
Pharmacokinetics and Enterohepatic Circulation of 2-(Quinoline-8-carboxamido)benzoic Acid (2-QBA) in Mice

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Development of 2-QBA Analysis Using LC-MS/MS
2.2.1. Instrument Conditions
2.2.2. Preparation of Stock and Working Solutions of 2-QBA
2.2.3. Sample Preparation
2.3. Validation of Analytical Method Validation
2.3.1. Selectivity and Linearity
2.3.2. Intra- and Interday Precision and Accuracy
2.3.3. Matrix Effect and Recovery
2.3.4. Stability
2.3.5. Dilution Integrity
2.4. Dose-Dependent Pharmacokinetics of 2-QBA
2.5. Effect of Activated Charcoal on the Pharmacokinetics of 2-QBA
2.6. Identification of Glucuronide Metabolites of 2-QBA
2.7. Intestinal Permeability of 2-QBA
2.8. Data Analysis and Statistics
3. Results
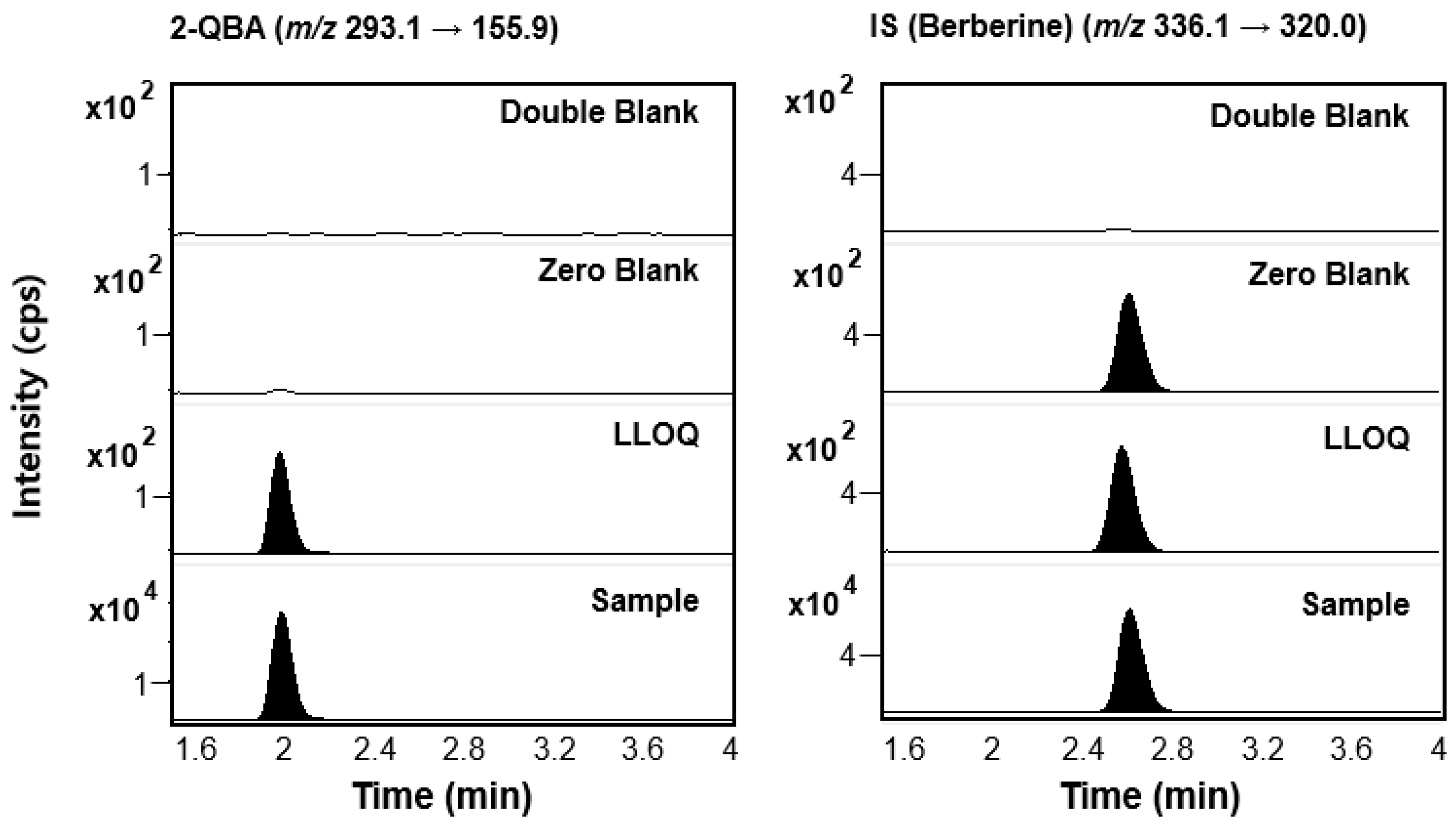
3.1. LC-MS/MS Analysis of 2-QBA
3.1.1. Optimization of Analysis
3.1.2. Linearity
3.1.3. Precision and Accuracy
3.1.4. Matrix Effect and Recovery
3.1.5. Stability
3.1.6. Dilution Integrity
3.2. Pharmacokinetics of 2-QBA
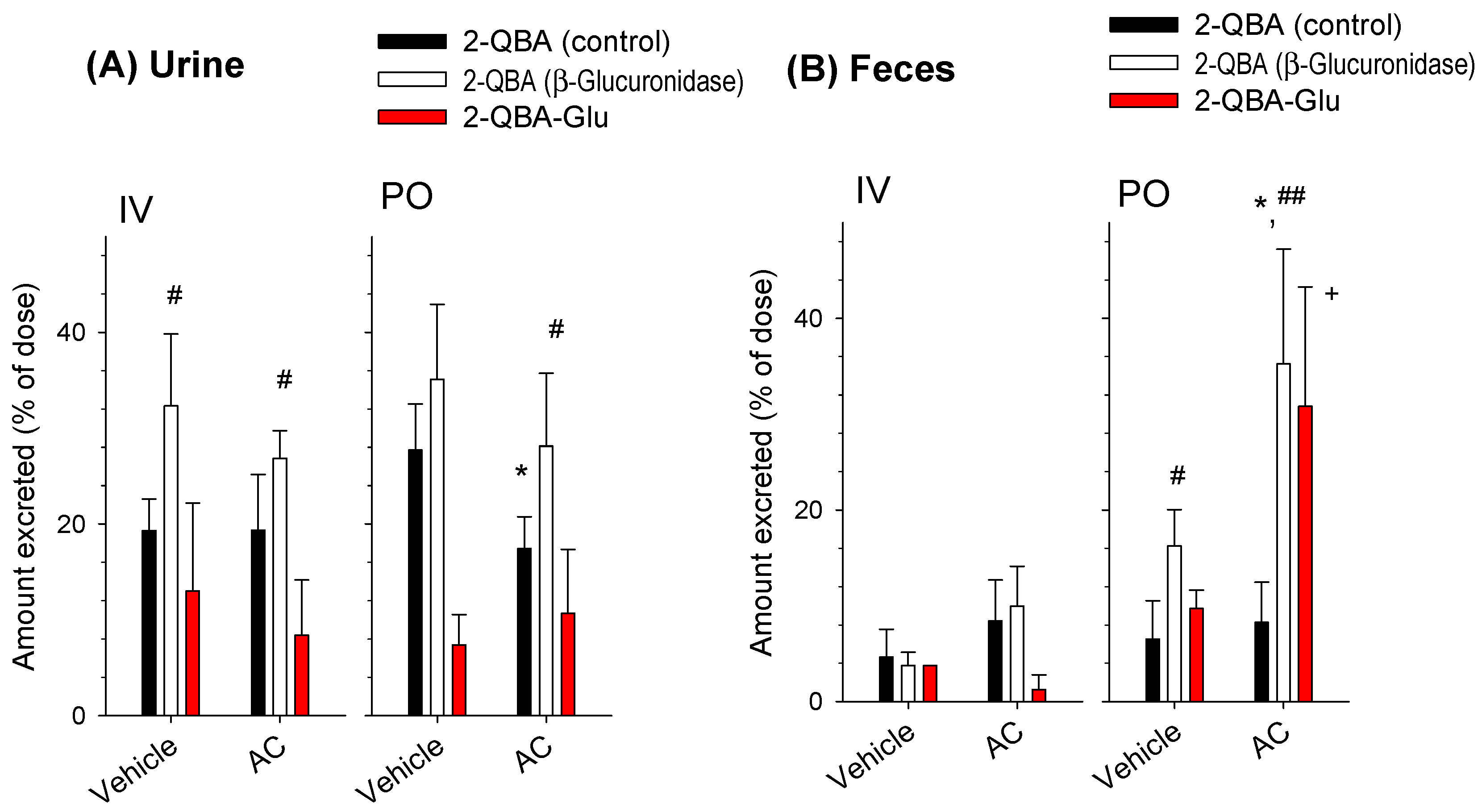
3.3. Effect of AC on the Pharmacokinetics of 2-QBA
3.4. Identification of Glucuronide Metabolites of 2-QBA
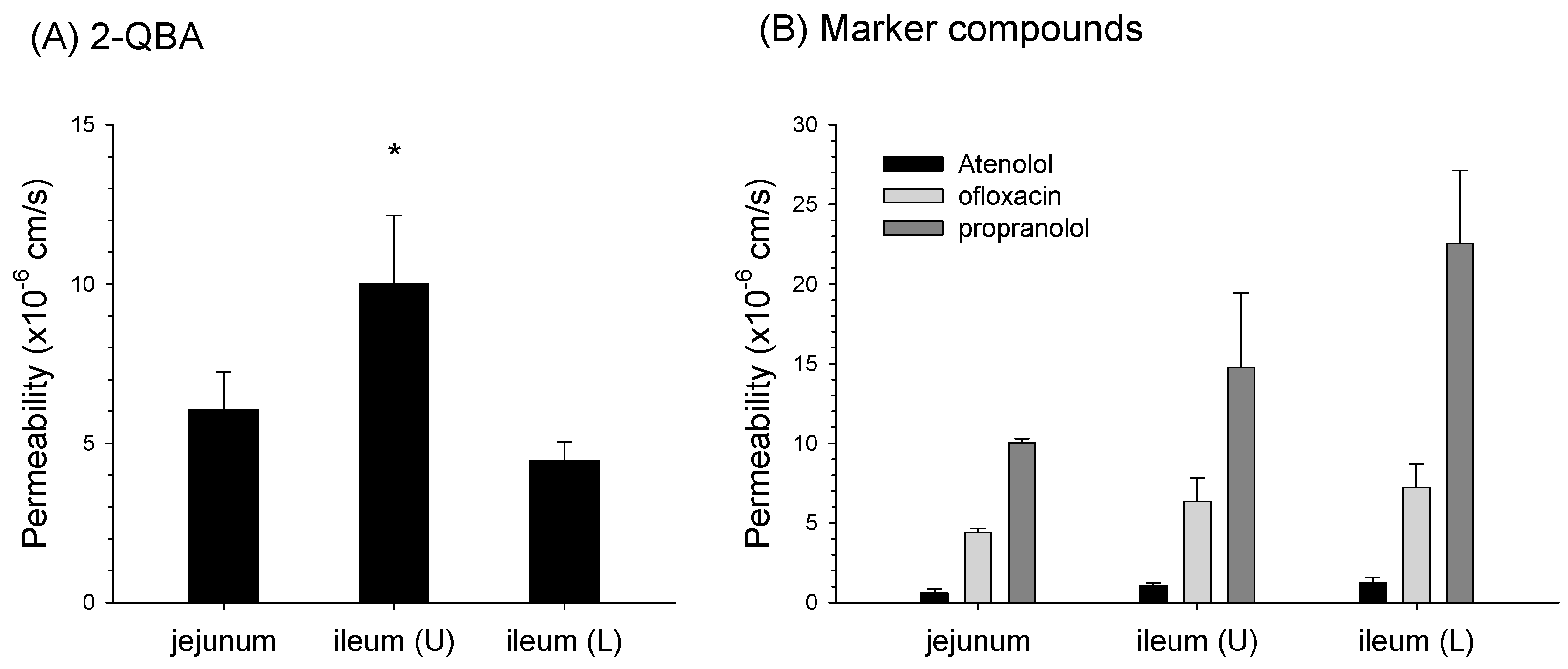
3.5. Absorptive Permeability of 2-QBA in Intestinal Segments
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, T.Y.; Yang, W.; Cha, D.S.; Han, Y.T. Synthesis of a natural quinoline alkaloid isolated from the deep-sea-derived fungus and its potential as a therapeutic for Parkinson’s disease. J. Asian Nat. Prod. Res. 2023, 25, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- Pålhagen, S.; Heinonen, E.; Hägglund, J.; Kaugesaar, T.; Mäki-Ikola, O.; Palm, R. Selegiline slows the progression of the symptoms of Parkinson disease. Neurology 2006, 66, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Rascol, O.; Fabbri, M.; Poewe, W. Amantadine in the treatment of Parkinson’s disease and other movement disorders. Lancet Neurol. 2021, 20, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Paterson, L.M.; Kornum, B.R.; Nutt, D.J.; Pike, V.W.; Knudsen, G.M. 5-HT radioligands for human brain imaging with PET and SPECT. Med. Res. Rev. 2013, 33, 54–111. [Google Scholar] [CrossRef]
