
Exploring a New Generation of Pyrimidine and Pyridine Derivatives as Anti-Influenza Agents Targeting the Polymerase PA–PB1 Subunits Interaction
 , ,
, ,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
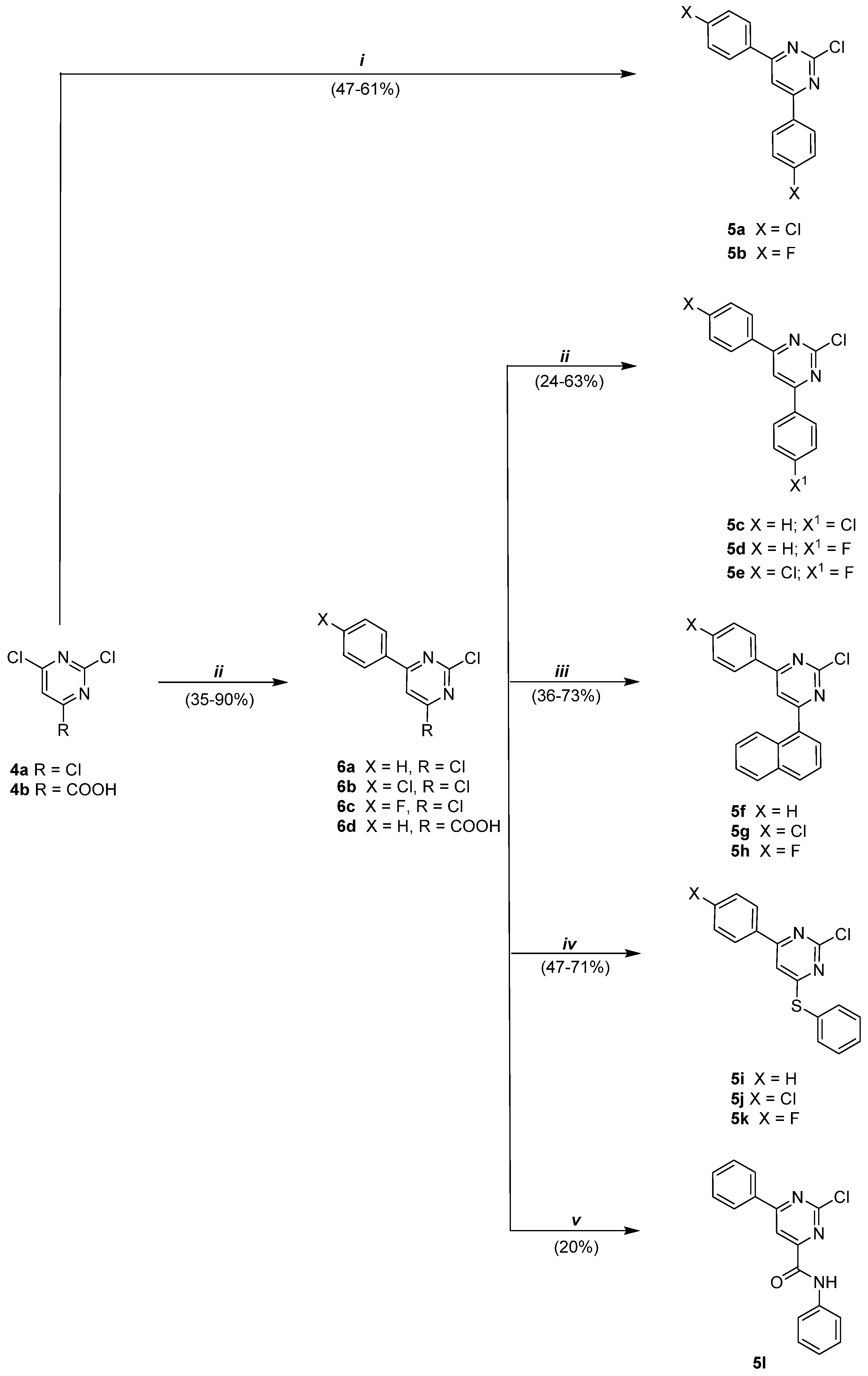
2.1.1. General Procedure for the Synthesis of Compounds 5a,b
- 2-Chloro-4,6-bis(4-chlorophenyl)pyrimidine (5a). Yield: 61%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.52–7.56 (m, 4H, Ar), 7.99 (s, 1H, pyr), 8.04–8.15 (m, 4H, Ar). Anal. calcd. for C16H9Cl3N2: C 57.26, H 2.70, N 8.35; found C 57.19, H 2.64, N 8.20.
- 2-Chloro-4,6-bis(4-fluorophenyl)pyrimidine (5b). Yield: 47%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.21–7.28 (m, 4H, Ar), 7.95 (s, 1H, pyr), 8.15–8.22 (m, 4H, Ar). Anal. calcd. for C16H9ClF2N2: C 63.49, H 3.00, N 9.25; found C 63.47, H 3.28, N 9.44.
2.1.2. General Procedure for the Synthesis of 2,4-Dichloro-6-phenylpyrimidines 6a–c
- 2,4-Dichloro-6-phenylpyrimidine (6a) [27]. Yield: 90%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.55–7.61 (m, 3H, Ar), 7.71 (s, 1H, pyr), 8.07–8.13 (m, 2H, Ar). Anal. calcd. for C10H6Cl2N2: C 53.37, H 2.69, N 12.45; found C 53.65, 2.58, N 12.37.
- 2,4-Dichloro-6-(4-chlorophenyl)pyrimidine (6b). Yield: 83%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.53 (d, J = 8.0 Hz, 2H, Ar), 7.68 (s, 1H, pyr), 8.06 (d, J = 8.0 Hz, 2H, Ar). Anal. calcd. for C10H5Cl3N2: C 46.28, H 1.94, N 10.79; found C 46.47, H 2.14, N 10.51.
- 2,4-Dichloro-6-(4-fluorophenyl)pyrimidine (6c). Yield: 35%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.20–7.29 (m, 2H, Ar), 7.67 (s, 1H, pyr), 8.10–8.17 (m, 2H, Ar). Anal. calcd. for C10H5Cl2FN2: C 49.42, H 2.07, N 11.53; found C 49.59, H 1.96, N 11.63.
2.1.3. Synthesis of 2-Chloro-6-phenylpyrimidine-4-carboxylic Acid 6d
2.1.4. General Procedure for the Synthesis of Compounds 5c–e
- 2-Chloro-4-(4-chlorophenyl)-6-phenylpyrimidine (5c). Yield: 45%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.57–7.60 (m, 5H, Ar), 8.02 (s, 1H, pyr), 8.15–8.19 (m, 4H, Ar). Anal. calcd. for C16H10Cl2N2: C 63.81, H 3.35, N 9.30; found C 63.65, H 3.37, N 9.31.
- 2-Chloro-4-(4-fluorophenyl)-6-phenylpyrimidine (5d). Yield: 63%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.24–7.30 and 7.56–7.60 (2m, 5H, Ar), 8.00 (s, 1H, pyr), 8.15–8.21 (m, 4H, Ar). Anal. calcd. for C16H10ClFN2: C 67.50, H 3.54, N 9.84; found C 67.44, H 3.84, N 9.89.
- 2-Chloro-4-(4-chlorophenyl)-6-(4-fluorophenyl)pyrimidine (5e). Yield: 24%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.26–7.30 and 7.52–7.57 (2m, 4H, Ar), 7.97 (s, 1H, pyr), 8.11–8.21 (m, 4H, Ar). Anal. calcd. for C16H9Cl2FN2: C 60.21, H 2.84, N 8.78; found C 60.09, H 2.78, N 8.98.
2.1.5. General Procedure for the Synthesis of 2-Chloro-4-(naphthalen-1-yl)-6-phenylpyrimidines 5f–h
- 2-Chloro-4-(1-naphthyl)-6-phenylpyrimidine (5f). Yield: 43%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.58–7.65 (m, 6H, Ar), 7.75 (s, 1H, pyr), 7.96–7.98 and 8.00–8.20 (2m, 6H, Ar). Anal. calcd. for C20H13ClN2: C 75.83, H 4.14, N 8.84; found C 75.93, H 4.21, N 8.69.
- 2-Chloro-4-(4-chlorophenyl)-6-(1-naphthyl)pyrimidine (5g). Yield: 36%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.52–7.63 (m, 5H, Ar), 7.94 (s, 1H, pyr), 7.96–8.17 (m, 6H, Ar). Anal. calcd. for C20H12Cl2N2: C 68.39, H 3.44, N 7.98; found C 68.30, H 3.31, N 8.33.
- 2-Chloro-4-(4-fluorophenyl)-6-(1-naphthyl)pyrimidine (5h). Yield: 73%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.21–7.25, 7.58–7.66 and 7.75–7.79 (3 m, 7H, Ar) 7.92 (s, 1H, pyr), 7.93–8.06 and 8.18–8.25 (2m, 4H, Ar). Anal. calcd. for C20H12ClFN2: C 71.75, H 3.61, N 8.37; found C 71.63, H 3.46, N 8.30.
2.1.6. Synthesis of 2-Chloro-4-phenyl-6-(phenylthio)pyrimidine 5i
2.1.7. General Procedure for the Synthesis of 2-Chloro-4-(4-substitued phenyl)-6-(phenylthio)pyrimidines 5j,k
- 2-Chloro-4-(4-chlorophenyl)-6-(phenylthio)pyrimidine (5j). Yield: 47%. 1H NMR (200 MHz, CDCl3): δ (ppm) 6.86 (s, 1H, pyr), 7.33–7.48 and 7.57–7.69 (2m, 9H, Ar). Anal. calcd. for C16H10Cl2N2S: C 57.67, H 3.02, N 8.41, S 9.62; found C 57.55, H 3.31, N 8.76, S 9.92.
- 2-Chloro-4-(4-fluorophenyl-6-(phenylthio)pyrimidine (5k). Yield: 71%. 1H NMR (200 MHz, CDCl3): δ (ppm) 6.85 (s, 1H, pyr), 7.41–7.43, 7.57–7.62 and 7.65–7.75 (3 m, 9H, Ar). Anal. calcd. for C16H10ClFN2S: C 60.67, H 3.18, N 8.84, S 10.12; found C 60.52, H 3.30, N 8.51, S 10.45.
