Abstract
Fluoropyrimidines (FPs) are commonly prescribed in many cancer streams. The EMA and FDA-approved drug labels for FPs recommend genotyping the DPYD*2A (rs3918290), *13 (rs55886062), *HapB3 (rs56038477), alleles, and DPYD rs67376798 before treatment starts. We implemented the DPYD genotyping in our daily clinical routine, but we still found patients showing severe adverse drug events (ADEs) to FPs. We studied among these patients the DPYD rs1801265, rs17376848, rs1801159, rs1801160, rs1801158, and rs2297595 as explanatory candidates of the interindividual differences for FP-related toxicities, examining the association with the response to FPs . We also studied the impact of DPYD testing for FP dose tailoring in our clinical practice and characterized the DPYD gene in our population. We found a total acceptance among physicians of therapeutic recommendations translated from the DPYD test, and this dose tailoring does not affect the treatment efficacy. We also found that the DPYD*4 (defined by rs1801158) allele is associated with a higher risk of ADEs (severity grade ≥ 3) in both the univariate (O.R. = 5.66; 95% C.I. = 1.35–23.67; p = 0.014) and multivariate analyses (O.R. = 5.73; 95% C.I. = 1.41–28.77; p = 0.019) among FP-treated patients based on the DPYD genotype. This makes it a candidate variant for implementation in clinical practice.
1. Introduction
Fluorouracil and its oral prodrug capecitabine, both fluoropyrimidines (FPs), are anticancer drugs used in the treatment of many solid tumors, such as breast and gastric, but especially in colorectal cancer. Despite the wide use of these drugs, among FP-treated patients with standard doses, up to 30% have severe (grade ≥ 3) treatment-related toxicity [1,2], which can lead to treatment-related death in up to 1% of patients [2,3,4]. The most common severe treatment-related toxicity includes diarrhea, oropharyngeal mucositis, hand–foot syndrome, and myelosuppression [2,5].
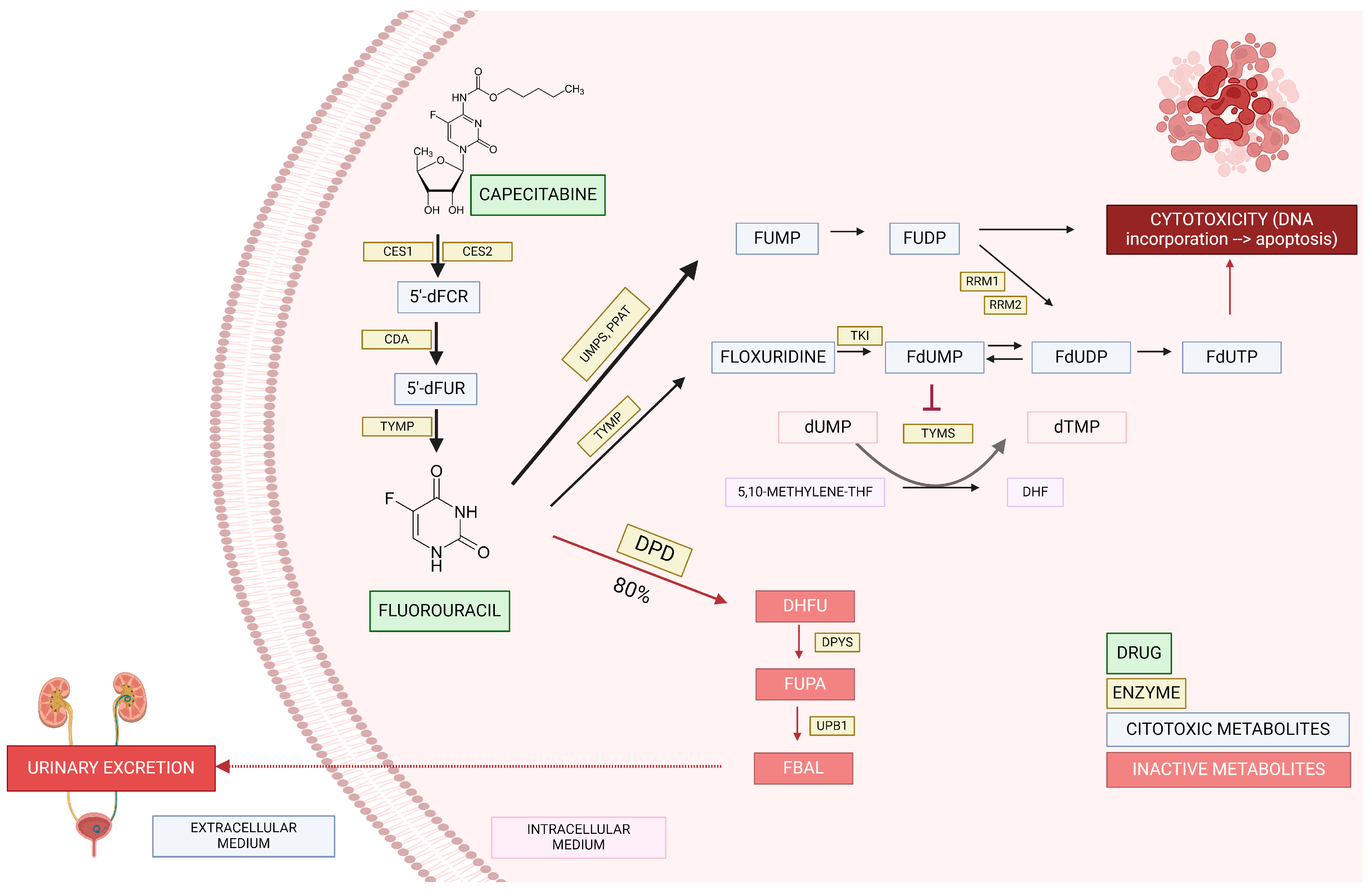
Fluorouracil is administered intravenously, while capecitabine is an oral prodrug metabolized by carboxylesterase, cytidine deaminase, and thymidine phosphorylase resulting, respectively, in 5′-deoxy-5-fluorocytidin (5′dFCR), 5′-deoxy-5-fluorouridine (5′dFUR), and finally 5-fluorouracil (5-FU) [6] (Figure 1). After this, around 80% of fluorouracil is inactivated, and the remaining 20% is converted into cytotoxic metabolites or excreted in the urine [5]. FPs are catabolized by different enzymes, leading to their inactivation, and dihydropyrimidine dehydrogenase (DPD) is the main enzyme responsible for this [7].