- Pyka, P.; Haberek, W.; Więcek, M.; Szymanska, E.; Ali, W.; Cios, A.; Jastrzębska-Więsek, M.; Satała, G.; Podlewska, S.; Di Giacomo, S.; et al. First-in-Class Selenium-Containing Potent Serotonin Receptor 5-HT6 Agents with a Beneficial Neuroprotective Profile against Alzheimer’s Disease. J. Med. Chem. 2024, 67, 1580–1610. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.M.; Mo, Y.; Sabbagh, M.; Solomon, P.; Boada, M.; Jones, R.W.; Frisoni, G.B.; Grimmer, T.; Dubois, B.; Harnett, M.; et al. Intepirdine as adjunctive therapy to donepezil for mild-to-moderate Alzheimer’s disease: A randomized, placebo-controlled, phase 3 clinical trial (MINDSET). Alzheimers Dement. 2021, 7, e12136. [Google Scholar] [CrossRef] [PubMed]
- Maher-Edwards, G.; Dixon, R.; Hunter, J.; Gold, M.; Hopton, G.; Jacobs, G.; Hunter, J.; Williams, P. SB-742457 and donepezil in Alzheimer disease: A randomized, placebo-controlled study. Int. J. Geriatr. Psychiatry 2011, 26, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Meneses, A.; Pérez-García, G.; Ponce-Lopez, T.; Castillo, C. 5-HT6 receptor memory and amnesia: Behavioral pharmacology--learning and memory processes. Int. Rev. Neurobiol. 2011, 96, 27–47. [Google Scholar] [CrossRef]
- Parker, C.A.; Gunn, R.N.; Rabiner, E.A.; Slifstein, M.; Comley, R.; Salinas, C.; Johnson, C.N.; Jakobsen, S.; Houle, S.; Laruelle, M.; et al. Radiosynthesis and characterization of 11C-GSK215083 as a PET radioligand for the 5-HT6 receptor. J. Nucl. Med. 2012, 53, 295–303. [Google Scholar] [CrossRef]
- Lord, A.M.; Mahon, M.F.; Lloyd, M.D.; Threadgill, M.D. Design, synthesis, and evaluation in vitro of quinoline-8-carboxamides, a new class of poly(adenosine-diphosphate-ribose)polymerase-1 (PARP-1) inhibitor. J. Med. Chem. 2009, 52, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Comley, R.A.; Salinas, C.; Mizrahi, R.; Vitcu, I.; Ng, A.; Hallett, W.; Keat, N.; Wilson, A.A.; Rabiner, E.A.; Laruelle, M.; et al. Biodistribution and radiation dosimetry of the serotonin 5-HT6 ligand [11C]GSK215083 determined from human whole-body PET. Mol. Imaging Biol. 2012, 14, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ayala, L.F.; Guzmán-López, E.G.; Galano, A. Quinoline Derivatives: Promising Antioxidants with Neuroprotective Potential. Antioxidants 2023, 12, 1853. [Google Scholar] [CrossRef] [PubMed]