2.1.8. Synthesis of 2-Chloro-N,6-diphenylpyrimidine-4-carboxamide 5l
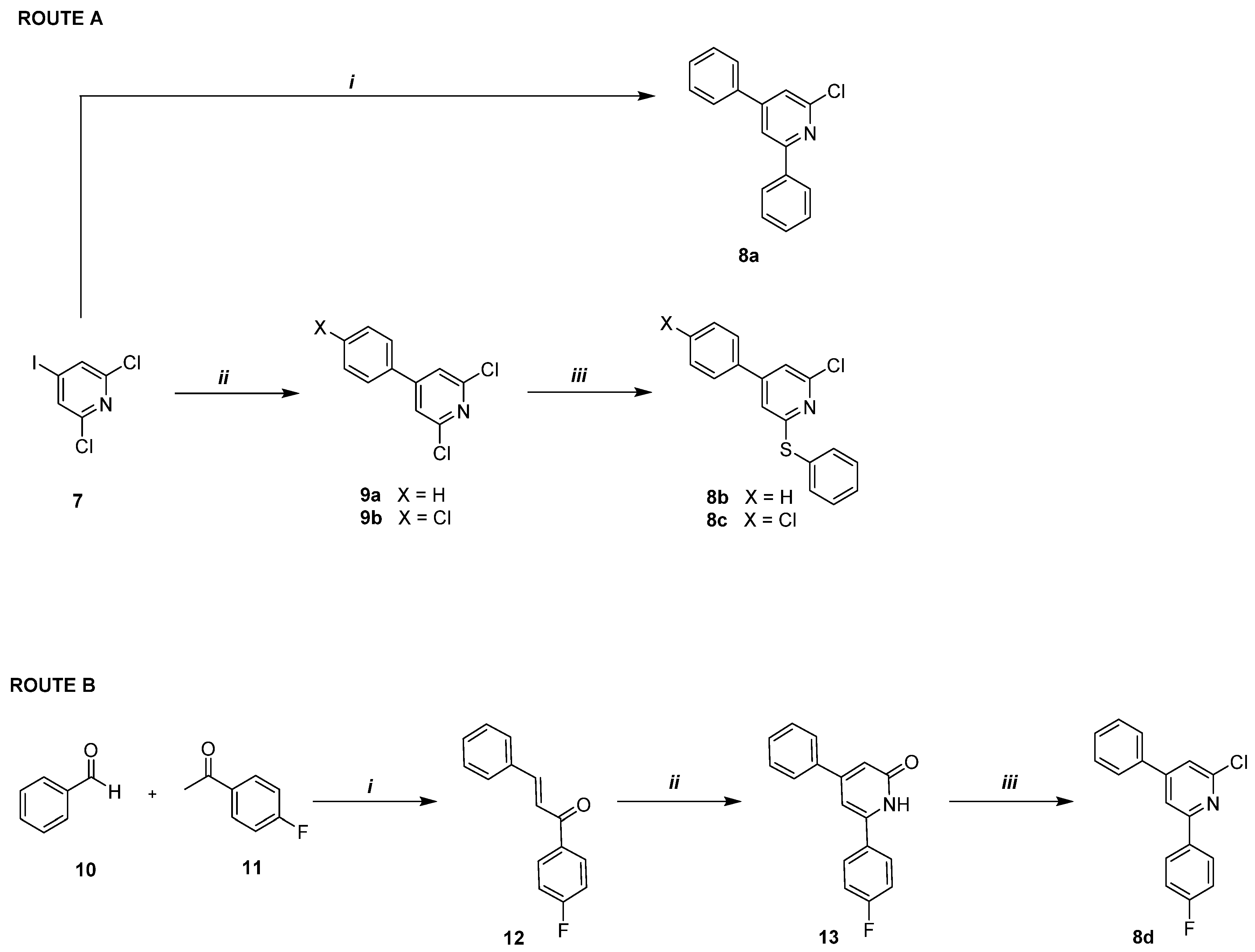
2.1.9. Synthesis of 2-Chloro-4,6-diphenylpyridine 8a
2.1.10. General Procedure for the Synthesis of 2,6-Dichloro-4-phenylpyridines 9a,b
- 2,6-Dichloro-4-phenylpyridine (9a). Yield: 73%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.49 (s, 2H, pyr), 7.50–7.54 and 7.60–7.63 (2m, 5H, Ar). Anal. calcd. for C11H7Cl2N: C 58.96, H 3.15, N 6.25; found C 58.88, H 3.12, N 5.96.
- 2,6-Dichloro-4-(4-chlorophenyl)pyridine (9b). Yield: 74%. 1H NMR (200 MHz, CDCl3): δ (ppm) 7.47 (s, 2H pyr), 7.54–7.55 (m, 4H Ar). Anal. calcd. for C11H6Cl3N: C 51.11, H 2.34, N 5.42; found C 51.14, H 2.25, N 5.07.
2.1.11. General Procedure for the Synthesis of 2-Chloro-4-phenyl-6-(phenylthio)pyridines 8b,c
- 2-Chloro-4-phenyl-6-(phenylthio)pyridine (8b). Yield: 95%. 1H NMR (200 MHz, CDCl3): δ (ppm) 6.94 (s, 1H, pyr), 7.25 (s, 1H, pyr), 7.44–7.51 and 7.63–7.70 (2m, 10H, Ar). Anal. calcd. for C17H12ClNS: C 68.56, H 4.06, N 4.70, S 10.77; found C 68.41, H 3.90, N 5.00, S 11.11.
- 2-Chloro-4-(4-chlorophenyl)-6-(phenylthio)pyridine (8c). Yield: 82%. 1H NMR (200 MHz, CDCl3): δ (ppm) 6.88 (s, 1H, pyr), 7.21 (s, 1H, pyr), 7.37–7.52 and 7.65–7.68 (2m, 9H, Ar). Anal. calcd. for C17H11Cl2NS: C 61.46, H 3.34, N 4.22, S 9.65; found C 61.40, H 3.54, N 4.45, S 9.82.
2.1.12. Synthesis of (E)-1-(4-Fluorophenyl)-3-phenylprop-2-en-1-one 12 [28]
2.1.13. Synthesis of 6-(4-Fluorophenyl)-4-phenylpyridin-2(1H)-one 13
2.1.14. Synthesis of 2-Chloro-6-(4-fluorophenyl)-4-phenylpyridine 8d
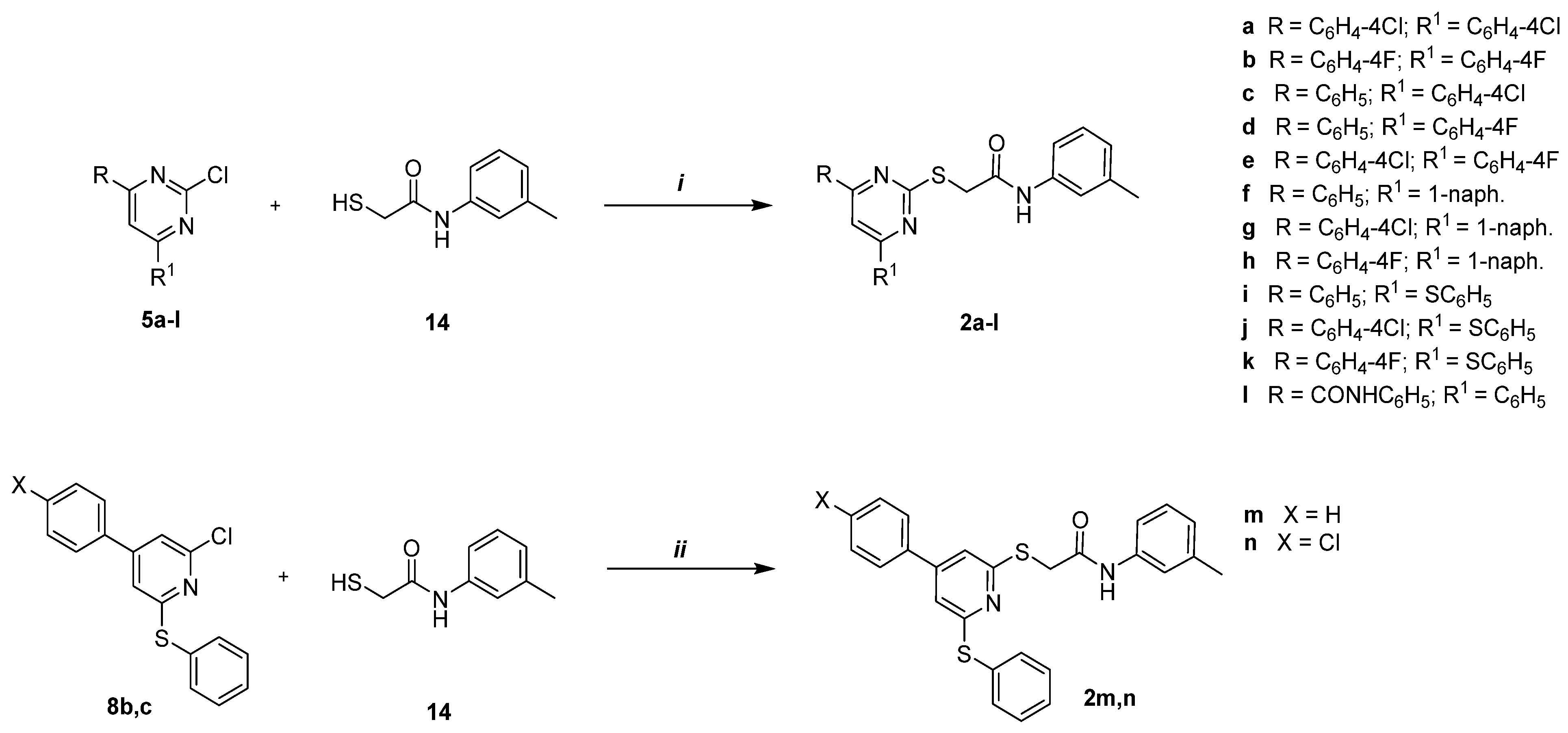
2.1.15. General Procedure for the Synthesis of Compounds 2a–l
- 2-{[4,6-Bis(4-chlorophenyl)pyrimidin-2-yl]thio}-N-(m-tolyl)acetamide (2a). Yield: 20%. Mp: 225–227 °C. 1H NMR (200 MHz, CDCl3): δ (ppm) 2.19 (s, 3H, CH3), 4.03 (s, 2H, CH2), 6.83 (d, J = 8.0 Hz, 2H, Ar), 6.96 (s, 1H, Ar), 7.08 and 7.15 (2 t, J = 8.0 Hz, 2H, Ar), 7.52 (d, J = 8.4 Hz, 4H, Ar), 7.78 (s, 1H, H-5), 8.09 (d, J = 8.4 Hz, 4H, Ar), 8.96 (br s, 1H, NH). 13C NMR (101 MHz, DMSO-d6): δ (ppm) 21.68, 36.38, 108.86, 116.77, 120.08, 124.52, 129.19, 129.41, 129.87, 135.12, 138.46, 139.62, 163.80, 166.87, 171.56. Anal. calcd. for C25H19Cl2N3OS: C 62.50, H 3.99, N 8.75, S 6.67; found C 62.43, H 4.25, N 8.83, S 6.60.
- 2-{[4,6-Bis(4-fluorophenyl)pyrimidin-2-yl]thio}-N-(m-tolyl)acetamide (2b). Yield: 23%. Mp: 210–211 °C. 1H NMR (200 MHz, CDCl3): δ (ppm) 2.18 (s, 3H, CH3), 4.04 (s, 2H, CH2), 6.82 (d, J = 7.2 Hz, 1H, Ar), 6.99 (s, 1H, Ar), 7.07 (t, J = 7.8 Hz, 1H, Ar), 7.13 (d, J = 8.0 Hz, 1H, Ar), 7.21–7.26 (m, 4H, Ar), 7.76 (s, 1H, H-5), 8.14–8.17 (m, 4H, Ar) 9.03 (br s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ (ppm) 21.42, 35.96, 108.37, 116.30, 116.52, 116.71, 120.28, 125.09, 128.86, 129.60, 129.69, 129.69, 132.37, 132.40, 137.79, 138.85, 164.53, 165.07 (d, JC-F = 263 Hz), 165.83 (d, JC-F = 263 Hz), 170.99. Anal. calcd. for C25H19F2N3OS: C 67.10, H 4.28, N 9.39, S 7.16; found C 67.35, H 4.11, N 9.34, S 6.94.