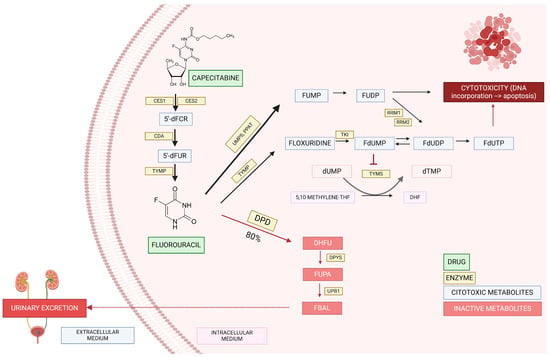
Figure 1.
Capecitabine/fluorouracil pathway. Carboxylesterase 1 (CES1); carboxylesterase 2 (CES2); cytidine deaminase (CDA); thymidine phosphorylase (TYMP); 5′-deoxy-5-fluorocytidin (5′dFCR); 5′-deoxy-5-fluorouridine (5′dFUR); uridine-5-monophosphate synthase (UMPS); amidophosphoribosyltransferase (PPAT); dihydropyrimidine dehydrogenase (DPD); fluorouridine monophosphate (FUMP); fluorouridine diphosphate (FUDP); ribonucleoside diphosphate reductase (RRM1 y 2); thymidine kinase (TKI); fluorodeoxyuridine monophosphate (FdUMP); fluorodeoxyuridine diphosphate (FdUDP); fluorodeoxyuridine triphosphate (FdUTP); deoxyuridine monophosphate (dUMP); deoxythymidine monophosphate (dTMP); 5,10 methylene tetrahydrofolate (5–10 methylene THF); dihidrofolato (DHF); dihydrofluorouracil (DHFU); dihidropirimidinasa (DPYS); 5-fluoro-ureidopropionic acid (FUPA); β-ureidopropionase (UPB1); α-fluoro-β-alanina (FBAL).
Patients with reduced DPD activity catabolize less fluorouracil, leading to more than 20% of cytotoxic/therapeutic metabolites, thus increasing the risk of supratherapeutic drug availability toxicity. In European and North American patients, reduced and complete lack of DPD activity are present in 3–5% and 0.01–0.1%, respectively [2].
1.1. The Dihydropyrimidine Dehydrogenase Gene (DPYD)
Many genetic variants in the gene encoding the DPD enzyme (DPYD gene) partially explained the interindividual differences in the toxicity of FPs. The most relevant genetic variants in this regard are the DPYD rs67376798 (c.2846A>T, D949V), the rs3918290 (c.1905+1G>A, IVS14+1G>A) defining the DPYD*2A allele, the rs55886062 (c.1679T>G, I560S) defining the DPYD*13, and the rs75017182 (c.1129-5923C>G) and rs56038477 (c.1236G>A, E412E) characterizing the DPYD*haplotype (Hap) B3. All of them have been associated with a reduction in DPD activity, resulting in higher concentrations of fluorouracil cytotoxic metabolites and an increased risk of severe or fatal toxicities.
The DPYD*2A (rs3918290) was the first allele known to have a functional impact on DPD activity [8,9]. It was stated that homozygous carriers of this variant have a complete loss of function of DPD while heterozygous carriers showed a significant reduction in mean DPD activity [9,10]. Several studies reported a significant association of this allele with the toxicity of FPs, even though the FP dose tailoring based on DPYD*2A genotype was demonstrated to be an effective strategy for the prevention of severe or fatal toxicity events [11]. In their study, Deene et al. showed in a cohort of n = 1631 patients that a 50% dose reduction in DPYD*2A (rs3918290) carriers may decrease the FP-related toxicity from 73% to 28% (p < 0.001). Thereafter, when studying DPYD*2A (defined by rs3918290) combined with DPYD*13 (defined by rs55886062), DPYD*Hap B3 (defined by rs56038477), and rs67376798, a 25.6-fold increased risk of death in FP-treated patients was found [12], and a systematic review and meta-analysis including n = 7365 patients concluded that DPYD genotyping for these four DPYD variants can identify a significant ratio of patients who are DPD deficient [13]. Finally, Henricks L.M. et al. [2] found the prospective DPYD genotyping feasible in routine clinical practice, and dose reductions based on DPYD genotype improved patient safety in FP-treated patients. In this prospective multicenter study, they tailored the FP treatment dose based on DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798 in n = 1103 patients. DPYD*2A and *13 carriers received a 50% dose reduction, while rs67376798 and DPYD*HapB3 carriers received a dose reduction of 25%. Results indicated that FP-related severe toxicity (grade ≥ 3) was more prevalent among DPYD variant carriers compared to wild-type patients (p = 0.0013), and a significantly lower relative risk for severe FP-related toxicity for DPYD genotype-guided dosing was observed compared with historical cohorts for each genetic variant carrier.
On the other hand, DPYD rs3918290, rs55886062, and rs56038477, characterizing the DPYD*2A, *13, and *HapB3 alleles, respectively, and rs67376798, are not the only DPYD variants associated with the toxicity of FPs. There are many others that showed the highest level of evidence according to the PharmGKB [14] with this association (Table 1).

Table 1.
DPYD variants associated with any fluoropyrimidine phenotype with a 1A level of evidence (obtained from PharmGKB [14]).
