide Fine Components. ACS Omega 2020, 5, 8572–8578. [Google Scholar] [CrossRef] [PubMed]
- Anita, S.H.; Zulfiana, D.; Digita, A.; Nuha, N.; Syafriana, V.; Febriani, A.; Yanto, D.H.Y. The First Evidence of Potential Antibacterial Activity of Laccase Enzyme from Indonesian White Rot Fungi against Pathogenic Bacteria. HAYATI J. Biosci. 2023, 31, 133–144. [Google Scholar] [CrossRef]
- Sharma, K.; Nilsuwan, K.; Ma, L.; Benjakul, S. Effect of Liposomal Encapsulation and Ultrasonication on Debittering of Protein Hydrolysate and Plastein from Salmon Frame. Foods 2023, 12, 761. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Murthy, B.N.; Shapter, J.G.; Constantopoulos, K.T.; Voelcker, N.H.; Ellis, A.V. Benzene Carboxylic Acid Derivatized Graphene Oxide Nanosheets on Natural Zeolites as Effective Adsorbents for Cationic Dye Removal. J. Hazard. Mater. 2013, 260, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Sasani, M. Evaluation of 3A zeolite as an adsorbent for the decolorization of rhodamine B dye in contaminated waters. J. Appl. Chem. 2017, 11, 83–90. [Google Scholar]
- Chenthamara, D.; Subramaniam, S.; Ramakrishnan, S.G.; Krishnaswamy, S.; Essa, M.M.; Lin, F.-H.; Qoronfleh, M.W. Therapeutic Efficacy of Nanoparticles and Routes of Administration. Biomater. Res. 2019, 23, 20. [Google Scholar] [CrossRef] [PubMed]
- Steichen, S.D.; Caldorera-Moore, M.; Peppas, N.A. A Review of Current Nanoparticle and Targeting Moieties for the Delivery of Cancer Therapeutics. Eur. J. Pharm. Sci. 2013, 48, 416–427. [Google Scholar] [CrossRef]
- Raza, S.A.; Naqvi, S.Q.; Usman, A.; Jennings, J.R.; Soon, Y.W. Spectroscopic Study of the Interaction between Rhodamine B and Graphene. J. Photochem. Photobiol. A Chem. 2021, 418, 113417. [Google Scholar] [CrossRef]
- Bartasun, P.; Cieśliński, H.; Bujacz, A.; Wierzbicka-Woś, A.; Kur, J. A Study on the Interaction of Rhodamine B with Methylthioadenosine Phosphorylase Protein Sourced from an Antarctic Soil Metagenomic Library. PLoS ONE 2013, 8, e55697. [Google Scholar] [CrossRef]
- Lopez Arbeloa, F.; Lopez Arbeloa, T.; Tapia Estevez, M.J.; Lopez Arbeloa, I. Photophysics of Rhodamines: Molecular Structure and Solvent Effects. J. Phys. Chem. 1991, 95, 2203–2208. [Google Scholar] [CrossRef]
- Tao, S.; Song, Y.; Zhu, S.; Shao, J.; Yang, B. A New Type of Polymer Carbon Dots with High Quantum Yield: From Synthesis to Investigation on Fluorescence Mechanism. Polymer 2017, 116, 472–478. [Google Scholar] [CrossRef]
- Yamashita, H.; Tanaka, A.; Nishimura, M.; Koyano, K.; Tatsumi, T.; Anpo, M. Photochemical Properties of Rhodamine-B Dye Molecules Included within Mesoporous Molecular Sieves. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1998; Volume 117, pp. 551–558. ISBN 978-0-444-82826-2. [Google Scholar]
- Pham, M.T.; Nguyen, T.V.; Vu Thi, T.D.; Nghiem Thi, H.L.; Tong, K.T.; Tran, T.T.; Chu, V.H.; Brochon, J.-C.; Tran, H.N. Synthesis, Photophysical Properties and Application of Dye Doped Water Soluble Silica-Based Nanoparticles to Label Bacteria E. Coli O157:H7. Adv. Nat. Sci. Nanosci. Nanotechnol. 2012, 3, 045013. [Google Scholar] [CrossRef]