- 2-{[4-(4-Chlorophenyl)-6-phenylpyrimidin-2-yl]thio}-N-(m-tolyl)acetamide (2c). Yield: 20%. Mp: 204–206 °C. 1H NMR (200 MHz, CDCl3): δ (ppm) 2.19 (s, 3H, CH3), 4.09 (s, 2H, CH2), 6.81 (d, J = 7.6 Hz, 1H, Ar), 6.91 (s, 1H, Ar), 7.03–7.14 (m, 2H, Ar), 7.51–7.57 (m, 5H, Ar), 7.82 (s, 1H, H-5), 8.09–8.15 (m, 4H, Ar), 9.14 (br s, 1H, NH). 13C NMR (100 MHz, CDCl3): δ (ppm) 21.40, 35.95, 108.90, 116.68, 120.24, 124.98, 127.51, 128.80, 129.53, 131.73, 134.71, 136.25, 138.78, 164.43, 165.79, 167.27,171.19. Anal. calcd. for C25H20ClN3OS: C 67.33, H 4.52, N 9.42, S 7.19; found C 67.25, H 4.54, N 9.10, S 7.40.
- 2-{[4-(4-Fluorophenyl)-6-phenylpyrimidin-2-yl]thio}-N-(m-tolyl)acetamide (2d). Yield: 20%. Mp: 186–190 °C. 1H NMR (400 MHz, CDCl3): 2.14 (s, 3H, CH3), 4.03 (s, 2H, CH2), 6.79 (d, J = 7.5 Hz, 1H, Ar), 6.88 (s, 1H, Ar), 7.02–7.12 (m, 2H, Ar), 7.19–7.25 (m, 2H, Ar), 7.54–7.56 (m, 3H, Ar), 7.80 (s, 1H, H-5), 8.11–8.17 (m, 4H, Ar), 9.18 (br s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ (ppm) 21.40, 35.93, 108.80, 116.26, 116.48, 116.65, 120.22, 124.94, 127.48, 128.80, 129.35, 129.61, 129.70, 131.66, 136.30, 137.86, 138.76, 164.50, 165.66, 167.05 (d, JC-F = 254 Hz), 167.32, 171.09. Anal. calcd. for C25H20FN3OS: C 69.91, H 4.69, N 9.78, S 7.46; found C 69.75, H 4.82, N 10.05, S 7.41.
- 2-{[4-(4-Chlorophenyl)-6-(4-fluorophenyl)pyrimidin-2-yl]thio}-N-(m-tolyl)acetamide (2e). Yield: 23%. Mp: 213–217 °C. 1HNMR (400 MHz, DMSO-d6): 2.22 (s, 3H, CH3), 4.14 (s, 2H, CH2), 6.83 (d, J = 7.5 Hz, 1H, Ar), 7.16 (t, J = 7.8 Hz, 1H, Ar), 7.25 (m, 2H, Ar), 7.39 (d, J = 8.1 Hz, 1H, Ar), 7.42 (s, 1H, Ar), 7.48 (d, J = 8.6 Hz, 2H, Ar), 8.28–8.32 (m, 3H, 2H Ar + H-5), 8.34–8.38 (m, 2H, Ar), 10.29 (br s, 1H, NH). 13CNMR (101 MHz, DMSO-d6): δ (ppm) 21.68, 36.37, 108.66, 116.41, 116.76, 120.07, 124.50, 129.17, 129.38, 129.83, 130.65, 132.78, 135.16, 136.77, 138.45, 139.61, 163.64, 163.91, 164.71 (d, JC-F = 248 Hz), 166.88, 171.44. Anal. Calcd. For C25H19ClFN3OS: C 64.72, H 4.13, N 9.06, S 6.91; found C 64.88, H 4.39, N 9.38, S 7.18.
- 2-{[4-(Naphthalen-1-yl)-6-phenylpyrimidin-2-yl)thio)-N-(m-tolyl)acetamide (2f). Yield: 20%. Mp: 147–148 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 2.02 (s, 3H, CH3), 3.99 (s, 2H, CH2), 6.56 (s, 1H, Ar), 6.71–6.73 (m, 1H, Ar), 6.93–7.00 (m, 2H, Ar), 7.51–7.57 (m, 5H, Ar), 7.75 (dd, J = 7.1, 1.2 Hz, 1H, Ar), 7.77 (s, 1H, H-5), 7.96 (dd, J = 7.4, 2.1 Hz, 1H, Ar), 8.03 (d, J = 8.2 Hz, 1H, Ar), 8.13–8.16 (m, 2H, Ar,), 8.18–8.22 (m, 2H, Ar,), 9.32 (br s, 1H, NH).13C NMR (101 MHz, CDCl3): δ (ppm) 21.26, 36.04, 113.98, 116.49, 119.91, 124.72, 124.84, 125.43, 126.68, 127.54, 127.62, 128.13, 128.69, 128.90, 129.35, 130.55, 130.93, 131.76, 134.09, 135.60, 136.10, 137.93, 138.61, 165.24, 167.45, 167.79, 171.41. Anal. calcd. for C29H23N3OS: C 75.46, H 5.02, N 9.10, S 6.95; found C 75.23, H 5.29, N 8.81, S 6.68.
- 2-{[4-(4-Chlorophenyl)-6-(naphthalen-1-yl)pyrimidin-2-yl]thio}-N-(m-tolyl)acetamide (2g). Yield: 20%. Mp: 103–107 °C. 1H NMR (200 MHz, CDCl3): δ (ppm) 2.06 (s, 3H, CH3), 4.00 (s, 2H, CH2), 6.60 (s, 1H, Ar), 6.75–6.76 (m, 1H, Ar), 6.96–6.99 (m, 2H, Ar), 7.51–7.65 and 7.75–7.78 (2m, 7H, 6Ar + H-5), 7.98 (d, J = 8.0 Hz, 1H, Ar), 8.04–8.06 and 8.10–8.12 (2 m, 3H, Ar), 8.19 (d, J = 7.6 Hz, Ar), 9.25 (br s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ (ppm) 21.26, 36.05, 113.63, 116.49, 119.88, 124.73, 124.82, 125.43, 126.74, 127.61, 128.16, 128.73, 128.89, 128.95, 129.57, 130.49, 131.05, 134.09, 134.44, 135.46, 137.82, 138.13, 138.67, 164.04, 167.25, 167.91, 171.45. Anal. calcd. for C29H22ClN3OS: C 70.22, H 4.47, N 8.47, S 6.46; found 70.45, H 4.73, N 8.15, S 6.80.
- 2-{[4-(4-Fluorophenyl)-6-(1-naphthyl)pyrimidin-2-yl]thio}-N-(3-methylphenyl)acetamide (2h). Yield: 55%. Mp: 145–147 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 2.04 (s, 3H, CH3), 3.97 (s, 2H, CH2), 6.58 (br s, 1H, Ar), 6.74 (d, J = 6.0 Hz, 1H, Ar), 6.92–7.00 (m, 2H, Ar), 7.18–7.25 (m, 2H, Ar), 7.51–7.63 (m, 3H, Ar), 7.70–7.77 (m, 2H, 1H Ar + H-5), 7.94–8.00 (m, 1H, Ar), 8.03 (d, J = 8.2 Hz, 1H Ar), 8.13–8.21 (m, 3H Ar), 9.20 (br s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ (ppm) 21.26, 36.04, 113.50, 116.30, 116.49, 116.51, 119.89, 124.76, 124.79, 125.41, 126.70, 127.56, 128.11, 128.70, 128.93, 129.73, 129.82, 130.51, 130.98, 132.14, 132.18, 134.08, 135.52, 137.85, 138.64, 164.08, 165.13 (d, JC-F = 252 Hz), 167.27, 167.76, 171.34. Anal. calcd. for C29H22FN3OS: C 72.63, H 4.62, N 8.76, S 6.69; found C 72.49, H 4.36, N 8.42, S 6.93.
- 2-{[4-Phenyl-6-(phenylthio)pyrimidin-2-yl]thio}-N-(m-tolyl)acetamide (2i). Yield: 27%. Mp: 127–128 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 2.33 (s, 3H, CH3), 3.68 (s, 2H, CH2), 6.89 (d, J = 7.4 Hz, 1H, Ar), 7.18 (t, J = 7.8 Hz, 1H, Ar), 7.25–7.26 (m, 1H, Ar), 7.30–7.31 (m, 2H, 1H Ar + H-5), 7.35–7.49 (m, 6H, Ar), 7.63–7.70 (m, 2H Ar), 7.90 (dd, J = 8.3, 1.3 Hz, 2H Ar), 8.57 (br s, 1H, NH). 13C NMR (100 MHz, CDCl3): δ (ppm) 21.58, 33.57, 110.17, 117.15, 120.71, 125.12, 127.39, 128.77, 129.01, 129.17, 129.37, 129.76, 131.56, 135.42, 135.62, 137.79, 138.82, 162.84, 166.99, 169.30, 172.16. Anal. calcd. for C25H21N3OS2: C 67.69, H 4.77, N 9.47, S 14.46; found C 67.75, H 4.77, N 9.47, S 14.10.
- 2-{[4-(4-Chlorophenyl)-6-(phenylthio)pyrimidin-2-yl]thio}-N-(m-tolyl)acetamide (2j). Yield: 20%. Mp: 150–154 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 2.34 (s, 3H, CH3), 3.70 (s, 2H, CH2), 6.91 (d, J = 7.2 Hz, 1H, Ar), 7.17–7.21 and 7.25–7.31 (2 m, 4H, 3Ar + H-5), 7.37–7.46 (m, 5H, Ar), 7.67 (d, J = 7.2 Hz, 2H, Ar), 7.85 (d, J = 8.4 Hz, 2H, Ar), 8.61 (br s, 1H, NH).13C NMR (101 MHz, CDCl3): δ (ppm) 21.59, 33.58, 109.91, 117.15, 120.71, 125.17, 128.65, 128.80, 129.30, 129.42, 129.86, 133.87, 135.66, 137.75, 137.89, 138.86, 161.57, 166.88, 169.61, 172.35. Anal. calcd. for C25H20ClN3OS2: C 62.82, H 4.22, N 8.79, S 13.41; found C 62.51, H 4.38, N 9.00, S 13.46.
- 2-{[4-(4-Fluorophenyl)-6-(phenylthio)pyrimidin-2-yl]thio}-N-(m-tolyl))acetamide (2k). Yield: 20%. Mp: 146–147 °C. 1H NMR (200 MHz, CDCl3): δ (ppm) 2.34 (s, 3H, CH3), 3.70 (s, 2H, CH2), 6.91 (d, J = 7.2 Hz, 1H, Ar), 7.10–7.13 and 7.17–7.21 (2m, 3H, Ar), 7.25–7.27 (m, 2H, Ar), 7.31 (s, 1H, H-5), 7.37–7.46 (m, 3H, Ar), 7.67 (d, J = 7.2 Hz, 2H, Ar), 7.90–7.93 (m, 2H, Ar), 8.53 (br s, 1H, NH).13C-NMR (101 MHz, CDCl3): δ (ppm) 21.59, 33.57, 116.03, 116.25,. 117.15, 120.71, 125.16, 128.80, 129.07, 129.42, 129.50, 129.59, 129.84, 130.99, 131.59, 135.66, 137.77, 138.86, 161.68, 165.00 (d, JC-F = 254 Hz), 166.94, 169.45, 172.25. Anal. calcd. for C25H20FN3OS2: C 65.05, H 4.37, N 9.10, S 13.89; found C 65.03, H 4.31, N 9.28, S 13.89.