1.2. Pharmacogenetics of Fluoropyrimidines
All the evidence regarding the association of DPYD variants with FP-related toxicity, including the reduction in toxicity with dose tailoring based on the DPYD genotype and the clinical impact of this practice, has led to updates for the drug labels approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA), incorporating this information, as well as the development of pharmacogenetic (PGx) dosing guidelines for these drugs.
The European Public Assessment Report (EPAR) [16] for capecitabine by the EMA states that capecitabine is contraindicated in patients with no DPD activity and recommends a reduction in starting dose in reduced DPD activity patients to avoid serious toxicity. The EPAR also reports that the reduced DPD activity may be stated considering the DPYD genotype and highlights DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798 (also known as c.2846A>T) as being mainly responsible for DPD reduced/complete absence activity, while considering that there may be other DPYD variants associated with an increased risk of life-threatening toxicity. The FDA table of PGx [17] includes information about the capecitabine and fluorouracil/DPYD drug–gene interactions, reporting that genotype-translated DPD intermediate or poor metabolizer (IM or PM) phenotypes result in higher severe or life-threatening risk toxicities. It considers that there is no dosage safe in PMs and recommends withholding or discontinuing treatment in the presence of early-onset or unusually severe toxicity.
Carrying these DPYD variants alone or in combination results in different translated phenotypes for DPD activity, thus different dosing recommendations. As commented above, there are available PGx dosing guidelines including this information. Both the Clinical Pharmacogenetics Implementation Consortium (CPIC) [18] and Dutch Pharmacogenomics Working Group (DPWG) guidelines [19] categorize patients’ DPYD gene activity score (GAS) depending on DPYD genotype, as shown in Table 2. Furthermore, CPIC guidelines refer to the DPD metabolizing status and categorize patients as normal metabolizers (NM) if they are not carrying any of these variants (GAS = 2), as IM if DPYD GAS = 1 or 1.5, and as PM when the GAS is lower than 1.
Table 2.
Phenotype translation from the considered DPYD allele combinations for fluoropyrimidine dose tailoring.
Also, both the DPWG and CPIC guidelines (Table 3), recommend an alternative drug, if possible, in patients with a DPYD GAS lower than 1 (DPD PMs, in CPIC guidelines), and to start with 50% of the standard dose in patients with a GAS of 1 or 1.5 (DPD IMs). Also, the CPIC guideline reports that IM patients carrying the c.[2846A>T]/[2846A>T] genotype (GAS = 1) may require a dose reduction higher than 50%. The main difference between the CPIC and DPWG guidelines is the DPYD alleles considered for analysis. While the DPWG considers only the DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798 (also known as c.2846A>T), the CPIC guideline for FPs and DPYD provides a list with n = 83 variants, resulting in diplotypes translated into IM or PM (GAS < 2) phenotypes for reduced DPD activity and FP dose tailoring.
Table 3.
CPIC and DPWG PGx guidelines recommendations for DPYD/fluoropyrimidine drug–gene interactions.
1.3. Hypothesis and Objectives
The EMA, AEMPS, SEFF, SEOM, and the drug labels for capecitabine and 5-FU recommend DPYD genotyping to state the DPD metabolizing status before treatment starts with these drugs. Furthermore, there are available dosing guidelines based on PGx information, such as those from the DPWG [19] and CPIC [18].
We have implemented DPYD genotyping in our daily clinical practice before treatment starts with FPs. Despite this, many patients still show severe adverse drug events.
With this study, we aim to state the impact in our clinical routine of capecitabine and 5-FU dose tailoring based on DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798 (also known as c.2846A>T), studying its association with the treatment efficacy and toxicity.
Also, we aim to explain the toxicity events in the cohort of patients receiving the FP treatment tailored by DPYD genotype. In this regard, we studied the association with the FP toxicity and efficacy of other relevant DPYD variants, previously associated with FP response, but not recommended by the scientific societies and sanitary authorities.
Finally, we aimed to characterize the DPYD gene in our population, studying its genotypes and phenotypes distribution, the minor allele frequency (MAF), Hardy–Weinberg (H-W) equilibrium of included variants, and the possible linkage disequilibrium (LD).
In Spain, the Spanish Agency for Medicine and Health Products (Agencia Española del Medicamento y Productos Sanitarios, AEMPS), Spanish Society of Pharmacogenetics and Pharmacogenomics (Sociedad Española de Farmacogenética y Farmacogenómica, SEFF), and Spanish Society of Medical Oncology (Sociedad Española de Oncología Médica, SEOM), recommend genotyping the DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798 (also known as c.2846A>T) prior to FP treatment, avoiding its use in DPD PM patients (GAS < 1), and using a 50% dose reduction in DPD IM patients (1 < GAS < 2).
Since 2021, we have implemented the PGx test of DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798 (also known as c.2846A>T) in our hospital before capecitabine and fluorouracil treatment initiation, as well as dose tailoring based on guidelines from the DPWG. Despite this implementation in clinical practice and its acceptance by sanitary authorities and physicians in daily routine, we still have patients experiencing severe or life-threatening FP-related toxicities.
2. Materials and Methods
2.1. Study Design
The observational retrospective study including patients treated with capecitabine or fluorouracil and tested for DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798 (also known as c.2846A>T) before treatment began in our hospital between 1 March 2021 and 31 December 2021.
The inclusion criteria were patients diagnosed with cancer and prescribed capecitabine or fluorouracil, those tested for DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798, with a capecitabine or fluorouracil dose tailored based on DPWG guidelines, with an available 6-month follow-up period based on medical records, and non-previously treated with FPs. Patients who did not sign the informed consent form or who asked for to withdraw from the study were excluded.
The Research Ethics Committee of Granada approved the study (Code: 1605-N_22; date of approval: 14 September 2022) and the principles of the Declaration of Helsinki were followed.
The main endpoints were the toxicity and efficacy of capecitabine or 5-FU. These data was obtained from electronic medical records and confirmed by physicians in case of discrepancies.