- Summers, K.L.; Roseman, G.P.; Sopasis, G.J.; Millhauser, G.L.; Harris, H.H.; Pickering, I.J.; George, G.N. Copper(II) binding to PBT2 differs from that of other 8-hydroxyquinoline chelators: Implications for the treatment of neurodegenerative protein misfolding diseases. Inorg. Chem. 2020, 59, 17519–17534. [Google Scholar] [CrossRef] [PubMed]
- Muruganantham, N.; Sivakumar, R.; Anbalagan, N.; Gunasekaran, V.; Leonard, J.T. Synthesis, anticonvulsant and antihypertensive activities of 8-substituted quinoline derivatives. Biol. Pharm. Bull. 2004, 27, 1683–1687. [Google Scholar] [CrossRef] [PubMed]
- Atwell, G.J.; Bos, C.D.; Baguley, B.C.; Denny, W.A. Potential antitumor agents. 56. “Minimal” DNA-intercalating ligands as antitumor drugs: Phenylquinoline-8-carboxamides. J. Med. Chem. 1988, 31, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Lin, X.; Zhou, X.; Yang, B.; Tian, X.; Wang, J.; Xu, S.; Liu, Y. New quinoline alkaloid and bisabolane-type sesquiterpenoid derivatives from the deep-sea-derived fungus Aspergillus sp. SCSIO06786. Fitoterapia 2020, 140, 104406. [Google Scholar] [CrossRef] [PubMed]
- Lücking, C.B.; Brice, A. Alpha-synuclein and Parkinson’s disease. Cell. Mol. Life Sci. 2000, 57, 1894–1908. [Google Scholar] [CrossRef] [PubMed]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- McFarthing, K.; Rafaloff, G.; Baptista, M.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R.W. Parkinson’s disease drug therapies in the clinical trial pipeline: 2022 update. J. Parkinsons Dis. 2022, 12, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Antonschmidt, L.; Matthes, D.; Dervişoğlu, R.; Frieg, B.; Dienemann, C.; Leonov, A.; Nimerovsky, E.; Sant, V.; Ryazanov, S.; Giese, A.; et al. The clinical drug candidate anle138b binds in a cavity of lipidic α-synuclein fibrils. Nat. Commun. 2022, 13, 5385. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Sing, N.; Melbourne, S.; Morgan, A.; Mariner, C.; Spillantini, M.G.; Wegrzynowicz, M.; Dalley, J.W.; Langer, S.; Ryazanov, S. Safety, tolerability and pharmacokinetics of the oligomer modulator anle138b with exposure levels sufficient for therapeutic efficacy in a murine Parkinson model: A randomised, double-blind, placebo-controlled phase 1a trial. EBioMedicine 2022, 80, 104021. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M.; Subramanian, T.; Pagan, F.; Isaacson, S.; Gil, R.; Hauser, R.A.; Feldman, M.; Goldstein, M.; Kumar, R.; Truong, D.; et al. Oral ENT-01 targets enteric neurons to treat constipation in parkinson disease: A randomized controlled trial. Ann. Intern. Med. 2022, 175, 1666–1674. [Google Scholar] [CrossRef]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Fiske, B.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R.W. Parkinson’s disease drug therapies in the clinical trial pipeline: 2023 update. J. Parkinsons Dis. 2023, 13, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Lim, D.Y.; Lee, C.H.; Jeon, J.H.; Choi, M.K.; Song, I.S. Enhanced intestinal absorption and pharmacokinetic modulation of berberine and its metabolites through the inhibition of P-glycoprotein and intestinal metabolism in rats using a berberine mixed micelle formulation. Pharmaceutics. 2020, 12, 882. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.Y.; Lee, C.H.; Lee, J.; Jeon, J.H.; Jin, Y.; Song, I.S.; Choi, M.K. Bioanalysis of ursodeoxycholic acid and its metabolites and improved oral bioavailability using mixed micelles with poloxamer 407 and polysorbate 80. Biotechnol. Bioprocess. Eng. 2024, 29, 313–327. [Google Scholar] [CrossRef]
- Lim, D.Y.; Pang, M.; Lee, J.; Lee, J.; Jeon, J.H.; Park, J.H.; Choi, M.K.; Song, I.S. Enhanced bioavailability and hepatoprotective effect of silymarin by preparing silymarin-loaded solid dispersion formulation using freeze-drying method. Arch. Pharm. Res. 2022, 45, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Ji, H.K.; Goo, S.H.; Nam, S.J.; Kang, Y.J.; Lee, E.; Liu, K.H.; Choi, M.K.; Song, I.S. Involvement of intestinal efflux and metabolic instability in the pharmacokinetics of platycodin D in rats. Drug Metab. Pharmacokinet. 2017, 32, 248–254. [Google Scholar] [CrossRef]
- Jeon, J.H.; Park, J.H.; Jeon, S.Y.; Pang, M.; Choi, M.K.; Song, I.S. Concomitant administration of red ginseng extract with lactic acid bacteria increases the plasma concentration of deglycosylated ginsenosides in healthy human subjects. Biomolecules 2022, 12, 1896. [Google Scholar] [CrossRef]