- Viveiros, F.; Gomes, J.; Oliveira, A.; Neves, S.; Almeida, J. Topical Application of Mitomycin-C as an Adjuvant Treatment to Bronchoscopic Procedures in Post-Intubation Tracheal Stenosis. Rev. Port. Pneumol. 2013, 19, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Li, X.; Cui, M.-N.; Liang, Y.; Li, X.-P.; Zhao, J.; Wei, L.-N.; Zhang, X.-L.; Quan, X.H. Low-Dose Mitomycin C Decreases the Postoperative Recurrence Rate of Pterygium by Perturbing NLRP3 Inflammatory Signalling Pathway and Suppressing the Expression of Inflammatory Factors. J. Ophthalmol. 2019, 2019, 9472782. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Sahu, K.; Dube, A.; Gupta, P.K. Extracellular pH Influences the Mode of Cell Death in Human Colon Adenocarcinoma Cells Subjected to Photodynamic Treatment with Chlorin P6. J. Photochem. Photobiol. B Biol. 2005, 81, 107–113. [Google Scholar] [CrossRef]
- Yan, X.-Q.; Lv, J.-L.; Ai, F.; Yang, J.; Xu, P.; Qiu, P.-Y.; Jiang, Q.-F. Preparation of Magnetic Dextran-Mitomycin C Prodrug Conjugate and Its in Vivo Antitumor Activity. Asian J. Chem. 2013, 25, 3562–3568. [Google Scholar] [CrossRef]
- Kavaz, D.; Kirac, F.; Kirac, M.; Vaseashta, A. Low Releasing Mitomycin C Molecule Encapsulated with Chitosan Nanoparticles for Intravesical Installation. J. Biomater. Nanobiotechnol. 2017, 8, 203–219. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W. Nanoparticle-Based Targeted Drug Delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef]
- López-García, J.; Lehocký, M.; Humpolíček, P.; Sáha, P. HaCaT Keratinocytes Response on Antimicrobial Atelocollagen Substrates: Extent of Cytotoxicity, Cell Viability and Proliferation. J. Funct. Biomater. 2014, 5, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Mitragotri, S. Challenges Associated with Penetration of Nanoparticles across Cell and Tissue Barriers: A Review of Current Status and Future Prospects. Nano Today 2014, 9, 223–243. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amin, K.W.K.; Deák, Á.; Csanády, M., Jr.; Szemerédi, N.; Szabó, D.; Turcsányi, Á.; Ungor, D.; Spengler, G.; Rovó, L.; Janovák, L. pH-Triggered Hydrogel Nanoparticles for Efficient Anticancer Drug Delivery and Bioimaging Applications. Pharmaceutics 2024, 16, 931. https://doi.org/10.3390/pharmaceutics16070931
Amin KWK, Deák Á, Csanády M Jr., Szemerédi N, Szabó D, Turcsányi Á, Ungor D, Spengler G, Rovó L, Janovák L. pH-Triggered Hydrogel Nanoparticles for Efficient Anticancer Drug Delivery and Bioimaging Applications. Pharmaceutics. 2024; 16(7):931. https://doi.org/10.3390/pharmaceutics16070931
Chicago/Turabian StyleAmin, Keristina Wagdi K., Ágota Deák, Miklós Csanády, Jr., Nikoletta Szemerédi, Diána Szabó, Árpád Turcsányi, Ditta Ungor, Gabriella Spengler, László Rovó, and László Janovák. 2024. "pH-Triggered Hydrogel Nanoparticles for Efficient Anticancer Drug Delivery and Bioimaging Applications" Pharmaceutics 16, no. 7: 931. https://doi.org/10.3390/pharmaceutics16070931
APA StyleAmin, K. W. K., Deák, Á., Csanády, M., Jr., Szemerédi, N., Szabó, D., Turcsányi, Á., Ungor, D., Spengler, G., Rovó, L., & Janovák, L. (2024). pH-Triggered Hydrogel Nanoparticles for Efficient Anticancer Drug Delivery and Bioimaging Applications. Pharmaceutics, 16(7), 931. https://doi.org/10.3390/pharmaceutics16070931