The toxicity endpoint was adverse drug events (ADEs) to capecitabine or 5-FU. The causality and severity of ADEs were stated using the Liverpool Causality Assessment Tool (LCAT) [20], and Common Terminology Criteria for Adverse Events (CTCAE) [21], respectively. Only ADEs categorized as probable or definite and showing a severity grade 3 or higher were considered for the study.
The efficacy endpoint was achieved if patients received a positive clinical assessment from the oncologist regarding the progression of the illness, as recorded in the medical records, and the non-discontinuation of FP treatment during follow-up if it was not because of an ADE. For the positive clinical assessment of metastatic patients by the oncologists, the Response Evaluation Criteria in Solid Tumors (RECIST 1.1) measure was used within 6 months of initiating therapy [22]. Radiological and imaging techniques, such as computed tomography (CT), were employed to monitor the evolution of both target and non-target lesions in treated patients. These assessments were conducted every 3 to 4 months (1–2 times during follow-up), unless patients exhibited clinical signs of progression earlier (e.g., pain, dyspnea, sweating, elevated tumor markers in blood tests).
2.2. Procedures for the Inclusion of DPYD Variants in the Study
To include candidate DPYD variants as explanatory factors of remaining toxicity events in our study population, who were receiving dose-tailored FP treatment based on DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798, we searched in PharmGKB [14] for genetic variants of DPYD reported as clinical annotations with the highest level of evidence (1A) concerning their association with any phenotype for capecitabine, 5-FU, or tegafur (Table 1). We included those genetic variants related to the toxicity of these drugs with MAF values higher than 1% in the Iberian Peninsula population according to the 1000 Genomes project [15].
2.3. Management of Patients
As part of the clinical practice at our hospital, whenever a doctor considers prescribing capecitabine or 5-FU in patients diagnosed with whatever cancer, they may request a DPYD PGx test from the hospital pharmacy.
Once the request for the test is received, a nurse takes a saliva sample with sterile cotton swabs, the DNA is extracted, and DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798 are tested less than 48 h after the saliva sample collection. The pharmacists upload a PGx report in the electronic medical records, including the therapeutic recommendation translated from the PGx result, within 72 h of sample collection.
The genotype–phenotype–therapeutic recommendation translation process (dose tailoring) is performed following the instructions in the DPWG guidelines and is shown in Table 2 and Table 3. This means that patients not carrying any of the tested variants are assigned a DPYD GAS = 2 and treated with the standard dose. Those carrying a single mutated allele are assigned GAS = 1–1.5 and start the treatment with a 50% reduction in the standard dose. Patients with two mutated alleles are assigned GAS = 0, and the treatment is switched.
The remaining DNA and saliva samples are stored as a private biosamples collection registered with the Carlos III Health Institute (C.0007322). Once the follow-up of patients was finished, we retrospectively tested, using this remaining DNA, the DPYD variants meeting the criteria that are explained in Section 2.2.
Those patients not treated with capecitabine or fluorouracil after the PGx test were excluded from the analysis. The included patients were followed up for six months.
2.4. Data Management, Statistical Analysis, and Genotyping
First, a descriptive analysis of the population included in the study was performed. The impact and usefulness of the DPYD PGx test for capecitabine and 5-FU dose tailoring in our clinical practice was assessed by studying the association with the toxicity and efficacy endpoints of the dose tailoring based on DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798.
After that, to explain ADEs that occurred during follow-up in patients receiving an FP-DPYD-dose tailored treatment, we performed a genotype association study with the toxicity and efficacy endpoint.
Moreover, we carried out a multivariate analysis to discard possible confounding factors on the association of genetic variants with the toxicity and efficacy endpoints.
Finally, we characterized the DPYD variants in our population, including both those used in our daily clinical practice and those studied for their association with the toxicity endpoint after PGx FP dose tailoring. In this regard, the distribution (number, n, and percentage, %) of genotypes, phenotypes, and MAFs were calculated, an LD analysis was carried out, and the H-W equilibrium was tested.
The descriptive analysis, MAFs, genotype/phenotype distribution, and multivariate analysis were conducted using R commander. For the association studies of genetic variants with the endpoints, LD analysis, and H–W equilibrium analyses, we used the SNPstats online tool [23].
For the multivariate analysis, we considered for inclusion all the study variables recorded, including clinical parameters, genetic variants, and concomitant treatments. We built different multivariate models using the backward, forward, and stepwise methods for the association with the efficacy and toxicity endpoints. These models were upgraded and compared using the Akaike information criterion (AIC). The final model showing the lower AIC was chosen.
The chi-square test or Fisher exact test were used, and odds ratios (OR) and p-values were calculated, considering p < 0.05 to be statistically significant.
2.5. DNA Extraction and Genotyping
For genotyping, DNA was isolated from saliva samples using standard procedures. DNA extraction was carried out following the method by Freeman et al. [24], a non-organic (proteinase K and salting out) protocol including modifications from the method described by Gomez-Martín A. et al. [25]. The included genetic variants were genotyped using Taqman assay technology (Thermo Fisher Scientific, Waltham, MA, USA) and analyzed with QuantStudio 12K Flex de Applied Biosystems (Foster City, CA, USA).