- Heimer, G.M.; Englund, D.E. Enterohepatic recirculation of oestriol: Inhibition by activated charcoal. Acta Endocrinologica 1986, 113, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Frost, C.E.; He, K.; Rodrigues, A.D.; Wang, X.; Wang, L.; Goosen, T.C.; Humphreys, W.G. Investigating the enteroenteric recirculation of apixaban, a factor Xa inhibitor: Administration of activated charcoal to bile duct-cannulated rats and dogs receiving an intravenous dose and use of drug transporter knockout rats. Drug Metab. Dispos. 2013, 41, 906–915. [Google Scholar] [CrossRef]
- Qi, Z.; Breyer, M.D. Measurement of glomerular filtration rate in conscious mice. In Kidney Research: Experimental Protocols; Becker, G.J., Hewitson, T.D., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 61–72. [Google Scholar] [CrossRef]
- Qi, Z.; Whitt, I.; Mehta, A.; Jin, J.; Zhao, M.; Harris, R.C.; Fogo, A.B.; Breyer, M.D. Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. Am. J. Physiol. Renal Physiol. 2004, 286, F590–F596. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Iwama, R.; Sato, T.; Heishima, K.; Shimamura, S.; Ichijo, T.; Satoh, H.; Furuhama, K. Estimation of glomerular filtration rate in conscious mice using a simplified equation. Physiol. Rep. 2014, 2, e12135. [Google Scholar] [CrossRef] [PubMed]
- Han, D.G.; Seo, S.W.; Choi, E.; Kim, M.S.; Yoo, J.W.; Jung, Y.; Yoon, I.S. Impact of route-dependent phase-II gut metabolism and enterohepatic circulation on the bioavailability and systemic disposition of resveratrol in rats and humans: A comprehensive whole body physiologically-based pharmacokinetic modeling. Biomed. Pharmacother. 2022, 151, 113141. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, M.; Trocóniz, I.F.; Fagiolino, P. Enteric reabsorption processes and their impact on drug pharmacokinetics. Sci. Rep. 2021, 11, 5794. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, Y.; Maruyama, S.; Hachimura, K.; Ohwada, T. Activated charcoal interrupts enteroenteric circulation of phenobarbital. J. Toxicol. Clin. Toxicol. 1994, 32, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Uehara, M.; Suzuki, K.; Watanabe, S.; Ohta, A.; Sakai, K.; Adlercreutz, H. Dietary fructooligosaccharides modify intestinal bioavailability of a single dose of genistein and daidzein and affect their urinary excretion and kinetics in blood of rats. J. Nutr. 2001, 131, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Hu, Y.; Zhang, B.; Teng, Z.; Gan, H.; Yang, Z.; Wang, Q.; Huan, M.; Mei, Q. Dose-dependent absorption, metabolism, and excretion of genistein in rats. J. Agric. Food Chem. 2008, 56, 8354–8359. [Google Scholar] [CrossRef]