- 2-{[2-oxo-2-(m-tolylamino)ethyl]thio}-N,6-diphenylpyrimidine-4-carboxamide (2l). Yield: 62%. Mp: 181–182 °C. H1 NMR (400 MHz, DMSO-d6): δ (ppm) 2.19 (s, 3H, CH3), 4.21 (s, 2H, CH2), 6.83 (d, J = 7.6 Hz, 1H, Ar), 7.13–7.19 (m, 3H, Ar), 7.36–7.43 and 7.46–7.50 (2m, 5H, Ar), 7.57 (t, J = 7.2 Hz, 1H, Ar), 7.88 (d, J = 8.0 Hz, 2H, Ar), 8.24–8.26 (m, 3H, 2H Ar + H-5), 10.29 and 10.52 (2 br s, 1H, 2 NH). 13C NMR (101 MHz, DMSO-d6): δ (ppm) 21.63, 36.58, 110.30, 116.98, 120.34, 121.08, 124.70, 125.15, 128.13, 129.14, 129.36, 129.65, 132.54, 135.61, 138.31, 138.48, 139.36, 158.89, 161.51, 166.33, 167.41, 171.26. Anal. calcd. for C26H22N4O2S: C 68.70, H 4.88, N 12.33, S 7.05; found C 68.40, H 5.18, N 12.14, S 6.70.
2.1.16. General Procedure for the Synthesis of Compounds 2m,n
- 2-{[4-Phenyl-6-(phenylthio)pyridin-2-yl]thio}-N-(m-tolyl)acetamide (2m). Yield: 20%. Mp: 122–123 °C. 1H NMR (200 MHz, CDCl3): δ (ppm) 2.33 (s, 3H, CH3), 3.76 (s, 2H, CH2), 6.89 (d, J = 7.2 Hz, 1H, Ar), 7.00 (s, 1H, Ar), 7.17–7.21 (m, 2H, Ar), 7.39–7.45 and 7.62–7.63 (2m, 12H, 10H Ar + 2H pyr), 9.34 (br s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ (ppm) 21.41, 34.58, 116.31. 116.68, 116.88, 120.45, 124.65, 126.94, 128.58, 129.10, 129.60, 129.74, 129.87, 135.00, 136.98, 138.16, 138.59, 146.18, 150.29, 161.14, 167.50, 168.05. Anal. calcd. for C26H22N2OS2: C 70.56, H 5.01, N 6.33, S 14.49; found C 70.52, H 5.31, N 6.07, S 14.84.
- 2-{[4-(4-chlorophenyl)-6-(phenylthio)pyridin-2-yl]thio}-N-(m-tolyl)acetamide (2n). Yield: 20%. Mp: 93–95 °C1H NMR (200 MHz, CDCl3): δ (ppm) 2.32 (s, 3H, CH3), 3.71 (s, 2H, CH2), 6.87–6.91 (m, 2H, Ar), 7.12 (s, 1H, Ar), 7.17 (t, J = 7.16 Hz, 1H, Ar), 7.35–7.51 (m, 9H, 7H Ar + 2H pyr), 7.61 (dd, J = 7.6, 1.6 Hz, 2H, Ar) 9.19 (br s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ (ppm) 167.97, 161.68, 158.67, 148.98, 138.78, 138.27, 135.99, 135.63, 135.18, 129.90, 129.80, 129.49, 128.75, 128.43, 128.33, 124.81, 120.54, 118.49, 116.95, 116.34, 34.52, 21.60. Anal. calcd. for C26H21ClN2OS2 C 65.46, H 4.44, N 5.87, S 13.44, found C 65.13, H 4.64, N 6.20, S 13.58.
2.1.17. General Procedure for the Synthesis of Compounds 15a,b
- Ethyl 2-{[4,6-bis(4-fluorophenyl)pyrimidin-2-yl]thio}acetate (15a). Yield: 68%.1H NMR (400 MHz, CDCl3): δ (ppm) 1.25 (t, J = 7.1 Hz, 3H, CH3), 3.99 (d, J = 1.3 Hz, 2H, S-CH2), 4.19 (q, J = 7.1 Hz, 2H, O-CH2), 7.17–7.23 (m, 4H, pF-Ar), 7.67 (d, J = 1.3 Hz, 1H, CH-Pyr), 8.18–8.10 (m, 4H, pF-Ar). Anal. calcd. for C20H16F2N2O2S: C, 62.17; H, 4.17; N, 7.25; S, 8.30; found C, 62.20; H, 4.17; N, 7.24; S, 8.23.
- Ethyl 2-{[4-(4-fluorophenyl)-6-phenylpyrimidin-2-yl]thio}acetate (15b). Yield: 76%. 1H NMR (400 MHz, CDCl3): δ (ppm) 1.24 (t, J = 7.1 Hz, 3H, CH3), 4.01 (s, 2H, S-CH2), 4.19 (q, J = 7.1 Hz, 2H, O-CH2), 7.08–7.25 (m, 2H, Ar), 7.48–7.52 (m, 3H, Ar), 7.73 (s, 1H, CH-Pyr), 8.10–8.16 (m, 4H, pF-Ar). Anal. calcd. for C20H17FN2O2S: C, 65.20; H, 4.65; N, 7.60; S, 8.70; found C, 65.23; H, 4.62; N, 7.63; S, 8.85.
2.1.18. General Procedure for the Synthesis of Compounds 15c,d
- Ethyl 2-{[4,6-diphenylpyridin-2-yl]thio}acetate (15c). Yield: 25%. 1H NMR (400 MHz, CDCl3): δ (ppm) 1.25 (t, J = 7.1 Hz, 3H, CH3), 4.08 (s, 2H, S-CH2), 4.20 (q, J = 7.1 Hz, 2H, O-CH2), 7.37 (d, J = 1.4 Hz, 1H, CH-Py), 7.40–7.53 (m, 6H, Ar), 7.58–7.80 (m, 3H, Ar + CH-Py), 8.00–8.18 (m, 2H, Ar). Anal. calcd. for C21H19NO2S: C, 72.18; H, 5.48; N, 4.01; S, 9.17; found C, 72.20; H, 5.50; N, 3.98; S, 9.22.
- Ethyl 2-{[6-(4-fluorophenyl)-4-phenylpyridin-2-yl]thio}acetate (15d). Yield: 40%.1H NMR (400 MHz, DMSO-d6): δ (ppm) 1.12 (t, J = 7.1 Hz, 3H, CH3), 4.06 (q, J = 7.1 Hz, 2H, O-CH2), 4.10 (s, 2H, S-CH2), 7.30 (t, J = 8.8 Hz, 2H, Ar), 7.40–7.55 (m, 3H, Ar), 7.59 (d, J = 1.3 Hz, 1H, CH-Py), 7.88 (dd, J = 8.0, 1.6 Hz, 2H, pF-Ar), 7.94 (d, J = 1.3 Hz, 1H, CH-Py), 8.27–8.19 (m, 2H, pF-Ar). Anal. calcd. for C21H18FNO2S: C, 68.65; H, 4.94; N, 3.81; S, 8.73; found C, 68.67; H, 4.96; N, 3.84; S, 8.75.
2.1.19. General Procedure for the Synthesis of Compounds 16a–d
2.1.20. General Procedure for the Synthesis of Compounds 3a–n
- Methyl (2-((4-(4-fluorophenyl)-6-phenylpyrimidin-2-yl)thio)acetyl)-L-valinate (3a). Yield: 48%. Mp: 152–153 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 0.58 (d, J = 6.8 Hz, 3H, CH3-Val), 0.68 (d, J = 6.8 Hz, 3H, CH3-Val), 1.94–1.99 (m, 1H, CH-Val), 3.53 (s, 3H, OCH3), 3.92–4.03 (m, 2H, S-CH2), 4.47–4.51 (m, 1H, CH-Val), 7.18–7.23 (m, 2H, Ar), 7.37 (d, J = 9.0 Hz, 1H, C=O-NH), 7.51–7.54 (m, 3H, Ar), 7.80 (s, 1H, CH-Pyr), 8.11–8.18 (m, 4H, Ar). 13C NMR (101 MHz, DMSO-d6) δ18.67, 19.35, 30.66, 34.84, 52.20, 58.28, 108.61, 116.29, 116.50, 128.04, 129.44, 130.65, 131.99, 132.90, 136.32, 163.50, 163.71, 164.74 (d, JC-F = 248 Hz), 164.85, 165.98, 168.46, 171.19, 172.39. Anal. calcd. for C24H24FN3O3S: C, 63.56; H, 5.33; N, 9.27; S, 7.07; found C, 63.56; H, 5.33; N, 9.28; S, 7.07.
- Methyl (2-((4-(4-fluorophenyl)-6-phenylpyrimidin-2-yl)thio)acetyl)-L-isoleucinate (3b). Yield: 60%. Mp: 94–95 °C.1H NMR (400 MHz, CDCl3): δ (ppm) 0.57–0.65 (m, 6H, (CH3)2-Iso), 0.73–0.88(m, 2H, CH2-Iso), 1.08–1.18 (m,1H, CH-Iso), 3.53 (s, 3H, OCH3), 3.90 (d, J = 15.7 Hz, 1H, S-CH), 4.01 (d, J = 15.7 Hz, 1H, S-CH), 4.49–4.53 (m, 1H), 7.19–7.23 (m, 2H, Ar), 7.38 (d, J = 8.9 Hz, 1H, C=O-NH), 7.51–7.55 (m, 3H, Ar), 7.80 (s, 1H, CH-Pyr), 8.11–8.18 (m, 4H, pF-Ar). 13C NMR (101 MHz, DMSO-d6) δ 11.57, 15.77, 25.23, 34.83, 37.09, 52.16, 57.23, 108.62, 116.40, 128.04, 129.44, 130.61, 131.99, 132.87, 136.31, 163.72, 164.75 (d, J C-F = 249.0 Hz), 164.85, 168.37, 171.17, 172.37. Anal. calcd. for C25H26FN3O3S: C, 64.22; H, 5.61; N, 8.99; S, 6.86; found C, 64.27; H, 5.58; N, 9.01; S, 6.87.
- Methyl (2-((4-(4-fluorophenyl)-6-phenylpyrimidin-2-yl)thio)acetyl)-L-tyrosinate (3c). Yield: 30%. Mp: 167–168 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 2.79 (dd, J = 14.1, 6.8 Hz, 1H, CH-Tyr), 2.90 (dd, J = 14.1, 5.4 Hz, 1H, CH-Tyr), 3.56 (s, 3H, OCH3), 3.84–3.94 (m, 2H, S-CH2), 4.76–4.81 (m, 1H, CH), 6.33 (d, J = 8.5 Hz, 2H, Ar-Tyr), 6.65 (m, 2H, d, J = 8.5 Hz, 2H, Ar-Tyr), 7.17–7.21 (m, 2H, Ar), 7.32 (d, J = 8.0 Hz, 1H, C=O-NH), 7.52–7.53 (m, 3H, Ar), 7.77 (s, 1H, CH-Pyr), 8.14–8.09 (m, 4H, Ar). 13C NMR (101 MHz, CDCl3) δ 34.68, 36.86, 52.33, 53.48, 108.42, 115.30, 116.13, 116.35, 127.41, 129.20, 129.62, 130.01, 131.54, 132.39, 136.18, 154.55, 164.06, 164.95 (d, JC-F = 251.9 Hz), 165.24, 169.00, 170.37, 171.72. Anal. calcd. for C28H24FN3O4S: C, 64.98; H, 4.67; N, 8.12; S, 6.19; found C 65.00; H, 4.68; N, 8.12; S, 6.20.