3. Results
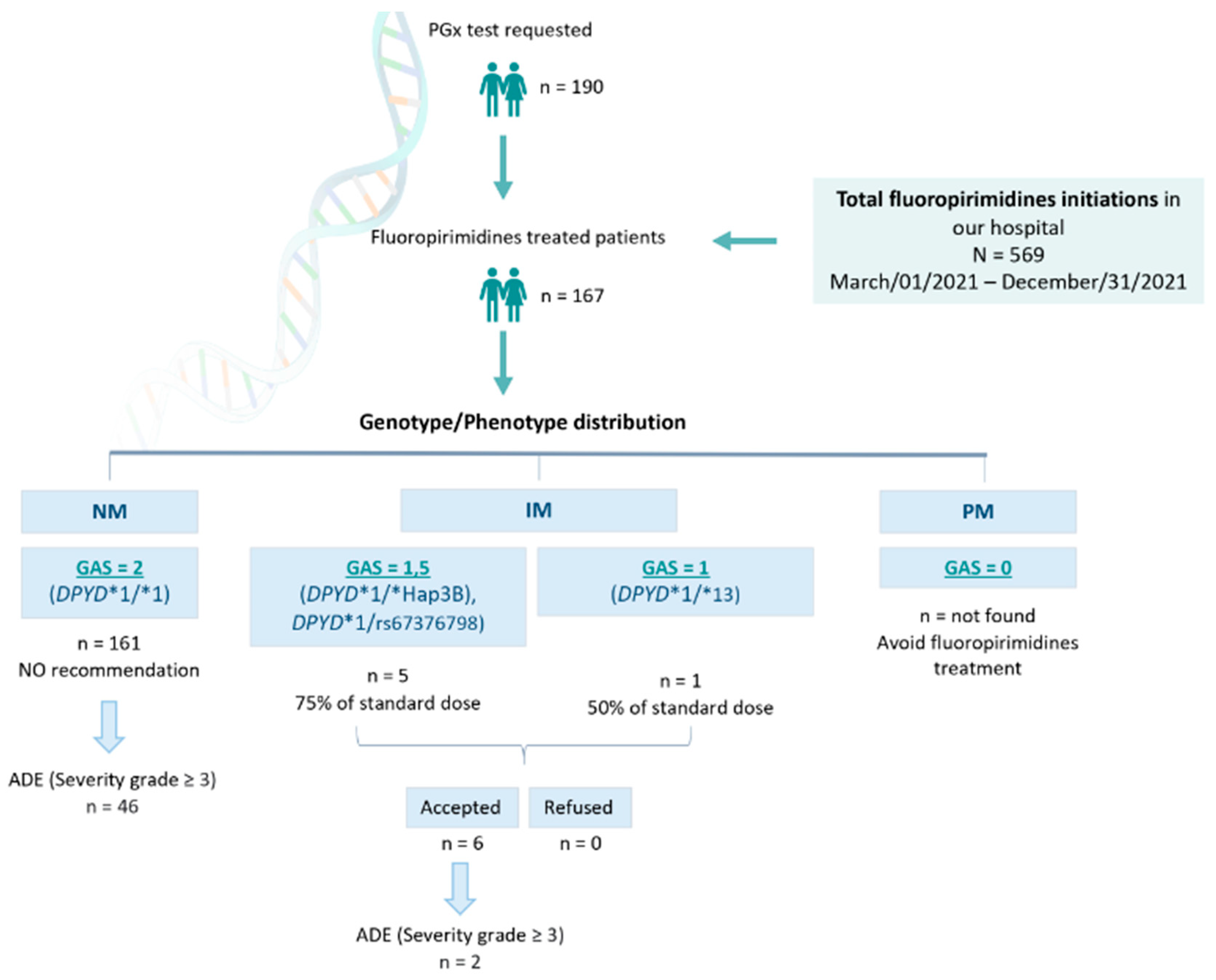
Between Mar/01/2021 and Dec/31/2021, N = 569 patients were prescribed capecitabine or 5-FU at the Hospital Universitario Clínico San Cecilio (Granada, Spain). In total, n = 190 DPYD PGx tests were requested, and n = 167 patients were finally treated with FPs (Figure 2). This means n = 402 patients were prescribed capecitabine or 5-FU in our hospital without being DPYD tested, and n = 23 were DPYD tested but not treated with FPs.
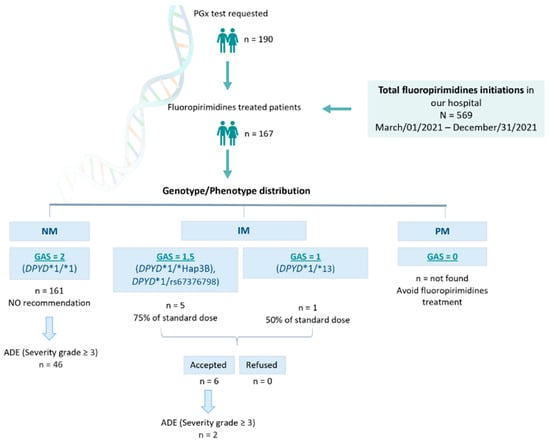
Figure 2.
Recruitment and management of patients. PGx: pharmacogenetic; NM: normal metabolizer; IM: intermediate metabolizer; PM: poor metabolizer; GAS: Gene Activity Score; ADE: adverse drug event.
Among the n = 167 DPYD-tested patients finally treated with capecitabine or 5-FU, we found n = 161 DPYD*1/* 1 (wild-type) patients, translated into a GAS = 2 phenotype receiving normal doses of FPs. We also found n = 1 DPYD*1/*13, n = 3 *1/*HapB3, and n = 2 *1/c.2846A>T genotypes, translated into n = 5 GAS = 1.5, and n = 1 GAS = 1, who were recommended to be treated with 50% of the standard dose. All these patients were dose-adjusted based on our recommendation (Figure 2). Among those n = 23 non-treated patients, we found n = 21 DPYD*1/*1 (wild-type) and n = 2 *1/*HapB3.
All the n = 167 FP-treated patients after dose tailoring based on DPYD genotyping were prescribed treatment because of digestive tumors, including colon (n = 81), stomach (n = 13), pancreas (n = 10), duodenum (n = 1), esophageal (n = 1), and rectal cancer (n = 45), although breast cancer (n = 12) and other kind of tumors (n = 4) affected some patients. All the patients received capecitabine or 5-FU as a first-line treatment, except the n = 12 breast cancer patients who were prescribed it after cytotoxic chemotherapy failure and a locally advanced tumor or metastasis. The mean age was 64.26 ± 10.89 years old, and 37.13% were women (Table 4).
Table 4.
Baseline characteristics of fluoropyrimidine-treated patients.