- Sfakianos, J.; Coward, L.; Kirk, M.; Barnes, S. Intestinal uptake and biliary excretion of the isoflavone genistein in Rats. J. Nutr. 1997, 127, 1260–1268. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Tsai, T.H. Enterohepatic circulation and pharmacokinetics of genistin and genistein in rats. ACS Omega 2019, 4, 18428–18433. [Google Scholar] [CrossRef] [PubMed]
- Marier, J.F.; Vachon, P.; Gritsas, A.; Zhang, J.; Moreau, J.P.; Ducharme, M.P. Metabolism and disposition of resveratrol in rats: Extent of absorption, glucuronidation, and enterohepatic recirculation evidenced by a linked-rat model. J. Pharmacol. Exp. Ther. 2002, 302, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, H.; Kobayashi, M.; Oda, M.; Nakasato, K.; Kobayashi, M.; Tadano, K. Characterization of intestinal absorption and enterohepatic circulation of mycophenolic acid and its 7-O-glucuronide in rats. Drug Metab. Pharmacokinet. 2006, 21, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Almousa, A.; Yonpiam, R.; Blakley, B.; Al-Dissi, A.N. Prolonged absorption and susceptibility to enterohepatic circulation after oral administration of ergot alkaloids in ewes. Can. J. Vet. Res. 2022, 86, 108–112. [Google Scholar]
- Ogiso, T.; Kitagawa, T.; Iwaki, M.; Tanino, T. Pharmacokinetic analysis of enterohepatic circulation of etodolac and effect of hepatic and renal injury on the pharmacokinetics. Biol. Pharm. Bull. 1997, 20, 405–410. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Nominal Concentration (ng/mL) | Slope | r2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 5 | 20 | 50 | 250 | 500 | |||
| Calculated concentration (ng/mL) | 1.0 | 2.0 | 5.1 | 19.6 | 51.8 | 256 | 485 | 0.246 | 0.997 |
| Accuracy (%) | 100.1 | 100.3 | 102.8 | 97.84 | 103.5 | 102.3 | 96.99 | - | - |
| Precision (CV, %) | 2.4 | 4.7 | 10.2 | 5.5 | 3.0 | 3.7 | 4.5 | 14.9 | 0.09 |
| Nominal Concentration (ng/mL) | Measured Concentration (ng/mL) | Precision (%) | Accuracy (%) | |
|---|---|---|---|---|
| Intra-day (n = 6) | 3 | 3.11 ± 0.14 | 4.57 | 103.56 |
| 100 | 105.5 ± 6.09 | 5.77 | 105.51 | |
| 400 | 401.5 ± 28.0 | 6.98 | 100.38 | |
| Inter-day (n = 5) | 3 | 3.04 ± 0.04 | 1.22 | 101.17 |