- Methyl (2-((4-(4-fluorophenyl)-6-phenylpyrimidin-2-yl)thio)acetyl)-L-histidinate (3d). Yield: 55%. Mp: 77–78 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 2.81–2.86 (m, 2H, CH2-His), 3.44 (s, 3H, OCH3), 4.01 (s, 2H, S-CH2), 4.42–4.55(m, 1H, CH), 6.73 (d, J = 1.2 Hz, 1H, CH-ArHis), 7.29–7.39 (m, 2H, Ar), 7.47 (d, J = 1.2 Hz, 1H, CH-ArHis), 7.51–7.56 (m, 3H, Ar), 7.63 (d, J = 8.4 Hz, 1H, C=O-NH), 7.90 (d, J = 7.5 Hz, 1H, NH-His tautomer), 8.24–8.33 (m, 3H, Ar and CH-Pyr), 8.35–8.43 (m, 2H, pF-Ar), 8.64 (d, J = 7.5 Hz, 1H, NH-His). 13C NMR (101 MHz, DMSO-d6) δ 29.50, 34.89, 52.29, 53.25, 108.63, 110.36, 116.29, 119.50, 124.69, 127.16, 128.03, 129.44, 130.58, 131.98, 135.37, 136.33, 163.69, 164.74 (d, JC-F = 249.0 Hz), 164.83, 168.16, 170.94, 172.23. Anal. calcd. for C25H22FN5O3S: C, 61.09; H, 4.51; N, 14.25; S, 6.52; found C, 61.10; H, 4.50; N, 14.27; S, 6.52.
- Methyl (2-((4,6-bis(4-fluorophenyl)pyrimidin-2-yl)thio)acetyl)-L-valinate (3e). Yield: 60%. Mp: 174–175 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 0.77 (t, J = 6.4 Hz, 6H, (CH3)2-Val), 1.92–2.01 (m, 1H, CH-Val), 3.53 (s, 3H, OCH3), 4.01–4.10 (m, 2H, S-CH2), 4.14–4.19 (m, 1H, CH), 7.33–7.38 (m, 4H, pF-Ar), 8.29 (s, 1H, CH-Pyr), 8.36–8.48 (m, 4H, Ar). 13C NMR (101 MHz, DMSO-d6) δ 18.65, 19.34, 30.66, 34.84, 52.19, 58.26, 108.44, 116.40, 130.61, 132.82, 163.75, 164.77 (d, JC-F = 249.5 Hz), 168.45, 171.19, 172.39. Anal. calcd. for C24H23F2N3O3S: C, 61.13; H, 4.92; N, 8.91; S, 6.80; found C, 61.00; H, 4.90; N, 8.89; S, 6.81.
- Methyl (2-((4,6-bis(4-fluorophenyl)pyrimidin-2-yl)thio)acetyl)-L-isoleucinate (3f). Yield: 64%. Mp: 150–151 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 0.61–0.64 (m, 6H, (CH3)2-Iso), 0.76–0.87 (m, 2H, CH2-Iso), 1.09–1.19 (m, 1H, CH-Iso), 3.54 (s, 3H, OCH3), 3.89 (d, J = 15.8 Hz, 1H, S-CH), 4.00 (d, J = 15.8 Hz, 1H, S-CH), 4.50–4.53 (m, 1H, CH), 7.18–7.24 (m, 4H, Ar), 7.33 (d, J = 9.1 Hz, 1H, C=O-NH), 7.75 (s, 1H, CH-Pyr), 8.13–8.18 (m, 4H, Ar). 13C NMR (101 MHz, DMSO-d6) δ 11.54, 15.76, 25.20, 34.83, 37.09, 52.16, 57.22, 116.29, 116.50, 130.58, 130.66, 132.82, 163.76, 164.77 (d, JC-F = 249.5 Hz), 168.37, 171.17, 172.37. Anal. calcd. for C25H25F2N3O3S: C, 61.84; H, 5.19; N, 8.65; S, 6.60; found C, 61.53; H, 5.25; N, 8.80; S, 6.40.
- Methyl (2-((4,6-bis(4-fluorophenyl)pyrimidin-2-yl)thio)acetyl)-L-tyrosinate (3g). Yield: 35%. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 2.72–2.85 (m, 2H, CH2-Tyr), 3.45 (s, 3H, OCH3), 3.99 (d, J = 2.9 Hz, 2H, S-CH2), 4.35–4.41 (m, 1H, CH), 6.5–6.52 (m, 2H, Ar-Tyr), 6.84–6.86 (m, 2H, Ar-Tyr), 7.34–7.38 (m, 4H, Ar), 8.30 (s, 1H, CH-Pyr), 8.38–8.41 (m,4H, Ar), 8.55 (d, J = 7.6 Hz, 1H, C=O-NH), 9.16 (d, J = 1.0 Hz, 1H, OH). 13C NMR (101 MHz, DMSO-d6) δ 34.77, 36.85, 52.37, 53.46, 108.06, 115.30, 116.18, 116.39, 127.47, 129.63, 130.03, 132.21, 154.53, 164.10, 165.00 (d, JC-F = 252.9 Hz), 168.84, 170.33, 171.73. Anal. calcd. for C28H23F2N3O4S C, 62.79; H, 4.33; N, 7.85; S, 5.99; found C, 62.83; H, 4.37; N, 7.86; S, 6.01.
- Methyl (2-((4,6-bis(4-fluorophenyl)pyrimidin-2-yl)thio)acetyl)-L-histidinate (3h). Yield: 43%. Mp: 107–108 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 2.76–2.92(m, 2H, CH2-His), 3.45 (s, 3H, OCH3), 4.00 (s, 2H, S-CH2), 4.44–4.54 (m, 1H, CH), 6.72 (d, J = 1.4 Hz, 1H, CH-ArHis), 7.36 (t, J = 8.9 Hz, 4H, Ar), 7.46 (d, J = 1.4 Hz, 1H, CH-ArHis), 7.61 (d, J = 8.3 Hz, 1H, C=O-NH), 7.89 (d, J = 8.4, 1H, NH-His tautomer), 8.30 (s, 1H, CH-Pyr), 8.35–8.47 (m, 4H, Ar). 13C NMR (101 MHz, DMSO-d6) δ 29.43, 34.89, 52.30, 53.21, 110.36, 116.40, 119.49, 124.68, 127.13, 130.58, 132.84, 135.36, 163.72, 164.75 (d, J = 249.0 Hz), 168.18, 170.93, 172.22. nal. calcd. for C25H21F2N5O3S: C, 58.93; H, 4.15; N, 13.74; S, 6.29; found C, 59.01; H, 4.17; N, 14.01; S, 6.34.
- Methyl (2-((4,6-diphenylpyridin-2-yl)thio)acetyl)-L-valinate (3i). Yield: 22%. Mp: 63–64 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 0.75–0.79 (m, 6H, (CH3)2), 1.90–1.99 (m, 1H, CH-Val), 3.53 (s, 3H, OCH3), 4.01–4.10 (m, 2H, S-CH2), 4.15–4.18 (m, 1H, CH), 7.42–7.52 (m, 6H, Ar), 7.59 (d, J = 1.4 Hz, 1H, CH-Py), 7.87–7.89 (m, 2H, Ar), 7.92 (d, J = 1.3 Hz, 1H, CH-Py), 8.18–8.20 (m, 2H, Ar), 8.40 (d, J = 8.2 Hz, 1H, C=O-NH). 13C NMR (101 MHz, DMSO-d6): δ (ppm) 17.94, 30.88, 34.53, 53.23, 56.29, 116.84, 122.47, 128.00, 128.16, 128.26, 129.42, 129.69, 129.91, 137.65, 138.57, 147.39, 156.02, 157.06, 168.68, 173.33. Anal. calcd. for C25H26N2O3S C, 69.10; H, 6.03; N, 6.45; S, 7.38; found C, 69.13; H, 6.02; N, 6.45; S, 7.40.
- Methyl (2-((4,6-diphenylpyridin-2-yl)thio)acetyl)-L-isoleucinate (3j). Yield: 25%. Mp: 70–71 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 0.65–0.71 (m, 6H, 2xCH3, 0.97–1.06 (m, 1H, CH-Iso), 1.19–1.29 (m, 1H, CH-Iso), 1.63–1.70 (m, 1H, CH-Iso), 3.52 (s, 3H, OCH3), 3.99–4.12 (m, 2H, S-CH2), 4.18–4.21 (m, Hz, 1H CH-Iso), 7.43–7.52 (m, 6H, Ar), 7.58 (d, J = 1.4 Hz, 1H, CH-Py), 7.87–7.89 (m, 2H, Ar), 7.93 (d, J = 1.4 Hz, 1H, CH-Py), 8.18–8.20 (m, 2H, Ar), 8.39 (d, J = 8.1 Hz, 1H, C=O-NH). 13C NMR (101 MHz, DMSO-d6) δ 11.60, 15.78, 25.19, 33.67, 37.07, 52.17, 57.18, 114.70, 117.79, 127.47, 127.74, 129.22, 129.64, 129.90, 130.02, 137.55, 138.66, 149.46, 157.02, 158.93, 168.72, 172.30. Anal. calcd. for C26H28N2O3S: C, 69.62; H, 6.29; N, 6.25; S, 7.15; found C, 69.64; H, 6.29; N, 6.23; S, 7.15.
- Methyl (2-((4,6-diphenylpyridin-2-yl)thio)acetyl)-L-tyrosinate (3k). Yield: 27%. Mp: 111–112 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 2.69–2.85 (m, 2H, CH2-Tyr), 3.46 (s, 3H, OCH3), 4.00 (s, 2H, S-CH2), 4.35–4.40 (m, 1H, CH-Tyr), 6.53 (d, J = 8.4 Hz, 2H, Ar-Tyr), 6.84(d, J = 8.4 Hz, 2H, Ar-Tyr), 7.41–7.53 (m, 6H, Ar), 7.57 (d, J = 1.4 Hz, 1H, CH-Py), 7.87–7.90 (m, 2H, Ar), 7.94 (d, J = 1.4 Hz, 1H, CH-Py), 8.18–8.20 (m, 2H, Ar), 8.53 (d, J = 7.6 Hz, 1H, C=O-NH), 9.17 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6) δ 33.64, 36.71, 52.28, 54.79, 114.72, 115.56, 117.85, 127.26, 127.45, 127.76, 129.23, 129.66, 129.92, 130.01, 130.45, 137.58, 138.65, 149.45, 156.53, 156.95, 158.71, 168.46, 172.33. Anal. calcd. for C29H26N2O4S: C, 69.86; H, 5.26; N, 5.62; S, 6.43; found C, 69.87; H, 5.26; N, 5.61; S, 6.42.