3.1. Association Study of DPYD Variants with the Response
3.1.1. Association with Response of Dose Tailoring Based on DPYD
We found no association between the DPYD variants used for FP dose tailoring and the toxicity endpoint. We also found no association with the efficacy endpoint (Table 5). This means that carrying a genotype translated into a GAS lower than 2 (DPD IM or PM) and receiving adjusted doses is not related to variable response to FPs.
Table 5.
Association study of DPYD variants used for fluoropyrimidine dose tailoring with the response.
3.1.2. Association with the Response of New DPYD Variants
Among treated patients based on DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798, we still found n = 47 patients meeting the toxicity endpoint.
As commented above, we retrospectively studied n = 6 DPYD variants that had been related to the toxicity of FPs and with an MAF higher than 1% in the Iberian Peninsula population according to the 1000 Genomes project.
We found that carrying the DPYD rs1801158 is associated with a higher risk of ADEs with a severity grade ≥ 3 (OR = 5.66; 95% C.I. = 1.35–23.67; p = 0.014) (Table 6). No other DPYD variant was associated with the toxicity endpoint.
Table 6.
Association study with the efficacy/toxicity endpoints of DPYD variants not used for FP dose tailoring.
We found no association of any of the included SNPs with the efficacy endpoint.
3.1.3. Multivariate Analysis
The following variables were included as possible explanatory parameters of the efficacy and toxicity endpoints in the multivariate analysis: clinical variables, including age, sex, and body surface area; concomitant drugs in the therapeutic scheme, including monoclonal antibodies, oxaliplatin, irinotecan, and radiotherapy; and all the DPYD genetic variants included in the study, i.e., those used and not used for FP dose tailoring.
For the multivariate analysis to explain the toxicity endpoint, the model using the stepwise (backward/forward) method showed the lower AIC, and this model was finally included. After adjustment, the model showed that the DPYD rs1801158 is associated with ADEs (severity grade ≥ 3) for FPs (OR = 5.73; 95% CI = 1.41–28.77; p = 0.019) in DPYD dose-tailored patients based on DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798 (Table 7).
Table 7.
Association study with the toxicity and efficacy endpoint of study variables included in the multivariate model after adjustment.
In the multivariate analysis to explain the efficacy endpoint we found that the concomitant treatment with irinotecan is associated with lower rates of efficacy (p ≤ 0.001).
3.2. DPYD Characterization
In total, n = 190 patients were requested to be tested and dose-tailored based on DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798. Among them, n = 189 were tested for DPYD variants considered candidates to explain differences in the response to FPs. This means that n = 1 patient could not be tested for these variants because we did not have enough stored DNA.
Among all the tested variants in our population, we found no differences with the MAF for the Iberian Peninsula population reported by the 1000 Genomes Project [15], and all the SNPs were in the Hardy–Weinberg equilibrium (Table 8).
Table 8.
DPYD characterization in our population.
We found many significant differences in the LD analysis. The DPYD rs1801265 was linked to the rs56038477 (r = 0.214; p < 0.001) and the rs2297595 (r = 0.604; p < 0.001) in our population (Table 9). In more detail, we found that all the patients carrying rs56038477 also carried rs1801265, and n = 31 (83.8%) of patients carrying rs2297595 also carried rs1801265. We found other p-values lower than 0.05 in the LD analyses but showing r values close to r = 1.
Table 9.
Linkage disequilibrium analysis.
4. Discussion
FPs, including 5-FU and the oral prodrug capecitabine, are commonly prescribed antimetabolite chemotherapies utilized across many cancer streams.
Among FP-treated patients with standard doses, up to 30% show severe (grade ≥ 3) treatment-related toxicity. Many genetic variants in the DPYD gene encoding the DPD enzyme partially explained these toxicities. The DPYD*2A (defined by rs3918290), *13 (defined by rs55886062), *HapB3 (defined by rs56038477), alleles, and DPYD rs67376798 had shown the highest level of evidence about their association with FPs response. The EMA- and FDA-approved drug labels for FPs, and the SEFF and SEOM in Spain, recommend genotyping these variants before treatment starts.
Depending on DPYD genotype, patients may be categorized as DPD NM, IM, or PM and receive a PGx dose-tailored treatment (50% of standard doses in DPD IM patients, and alternative therapies in DPD PMs).
Moreover, different studies concluded that dose adjustments based on DPYD genotype do not influence the treatment efficacy. Deenen M.J. et al. did not find a relationship between DPYD variants and progression-free survival or overall survival despite a 50% dose reduction in DPYD*2A carriers [26], and Lam S.W. et al. [27] observed no differences in response in seven more clinical studies examining DPYD polymorphisms with a dose reduction, time to progression, progression-free survival, and/or overall survival, concluding that there is no evidence that a priori dose adjustments for DPYD carriers decrease FP efficacy, and low-activity variant carriers treated with standard of care appear to have similar efficacy once an acceptable dose is found.
We implemented DPYD genotyping in our daily clinical routine, but we still find patients showing severe toxicities to FPs. Thus, we hypothesized that there might be other variants influencing the FP-related toxicities.
We identified six DPYD variants (rs1801265, rs17376848, rs1801159, rs1801160, rs1801158, and rs2297595) as explanatory candidates of the interindividual differences for the FP-related toxicities, since these had been related to the toxicity of FPs with the highest level of evidence, and they have an MAF higher than 1% in the Iberian Peninsula population.
In this study, we assessed the association with response to FPs of these novel candidate variants to explain suboptimal patient response for the first time in a cohort that received FP treatment based on DPYD*2A (defined by rs3918290), *13 (defined by rs55886062), *HapB3 (defined by rs56038477), and rs67376798.
This way, we could determine whether these new DPYD variants explain the remaining toxicities despite a PGx dose-tailored treatment and whether they would be potentially useful in daily clinical practice.
This study may also be used as a guide for the clinical implementation of the FP-DPYD drug–gene interaction.