| 100 | 108.9 ± 1.91 | 1.76 | 108.86 | |
| 400 | 417.0 ± 21.8 | 5.22 | 104.24 |
| Analyte | QC (ng/mL) | Extraction Recovery (%) | CV (%) | Matrix Effects (%) | CV (%) |
|---|---|---|---|---|---|
| 2-QBA | 3 | 89.48 ± 6.76 | 7.56 | 56.47 ± 6.25 | 11.1 |
| 400 | 91.71 ± 3.13 | 3.42 | 69.00 ± 6.03 | 8.74 | |
| IS | 1 | 97.46 ± 3.40 | 3.49 | 94.64 ± 3.36 | 3.55 |
| Nominal Concentration (ng/mL) | Measured Concentration (ng/mL) | Precision (%) | Accuracy (%) |
|---|---|---|---|
| Bench-top stability (at 25 °C for 6 h) | |||
| 3 | 2.96 ± 0.17 | 5.61 | 98.51 |
| 100 | 89.14 ± 8.92 | 10.0 | 89.14 |
| 400 | 366.22 ± 13.4 | 3.66 | 91.55 |
| Freeze-thaw cycle stability (3 cycles) | |||
| 3 | 3.43 ± 0.26 | 7.47 | 114.4 |
| 100 | 104.3 ± 13.8 | 13.2 | 104.3 |
| 400 | 425.7 ± 34.3 | 8.06 | 106.4 |
| Autosampler stability (at 6 °C for 24 h) | |||
| 3 | 3.29 ± 0.08 | 2.49 | 109.5 |
| 100 | 103.1 ± 10.1 | 9.83 | 103.1 |
| 400 | 434.7 ± 35.5 | 8.15 | 108.7 |
| Nominal Concentration (ng/mL) | Dilution | Measured Concentration (ng/mL) | Accuracy (%) | Precision (%) |
|---|---|---|---|---|
| 4000 | 10-fold | 376.7 ± 17.5 | 94.16 | 4.66 |
| 2000 | 5-fold | 387.2 ± 4.35 | 96.49 | 1.53 |
| Site | Parameters | Dose | p Value | |
|---|---|---|---|---|
| 0.5 mg/kg | 1.0 mg/kg | |||
| Plasma | AUC24h (ng·h/mL) | 412.3 ± 32.1 | 996.7 ± 146.6 | ND |
| AUCinf (ng·h/mL) | 417.8 ± 32.3 | 1044 ± 174.9 | ND | |
| AUC24h/dose (ng·h/mL/mg/kg) | 824.7 ± 64.1 | 996.7 ± 146.6 | 0.083 | |
| AUCinf/dose (ng·h/mL/mg/kg) | 835.6 ± 64.6 | 1044 ± 174.9 | 0.083 | |
| T1/2 (h) | 4.61 ± 2.19 | 5.26 ± 1.31 | 0.773 | |
| MRT (h) | 3.88 ± 1.58 | 3.03 ± 1.11 | 0.149 | |
| CL (mL/min/kg) | 20.03 ± 1.53 | 16.32 ± 2.81 | 0.059 | |
| Vdss (L/kg) | 4.73 ± 2.15 | 2.88 ± 0.72 | 0.153 | |
| Excretion | Amount excreted in urine for 96 h (% of dose) | - | 13.8 ± 4.24 | - |
| Amount excreted in feces for 96 h (% of dose) | - | 9.30 ± 6.17 | - | |
| CLrenal (mL/min/kg) | - | 5.47 ± 2.22 | - | |
| CLfeces (mL/min/kg) | - | 1.59 ± 1.14 | - | |
| CLmetabolism (mL/min/kg) | - | 9.26 ± 1.43 | - | |
| Parameters | Dose | p Value | ||
|---|---|---|---|---|
| 2 mg/kg | 5 mg/kg | 15 mg/kg | ||
| Cmax,1 (ng/mL) | 282.6 ± 173.9 | 752.1 ± 319.3 | 2457 ± 696.1 | ND |
| Tmax,1 (h) | 0.20 ± 0.11 | 0.25 ± 0.0 | 0.30 ± 0.11 | 0.246 |