- Methyl (2-((6-(4-fluorophenyl)-4-phenylpyridin-2-yl)thio)acetyl)-L-isoleucinate (3l). Yield: 27%. Mp: 76–77 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 0.55–0.75 (m, 6H (CH3)2), 0.95–1.07 (m, 1H, CH-Iso), 1.19–1.29 (m, 1H, CH-Iso), 1.63–1.70 (m, 1H, CH), 3.53 (s, 3H, OCH3), 4.00 (d, J = 15.1 Hz, 1H, S-CH), 4.10 (d, J = 15.1 Hz, 1H, S-CH), 4.19–4.21 (m, 1H, CH), 7.25–7.31 (m, 2H, Ar), 7.44–7.52 (m, 3H, Ar), 7.58 (d, J = 1.3 Hz, 1H, CH-Py), 7.80–7.92 (m, 2H, Ar), 7.94 (d, J = 1.3 Hz, 1H, CH-Py), 8.25–8.29 (m, 2H, pF-Ar), 8.40 (d, J = 8.2 Hz, 1H, C=O-NH). 13C NMR (101 MHz, DMSO-d6) δ 11.58, 15.76, 25.15, 33.67, 37.07, 52.18, 57.16, 114.53, 116.02, 117.69, 127.77, 129.63, 130.05, 135.18, 137.47, 149.49, 155.97, 158.96, 163.58 (d, J C-F = 246.6 Hz), 168.70, 172.34. Anal. calcd. for C26H27FN2O3S: C, 66.93; H, 5.83; N, 6.00; S, 6.87; found C, 67.00; H, 5.82; N, 6.05; S, 6.89.
- Methyl (2-((6-(4-fluorophenyl)-4-phenylpyridin-2-yl)thio)acetyl)-L-tyrosinate (3m). Yield: 30%. Mp: 151–152 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 2.71–2.83 (m, 2H, CH2-Tyr), 3.47 (s, 3H, OCH3), 3.99 (d, J = 1.5 Hz, 2H, S-CH2), 4.35–4.40(m, 1H, CH), 6.53 (d, J = 8.4 Hz, 2H, Ar-Tyr), 6.84 (d, J = 8.0 Hz, 2H, Ar-Tyr), 7.28 (t, J = 8.8 Hz, 2H, Ar), 7.44–7.53 (m, 3H, Ar), 7.57 (d, J = 1.3 Hz, 1H, CH-Py), 7.87–7.90 (m, 2H, Ar), 7.94 (d, J = 1.4 Hz, 1H, CH-Py), 8.24–8.27(m, 2H, pF-Ar), 8.51 (d, J = 7.6 Hz, 1H, C=O-NH), 9.17 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6) δ 33.64, 36.69, 52.28, 54.76, 114.56, 115.55, 115.92, 116.13, 117.75, 127.27, 127.78, 129.77, 130.04, 130.44, 135.14, 137.51, 149.50, 155.91, 156.53, 158.73, 163.58 (d, JC-F = 246.6 Hz), 168.46, 172.34. Anal. calcd. for C29H25FN2O4S: C, 67.43; H, 4.88; N, 5.42; S, 6.21; found C, 67.48; H, 4.92; N, 5.42; S, 6.23.
- Methyl (2-((6-(4-fluorophenyl)-4-phenylpyridin-2-yl)thio)acetyl)-L-histidinate (3n). Yield: 20%. Mp: 155–156 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 2.74–2.90 (m, 2H, CH2-His), 3.45 (s, 3H, OCH3), 4.00 (s, 2H, S-CH2), 4.28–4.60 (m, 1H, CH), 6.70 (s, 1H, Ar-His), 7.16–7.32 (m, 2H, Ar), 7.44 (d, J = 1.2 Hz, 1H, Ar-His), 7.46–7.54 (m, 3H, Ar), 7.57 (d, J = 1.3 Hz, 1H, CH-Py), 7.81–7.92 (m, 2H, pF-Ar), 7.93 (d, J = 1.3 Hz, 1H, CH-Py), 8.18–8.33 (m, 2H, Ar), 8.61 (d, J = 7.4 Hz, 1H, NH-His). 13C NMR (101 MHz, DMSO-d6): δ 28.76, 34.49, 52.44, 52.83, 116.14, 116.80, 119.18, 122.50, 128.16, 128.70, 129.35, 129.42, 129.91, 133.75, 135.28, 137.64, 147.09, 155.87, 157.47, 162.62 (d, JC-F = 246.6 Hz), 168.36, 172.40. Anal. calcd. for C26H23FN4O3S C, 63.66; H, 4.73; N, 11.42; S, 6.54; found C, 63.68; H, 4.74; N, 11.40; S, 6.52.
2.2. Biology
2.3. Molecular Modeling
3. Results and Discussion
3.1. Chemistry
3.2. Biology
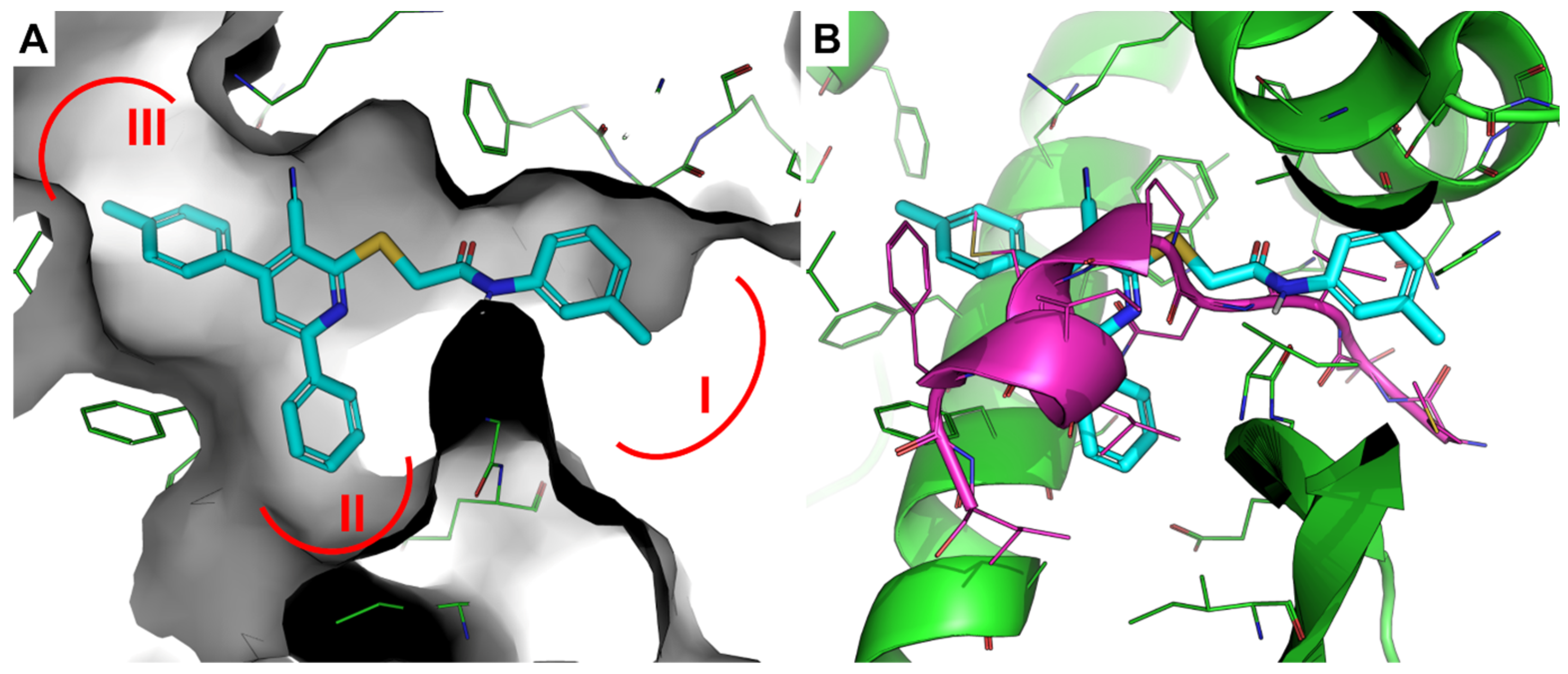
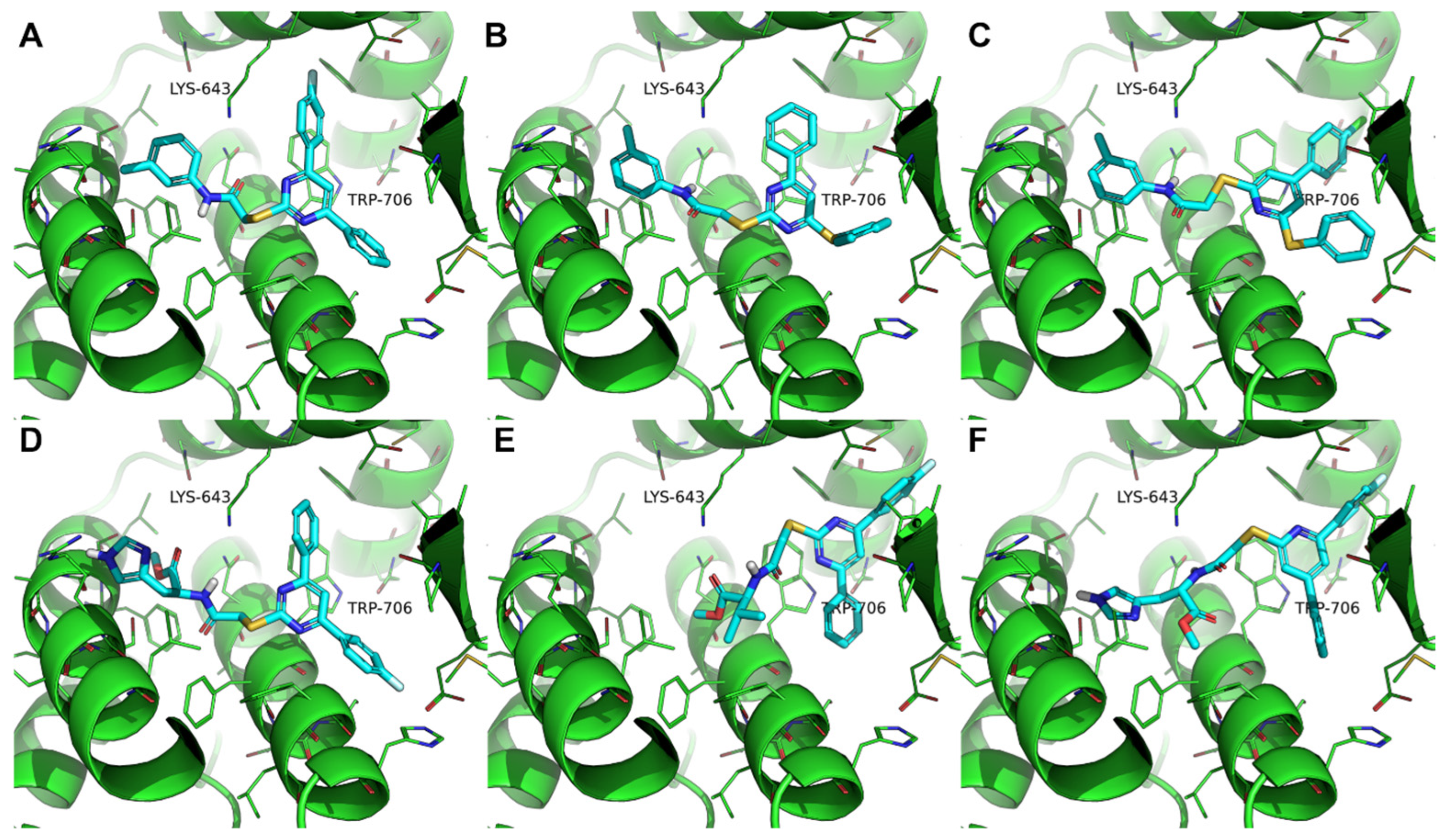
3.3. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Venkataraman, S.; Prasad, B.V.L.S.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Uyeki, T.M.; Hui, D.S.; Zambon, M.; Wentworth, D.E.; Monto, A.S. Influenza. Lancet 2022, 400, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F.; Smith, G.; Fouchier, R.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Asha, K.; Khanna, M.; Ronsard, L.; Meseko, C.A.; Sanicas, M. The Emerging Influenza Virus Threat: Status and New Prospects for Its Therapy and Control. Arch. Virol. 2018, 163, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Giacchello, I.; Musumeci, F.; D’Agostino, I.; Greco, C.; Grossi, G.; Schenone, S. Insights into RNA-Dependent RNA Polymerase Inhibitors as Antiinfluenza Virus Agents. Curr. Med. Chem. 2021, 28, 1068–1090. [Google Scholar] [CrossRef] [PubMed]
- Angeletti, D.; Yewdell, J.W. Is It Possible to Develop a “Universal” Influenza Virus Vaccine?: Outflanking Antibody Immunodominance on the Road to Universal Influenza Vaccination. Cold Spring Harb. Perspect. Biol. 2018, 10, a028852. [Google Scholar] [CrossRef] [PubMed]
- Nuwarda, R.F.; Alharbi, A.A.; Kayser, V. An Overview of Influenza Viruses and Vaccines. Vaccines 2021, 9, 1032. [Google Scholar] [CrossRef] [PubMed]
- Lampejo, T. Influenza and Antiviral Resistance: An Overview. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Michiels, B.; van Puyenbroeck, K.; Verhoeven, V.; Vermeire, E.; Coenen, S. The Value of Neuraminidase Inhibitors for the Prevention and Treatment of Seasonal Influenza: A Systematic Review of Systematic Reviews. PLoS ONE 2013, 8, e6034. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, N.S.H.N.; Poongavanam, V.; Pratheepa, V. Viral M2 Ion Channel Protein: A Promising Target for Anti-Influenza Drug Discovery. Mini-Rev. Med. Chem. 2022, 15, 819–830. [Google Scholar] [CrossRef]
- Pathania, S.; Rawal, R.K.; Singh, P.K. RdRp (RNA-Dependent RNA Polymerase): A Key Target Providing Anti-Virals for the Management of Various Viral Diseases. J. Mol. Struct. 2022, 1250, 131756. [Google Scholar] [CrossRef] [PubMed]
- Hengrung, N.; El Omari, K.; Serna Martin, I.; Vreede, F.T.; Cusack, S.; Rambo, R.P.; Vonrhein, C.; Bricogne, G.; Stuart, D.I.; Grimes, J.M.; et al. Crystal Structure of the RNA-Dependent RNA Polymerase from Influenza C Virus. Nature 2015, 527, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.; Guilligay, D.; Pflug, A.; Malet, H.; Berger, I.; Crepin, T.; Hart, D.; Lunardi, T.; Nanao, M.; Ruigrok, R.W.H.; et al. Structural Insight into Cap-Snatching and RNA Synthesis by Influenza Polymerase. Nature 2014, 516, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Loregian, A.; Mercorelli, B.; Nannetti, G.; Compagnin, C.; Palù, G. Antiviral Strategies against Influenza Virus: Towards New Therapeutic Approaches. Cell Mol. Life Sci. 2014, 71, 3659–3683. [Google Scholar] [CrossRef] [PubMed]