4.1. Association of Genetic Variants with Response to Fluoropyrimidines
In the study of the association of FP dose tailoring according to the DPYD genotype with the toxicity and efficacy endpoints, we found no significant differences (Table 5). These results make sense, since significant differences would have meant an underestimation of dose modifications resulting from the presence of the DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798 variants.
In the association study with the toxicity and efficacy endpoints of the new variants, which are not currently being used to guide treatment with FPs in our population, we have observed the following.
The DPYD*4 (defined by rs1801158) allele is associated with a higher risk of severe ADEs (severity grade ≥ 3) in both the univariate (OR = 5.66; 95% CI = 1.35–23.67; p = 0.014) and multivariate analyses (OR= 5.73; 95% CI= 1.41–28.77; p = 0.019) after adjusting the model.
This variant had been previously associated with FP response. The DPYD*4 CT genotype was associated with decreased catalytic activity of DPD [28], and an increased risk of drug toxicity when treated with capecitabine or fluorouracil in colorectal cancer patients [29], as compared to CC genotype. On the other hand, many other studies showed contradictory results [13] in this regard. An interesting study by André B. P. van Kuilenburg et al. [30] found that DPYD*4 allele T is associated with decreased activity of DPD when expressed in mammalian cells (HEK293 Flp-In) as compared to allele C, while highlighting the conflicting data about this association, as they found no association when DPD activity was assessed within a healthy cohort of n = 100 individuals.
These findings, and the results described below, reveal the need for further studies, especially considering the expression of genes, and not just categorizing patients as carriers/non-carriers of single or combined DPYD variants.
We found an association of chemotherapy schemes including irinotecan with a lower efficacy in the multivariate analysis (Table 7). We observed that the n = 10 patients treated with FOLFIRINOX (including irinotecan) are the same n = 10 patients with pancreatic cancer, with the worse baseline condition and prognosis among recruited patients.
Also, regarding our results, no other variants were significantly associated with either toxicity or efficacy endpoints. The DPYD rs17376848 showed a confounding association with the response. According to the significance criteria based on p-values, carriers of this variant (Genotype AG or GG vs. AA) were associated with a lower risk of the toxicity endpoint (p = 0.043) and a certain trend toward the efficacy of FPs (p = 0.079). However, upon closer examination of these results, we find that only n = 6 patients carry the DPYD rs17376848 variant, and none of them experienced an ADE (severity grade ≥ 3) during follow-up, preventing us from confirming this association. Furthermore, this statistical significance is lost in the multivariate analysis (p = 0.992; Table 7) when considering the influence of concomitant treatments, other clinical variables, and interactions with other DPYD variants.
As happens with DPYD*4 (defined by rs1801158), those other five variants suggested as candidate variants to explain the remaining toxicity events in our population showed inconclusive results in previous studies [13,31]. In any case, further studies with larger cohorts and treatment guidance based on these variants are necessary to confirm their utility or lack of influence in clinical practice.
Anyway, despite of the baseline characteristics of recruited patients, stage of the tumor, different fluoropyrimidines schemes, interactions between clinical and PGx variables, and receiving a PGx dose tailored treatment, the DPYD*4 (defined by rs1801158) showed an association with a higher risk of severe ADEs with FPs in our population. This makes it a candidate variant for potential implementation in clinical practice.
4.2. Insights on Clinical Practice
We have observed that in the clinical practice of our hospital, there is an important association between the profile of the health professional and the degree of acceptance and requests for PGx tests. As also happened to us in previous studies [32], there is a bias between the hospital departments prescribing the drugs and those requesting the PGx tests. In this case, we have observed that 96.3% (n = 183) of the n = 190 requests for the DPYD test were made by the medical oncology department, and n = 7 (3.7%) were made by other units of our hospital. Furthermore, we observed that, of the n = 170 patients who were prescribed capecitabine or 5-FU in the medical oncology unit during the time of recruitment, n = 167 (98.2%) received the treatment as guided by the PGx test. On the other hand, n = 402 patients in total in our hospital received treatment with FPs without having been genotyped for the variants recommended on the drug label by the EMA, FDA, SEFF, and SEOM. Among these, only n = 3 were treated by doctors assigned to the medical oncology service, and n = 399 were treated by other units.
As we can see, despite the level of evidence regarding the DPYD–FP interaction and the recommendations on the drug label by health authorities and scientific societies, the degree of implementation of these tests is strongly linked to the profile of the healthcare professional and the internal procedures or protocols of their hospital department.
On the other hand, it is true that until a few months ago, PGx tests, including the DPYD–FP interaction described in this study, had not been included in the portfolio of common services of the national health system in Spain, which is the set of health procedures that must be available to any citizen. The recent update of the service portfolio institutionally supports the performance of PGx tests and invites health professionals to perform them.
Another aspect that we observed in our results is that the level of acceptance of the therapeutic recommendations that emerge from the PGx results is total, and that dose reductions do not translate into a decrease in the treatment’s efficacy. Furthermore, based on our results and, as mentioned above, DPYD rs1801158 could be implemented in clinical practice. This variant has a relatively high frequency (MAF = 0.032; 6% carriers) and was the only genetic or clinical variant that was associated with an increased risk of toxicity secondary to FPs in the univariate and multivariate analyses.
The DPYD–FP interaction, regardless of many contradictory results, has been demonstrated to be useful in clinical practice in different studies. Furthermore, recent studies have demonstrated the cost-effectiveness of PGx tests [33,34]. The potential benefit, actual related costs, and support by sanitary authorities should be enough reasons for the large-scale implementation of PGx tests, especially the DPYD–FP interaction.
4.3. Limitations
This is not a comparative clinical trial; rather, it is an observational cohort study. We did not consider all the variants in the DPYD gene. However, for the recruited cohort, it was not useful to study further variants. In fact, we examined all variants with a MAG higher than 1% in our population. Rare variants do not make sense in a cohort of n = 167 patients.