| Cmax,2 (ng/mL) | 141.3 ± 67 | 194.2 ± 143.6 | 812.4 ± 221.7 | ND |
| Tmax,2 (h) | 5.60 ± 2.19 | 3.50 ± 1.00 | 5.60 ± 2.19 | 0.158 |
| AUC24h (ng·h/mL) | 1368 ± 680.5 | 3514 ± 846.2 | 10,583 ± 2734 | ND |
| AUCinf (ng·h/mL) | 1426 ± 695.9 | 4369 ± 934.5 | 11,021 ± 2645 | ND |
| AUC24h/dose (ng·h/mL/mg/kg) | 684.2 ± 340.3 | 702.9 ± 169.2 | 705.5 ± 182.3 | 0.755 |
| AUCinf/dose (ng·h/mL/mg/kg) | 712.7 ± 348.0 | 873.7 ± 186.9 | 734.7 ± 176.3 | 0.326 |
| T1/2 (h) | 7.82 ± 1.76 | 9.91 ± 1.92 | 5.58 ± 1.24 * | 0.015 |
| MRT (h) | 12.0 ± 4.91 | 13.7 ± 3.14 | 7.04 ± 0.76 * | 0.016 |
| BA (%) | 68.3 | 83.7 | 70.4 | |
| Administration | Parameters | Treatment | p Value | |
|---|---|---|---|---|
| Control | AC (0.8 g/kg) | |||
| IV (1 mg/kg) | AUC24h (ng·h/mL) | 1273.5 ± 360.8 | 1175.3 ± 264.4 | 0.637 |
| AUCinf (ng·h/mL) | 1325.8 ± 421.9 | 1212.5 ± 293.4 | 0.635 | |
| T1/2 (h) | 3.88 ± 1.72 | 3.18 ± 2.52 | 0.620 | |
| MRT (h) | 2.88 ± 1.14 | 2.48 ± 2.56 | 0.759 | |
| CL (mL/min/kg) | 13.88 ± 5.41 | 14.50 ± 3.95 | 0.841 | |
| Vdss (mL/kg) | 2.24 ± 0.88 | 1.89 ± 1.59 | 0.683 | |
| PO (2 mg/kg) | Cmax (ng/mL) | 399.0 ± 90.7 | 315.2 ± 105.9 | 0.343 |
| Tmax (h) | 0.25 ± 0.00 | 0.25 ± 0.00 | 1.00 | |
| AUC24h (ng·h/mL) | 1932 ± 453.2 | 649.6 ± 305.4 | 0.0032 | |
| AUCinf (ng·h/mL) | 2114 ± 608.1 | 835.7 ± 208.9 | 0.0082 | |
| T1/2 (h) | 6.05 ± 2.04 | 5.50 ± 2.24 | 0.726 | |
| MRT (h) | 8.49 ± 3.08 | 7.00 ± 2.48 | 0.486 | |
| BA (%) | 79.8 | 34.5 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, J.-H.; Jeon, S.-Y.; Baek, Y.-J.; Park, C.-E.; Choi, M.-K.; Han, Y.T.; Song, I.-S. Pharmacokinetics and Enterohepatic Circulation of 2-(Quinoline-8-carboxamido)benzoic Acid (2-QBA) in Mice. Pharmaceutics 2024, 16, 934. https://doi.org/10.3390/pharmaceutics16070934
Jeon J-H, Jeon S-Y, Baek Y-J, Park C-E, Choi M-K, Han YT, Song I-S. Pharmacokinetics and Enterohepatic Circulation of 2-(Quinoline-8-carboxamido)benzoic Acid (2-QBA) in Mice. Pharmaceutics. 2024; 16(7):934. https://doi.org/10.3390/pharmaceutics16070934
Chicago/Turabian StyleJeon, Ji-Hyeon, So-Yeon Jeon, Yeon-Ju Baek, Chan-E Park, Min-Koo Choi, Young Taek Han, and Im-Sook Song. 2024. "Pharmacokinetics and Enterohepatic Circulation of 2-(Quinoline-8-carboxamido)benzoic Acid (2-QBA) in Mice" Pharmaceutics 16, no. 7: 934. https://doi.org/10.3390/pharmaceutics16070934