- Palù, G.; Loregian, A. Inhibition of Herpesvirus and Influenza Virus Replication by Blocking Polymerase Subunit Interactions. Antivir. Res. 2013, 99, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Massari, S.; Goracci, L.; Desantis, J.; Tabarrini, O. Polymerase Acidic Protein-Basic Protein 1 (PA-PB1) Protein-Protein Interaction as a Target for next-Generation Anti-Influenza Therapeutics. J. Med. Chem. 2016, 59, 7699–7718. [Google Scholar] [CrossRef] [PubMed]
- Massari, S.; Desantis, J.; Nizi, M.G.; Cecchetti, V.; Tabarrini, O. Inhibition of Influenza Virus Polymerase by Interfering with Its Protein-Protein Interactions. ACS Infect. Dis. 2021, 7, 1332–1350. [Google Scholar] [CrossRef] [PubMed]
- Loregian, A.; Palù, G. How Academic Labs Can Approach the Drug Discovery Process as a Way to Synergize with Big Pharma. Trends Microbiol. 2013, 21, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Muratore, G.; Goracci, L.; Mercorelli, B.; Foeglein, Á.; Digard, P.; Cruciani, G.; Palù, G.; Loregian, A. Small Molecule Inhibitors of Influenza A and B Viruses That Act by Disrupting Subunit Interactions of the Viral Polymerase. Proc. Natl. Acad. Sci. USA 2012, 109, 6247–6252. [Google Scholar] [CrossRef] [PubMed]
- Stevaert, A.; Naesens, L. The Influenza Virus Polymerase Complex: An Update on Its Structure, Functions, and Significance for Antiviral Drug Design. Med. Res. Rev. 2016, 36, 1127–1173. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liu, T.; Zhang, J.; Zhan, P.; Liu, X. Influenza A Virus Polymerase: An Attractive Target for next-Generation Anti-Influenza Therapeutics. Drug Discov. Today 2018, 23, 503–518. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, I.; Giacchello, I.; Nannetti, G.; Fallacara, A.L.; Deodato, D.; Musumeci, F.; Grossi, G.; Palù, G.; Cau, Y.; Trist, I.M.; et al. Synthesis and Biological Evaluation of a Library of Hybrid Derivatives as Inhibitors of Influenza Virus PA-PB1 Interaction. Eur. J. Med. Chem. 2018, 157, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Trist, I.M.L.; Nannetti, G.; Tintori, C.; Fallacara, A.L.; Deodato, D.; Mercorelli, B.; Palù, G.; Wijtmans, M.; Gospodova, T.; Edink, E.; et al. 4,6-Diphenylpyridines as Promising Novel Anti-Influenza Agents Targeting the PA-PB1 Protein-Protein Interaction: Structure-Activity Relationships Exploration with the Aid of Molecular Modeling. J. Med. Chem. 2016, 59, 2688–2703. [Google Scholar] [CrossRef] [PubMed]
- Tintori, C.; Laurenzana, I.; Fallacara, A.L.; Kessler, U.; Pilger, B.; Stergiou, L.; Botta, M. High-Throughput Docking for the Identification of New Influenza A Virus Polymerase Inhibitors Targeting the PA-PB1 Protein-Protein Interaction. Bioorg. Med. Chem. Lett. 2014, 24, 280–282. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhou, J.; Bartlam, M.; Zhang, R.; Ma, J.; Lou, Z.; Li, X.; Li, J.; Joachimiak, A.; Zeng, Z.; et al. Crystal Structure of the Polymerase PA(C)-PB1(N) Complex from an Avian Influenza H5N1 Virus. Nature 2008, 454, 1123–1126. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.H.; Journet, M.; Humphrey, G. A Highly Regioselective Amination of 6-Aryl-2,4-Dichloropyrimidine. Org. Lett. 2006, 8, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.K.; Tsai, Y.L.; Chang, M.Y. Bi(OTf)3 Catalyzed Disproportionation Reaction of Cinnamyl Alcohols. Tetrahedron 2017, 73, 3368–3376. [Google Scholar] [CrossRef]
- Nannetti, G.; Massari, S.; Mercorelli, B.; Bertagnin, C.; Desantis, J.; Palù, G.; Tabarrini, O.; Loregian, A. Potent and Broad-Spectrum Cycloheptathiophene-3-Carboxamide Compounds That Target the PA-PB1 Interaction of Influenza Virus RNA Polymerase and Possess a High Barrier to Drug Resistance. Antivir. Res. 2019, 165, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Massari, S.; Desantis, J.; Nannetti, G.; Sabatini, S.; Tortorella, S.; Goracci, L.; Cecchetti, V.; Loregian, A.; Tabarrini, O. Efficient and Regioselective One-Step Synthesis of 7-Aryl-5-Methyl- and 5-Aryl-7-Methyl-2-Amino-[1,2,4]Triazolo[1,5-a]Pyrimidine Derivatives. Org. Biomol. Chem. 2017, 15, 7944–7955. [Google Scholar] [CrossRef] [PubMed]
- Loregian, A.; Appleton, B.A.; Hogle, J.M.; Coen, D.M. Residues of Human Cytomegalovirus DNA Polymerase Catalytic Subunit UL54 That Are Necessary and Sufficient for Interaction with the Accessory Protein UL44. J. Virol. 2004, 78, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Mercorelli, B.; Luganini, A.; Nannetti, G.; Tabarrini, O.; Palù, G.; Gribaudo, G.; Loregian, A. Drug Repurposing Approach Identifies Inhibitors of the Prototypic Viral Transcription Factor IE2 That Block Human Cytomegalovirus Replication. Cell Chem. Biol. 2016, 23, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Desantis, J.; Nannetti, G.; Massari, S.; Barreca, M.L.; Manfroni, G.; Cecchetti, V.; Palù, G.; Goracci, L.; Loregian, A.; Tabarrini, O. Exploring the Cycloheptathiophene-3-Carboxamide Scaffold to Disrupt the Interactions of the Influenza Polymerase Subunits and Obtain Potent Anti-Influenza Activity. Eur. J. Med. Chem. 2017, 138, 128–139. [Google Scholar] [CrossRef] [PubMed]
- OpenEye OpenEye QUACPAC 2.0.0.3. OpenEye, Cadence Molecular Sciences, Santa Fe, NM. Available online: https://docs.eyesopen.com/ (accessed on 12 December 2023).
- OpenEye OpenEye, SZYBKI 1.10.0.3. OpenEye, Cadence MolecularOpenEye Sciences, Santa Fe, NM. Available online: https://www.eyesopen.com/ (accessed on 23 February 2024).
- Milletti, F.; Storchi, L.; Sfoma, G.; Cross, S.; Cruciani, G. Tautomer Enumeration and Stability Prediction for Virtual Screening on Large Chemical Databases. J. Chem. Inf. Model. 2009, 49, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F.; Storchi, L.; Sforna, G.; Cruciani, G. New and Original PKa Prediction Method Using Grid Molecular Interaction Fields. J. Chem. Inf. Model. 2007, 47, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F.; Vulpetti, A. Tautomer Preference in PDB Complexes and Its Impact on Structure-Based Drug Discovery. J. Chem. Inf. Model. 2010, 50, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Massari, S.; Nannetti, G.; Desantis, J.; Muratore, G.; Sabatini, S.; Manfroni, G.; Mercorelli, B.; Cecchetti, V.; Palù, G.; Cruciani, G.; et al. A Broad Anti-Influenza Hybrid Small Molecule That Potently Disrupts the Interaction of Polymerase Acidic Protein-Basic Protein 1 (PA-PB1) Subunits. J. Med. Chem. 2015, 58, 3830–3842. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Chessari, G.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Nissink, J.W.M.; Taylor, R.D.; Taylor, R. Modeling Water Molecules in Protein-Ligand Docking Using GOLD. J. Med. Chem. 2005, 48, 6504–6515. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, K.; Mayer, D.; Ranadheera, C.; Holler, A.S.; Mänz, B.; Martin, A.; Chase, G.; Tegge, W.; Frank, R.; Kessler, U.; et al. Identification of a PA-Binding Peptide with Inhibitory Activity against Influenza A and B Virus Replication. PLoS ONE 2009, 4, e7517. [Google Scholar] [CrossRef]
- Sidwell, R.W.; Huffman, J.H.; Khare, G.P.; Allen, L.B.; Witkowski, J.T.; Robins, R.K. Broad-Spectrum Antiviral Activity of Virazole: 1-Beta-D-Ribofuranosyl-1,2,4-Triazole-3-Carboxamide. Science 1972, 177, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.; Small, I.; Smith, J.; Suter, P.; Dutkowski, R. Oseltamivir (Tamiflu) and Its Potential for Use in the Event of an Influenza Pandemic. J. Antimicrob. Chemother. 2005, 55 (Suppl. S1), i5–i21. [Google Scholar] [CrossRef] [PubMed]

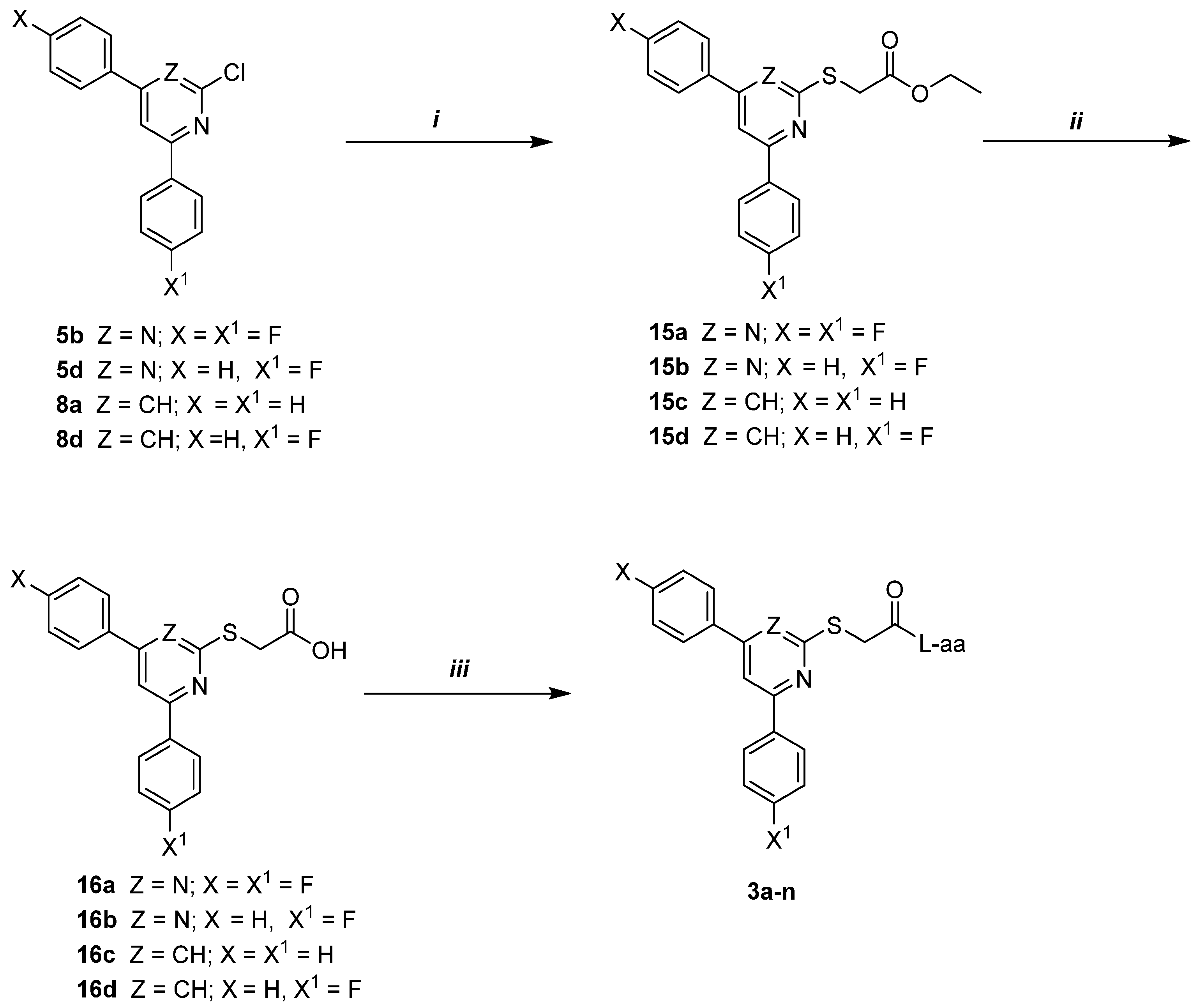

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Cpd | Z | R | R1 | L-Amino Acid | IC50 (ELISA PA–PB1) [µM] a | EC50 (PRA) [µM] b | CC50 (MDCK Cells) [µM] c |
| 1a [25] | CCN | C6H4-4CH3 | C6H5 | - | 80 | ND d | ND d |
| 1b [23] | CCN | C6H4-4CH3 | C6H5 | Ile-OCH3 | 36 ± 3 | 39 ± 2 | >250 |
| 1c [24] | N | C6H4-4CF3 | C6H4-4CF3 | - | >200 | 26.5 ± 4.3 | >250 |
| 1d [24] | N | C6H5 | C6H5 | - | 165 ± 18 | 3.5 ± 0.5 | 10.1 ± 1.7 |
| 1e [24] | CH | C6H5 | C6H5 | - | 52.6 ± 9.0 | 7.3 ± 1.3 | >250 |
| 2a | N | C6H4-4Cl | C6H4-4Cl | - | 70.8 ± 19.5 | >100 | >250 |
| 2b | N | C6H4-4F | C6H4-4F | - | 137 ± 35 | 62.5 ± 4.3 | >250 |
| 2c | N | C6H5 | C6H4-4Cl | - | 86.0 ± 5.3 | 45.5 ± 2.1 | >250 |
| 2d | N | C6H5 | C6H4-4F | - | 90.1 ± 30.2 | 2.8 ± 0.6 | >250 |
| 2e | N | C6H4-4Cl | C6H4-4F | - | 125 ± 37 | 29.5 ± 0.7 | 206 ± 12 |
| 2f | N | C6H5 | 1-naph | - | >200 | 21.1 ± 8.0 | >250 |
| 2g | N | C6H4-4Cl | 1-naph | - | >200 | >100 | >250 |
| 2h | N | C6H4-4F | 1-naph | - | >200 | 14.8 ± 1.6 | >250 |
| 2i | N | C6H5 | SC6H5 | - | 28.8 ± 1.5 | 4.6 ± 0.3 | 73.3 ± 9.1 |
| 2j | N | C6H4-4Cl | SC6H5 | - | >200 | 32.9 ± 4.1 | 16.0 ± 4.2 |
| 2k | N | C6H4-4F | SC6H5 | - | 159 ± 29 | 16.4 ± 1.9 | 48.2 ± 2.5 |
| 2l | N | CONHC6H5 | C6H5 | - | 125 ± 37 | 13.0 ± 5.5 | 46.3 ± 10.4 |
| 2m | CH | C6H5 | SC6H5 | - | 141 ± 44 | 32.3 ± 3.2 | >250 |
| 2n | CH | C6H4-4Cl | SC6H5 | - | 87.0 ± 15.7 | 62.9 ± 13.2 | >250 |
| 3a | N | C6H5 | C6H4-4F | Val-OCH3 | >200 | >100 | 234 ± 19 |
| 3b | N | C6H5 | C6H4-4F | Ile-OCH3 | >200 | >100 | >250 |
| 3c | N | C6H5 | C6H4-4F | Tyr-OCH3 | 172 ± 7 | >100 | >250 |
| 3d | N | C6H5 | C6H4-4F | His-OCH3 | 70.0 ± 2.8 | >100 | 98.0 ± 11.3 |
| 3e | N | C6H4-4F | C6H4-4F | Val-OCH3 | >200 | >100 | >250 |
| 3f | N | C6H4-4F | C6H4-4F | Ile-OCH3 | >200 | >100 | >250 |
| 3g | N | C6H4-4F | C6H4-4F | Tyr-OCH3 | >200 | >100 | >250 |
| 3h | N | C6H4-4F | C6H4-4F | His-OCH3 | 108 ± 14 | >100 | >250 |
| 3i | CH | C6H5 | C6H5 | Val-OCH3 | >200 | ND d | ND d |
| 3j | CH | C6H5 | C6H5 | Ile-OCH3 | >200 | ND d | ND d |
| 3k | CH | C6H5 | C6H5 | Tyr-OCH3 | >200 | ND d | ND d |
| 3l | CH | C6H5 | C6H4-4F | Ile-OCH3 | >200 | ND d | ND d |
| 3m | CH | C6H5 | C6H4-4F | Tyr-OCH3 | >200 | ND d | ND d |
| 3n | CH | C6H5 | C6H4-4F | His-OCH3 | >200 | ND d | ND d |
| PB1(1–15)-Tat peptide | 31.7 ± 10.8 | 49.7 ± 5.1 | >250 | ||||
| OSC | - | 0.04 ± 0.01 | >250 | ||||
| RBV | - | 12.8 ± 3.2 | >250 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacchello, I.; Cianciusi, A.; Bertagnin, C.; Bonomini, A.; Francesconi, V.; Mori, M.; Carbone, A.; Musumeci, F.; Loregian, A.; Schenone, S. Exploring a New Generation of Pyrimidine and Pyridine Derivatives as Anti-Influenza Agents Targeting the Polymerase PA–PB1 Subunits Interaction. Pharmaceutics 2024, 16, 954. https://doi.org/10.3390/pharmaceutics16070954
Giacchello I, Cianciusi A, Bertagnin C, Bonomini A, Francesconi V, Mori M, Carbone A, Musumeci F, Loregian A, Schenone S. Exploring a New Generation of Pyrimidine and Pyridine Derivatives as Anti-Influenza Agents Targeting the Polymerase PA–PB1 Subunits Interaction. Pharmaceutics. 2024; 16(7):954. https://doi.org/10.3390/pharmaceutics16070954
Chicago/Turabian StyleGiacchello, Ilaria, Annarita Cianciusi, Chiara Bertagnin, Anna Bonomini, Valeria Francesconi, Mattia Mori, Anna Carbone, Francesca Musumeci, Arianna Loregian, and Silvia Schenone. 2024. "Exploring a New Generation of Pyrimidine and Pyridine Derivatives as Anti-Influenza Agents Targeting the Polymerase PA–PB1 Subunits Interaction" Pharmaceutics 16, no. 7: 954. https://doi.org/10.3390/pharmaceutics16070954