Nevertheless, it is necessary to investigate rare variants in larger cohorts to study the different interactions between them in detail. In particular, conducting an association study between DPYD haplotypes and their response to FPs would be especially interesting. Also, studying the epigenetics of the DPYD gene as a key gene in the PGx of FPs would be also interesting. This might elucidate discrepancies between association studies of DPYD variants with their FP response.
We did not study all the clinical variables that influence the response to FPs, especially the baseline condition of recruited patients, as commented above. Furthermore, this study is based on real-world data obtained from daily clinical practice, and we are limited by the reliable information collected in the patients’ medical records.
In the same regard, we recruited a wide range of different patients, including metastatic/adjuvant-treated patients, different stages of tumors, chemotherapy schemes, etc. Anyway, the aim was to perform the study considering real-world data based on our daily clinical practice, and we found significant differences concerning the influence of the DPYD*4 alle on FP toxicities.
Also, considering the inclusion criteria of candidate DPYD variants as explanatory factors of FP-related toxicities, we should study the DPYD rs75017182 variant (Table 1). It was not genotyped because it has a lower MAF thanthe DPYD rs56038477,and both variants charactere the DPYD*HapB3.
5. Conclusions
Based on our results, we concluded that FP dose lowering based on the DPYD genotype does not affect the treatment efficacy. DPYD*4 (defined by the rs1801158) is associated with FP toxicity in patients receiving a PGx dose-tailored treatment based on DPYD*2A (defined by rs3918290), DPYD*13 (defined by rs55886062), DPYD*HapB3 (defined by rs56038477), and rs67376798. Based on this, DPYD*4 (defined by the rs1801158) is an explanatory factor of remaining ADEs among FP-treated and PGx dose-tailored patients, and its genotyping should be implemented in daily clinical practice.
Author Contributions
X.D.-V. and M.M.-P.: conceptualization, methodology, visualization, formal analysis, writing—original draft; E.F.-V. and A.J.I.: methodology, data curation, formal analysis, writing—review and editing; M.T.N.-S., G.R.-T., and A.T.-G.: software, investigation, resources; B.G.A. and I.B.: validation, data curation, writing—review and editing. J.C.-B.: supervision, project administration; R.M.: conceptualization, methodology, visualization, formal analysis, writing—original draft. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the “Fundación Andaluza de Farmacia Hospitalaria”, grant number 74/2024.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Research Ethics Committee of Granada (protocol code 1605-N_22 and date of approval: 14 September 2022).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to containing clinical and personal information.
Acknowledgments
We want to kindly thank patients participating in the study and sanitary personnel of the Hospital Universitario San Cecilio collaborating in the recruitment of patients.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Mikhail, S.E.; Sun, J.F.; Marshall, J.L. Safety of capecitabine: A review. Expert. Opin. Drug Saf. 2010, 9, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Henricks, L.M.; Lunenburg, C.A.T.C.; de Man, F.M.; Meulendijks, D.; Frederix, G.W.J.; Kienhuis, E.; Creemers, G.J.; Baars, A.; Dezentjé, V.O.; Imholz, A.L.T.; et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: A prospective safety analysis. Lancet Oncol. 2018, 19, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Henricks, L.M.; Opdam, F.L.; Beijnen, J.H.; Cats, A.; Schellens, J.H.M. DPYD genotype-guided dose individualization to improve patient safety of fluoropyrimidine therapy: Call for a drug label update. Ann. Oncol. 2017, 28, 2915–2922. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Twelves, C.; Cassidy, J.; Allman, D.; Bajetta, E.; Boyer, M.; Bugat, R.; Findlay, M.; Frings, S.; Jahn, M.; et al. Oral capecitabine compared with intravenous fluorouracil plus leucovorin in patients with metastatic colorectal cancer: Results of a large phase III study. J. Clin. Oncol. 2001, 19, 4097–4106. [Google Scholar] [CrossRef]
- White, C.; Scott, R.J.; Paul, C.; Ziolkowski, A.; Mossman, D.; Fox, S.B.; Michael, M.; Ackland, S. Dihydropyrimidine Dehydrogenase Deficiency and Implementation of Upfront DPYD Genotyping. Clin. Pharmacol. Ther. 2022, 112, 791–802. [Google Scholar] [CrossRef]
- Miwa, M.; Ura, M.; Nishida, M.; Sawada, N.; Ishikawa, T.; Mori, K.; Shimma, N.; Umeda, I.; Ishitsuka, H. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5 fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur. J. Cancer 1998, 34, 1274–1281. [Google Scholar] [CrossRef]
- van Kuilenburg, A.B.; Meinsma, R.; Zonnenberg, B.A.; Zoetekouw, L.; Baas, F.; Matsuda, K.; Tamaki, N.; van Gennip, A.H. Dihydropyrimidinase deficiency and severe 5-fluorouracil toxicity. Clin. Cancer Res. 2003, 9, 4363–4367. [Google Scholar]
- Meinsma, R.; Fernandez Salguero, P.; Van Kuilenburg, A.B.P.; Van Gennip, A.H.; Gonzalez, F.J. Human polymorphism in drug metabolism: Mutation in the dihydropyrimidine dehydrogenase gene results in exon skipping and thymine uracilurea. DNA Cell Biol. 1995, 14, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Van Kuilenburg, A.B.P.; Blom, M.J.; Van Lenthe, H.; Mul, E.; Van Gennip, A.H. The activity of dihydropyrimidine dehydrogenase in human blood cells. J. Inherit. Metab. Dis. 1997, 20, 331–334. [Google Scholar] [CrossRef]
- Wei, X.; McLeod, H.L.; McMurrough, J.; Gonzalez, F.J.; Fernandez-Salguero, P. Molecular basis of the human dihydropyrimidine dehydrogenase deficiency and 5-fluorouracil toxicity. J. Clin. Investig. 1996, 98, 610–615. [Google Scholar] [CrossRef]
- Deenen, M.J.; Meulendijks, D.; Cats, A.; Sechterberger, M.K.; Severens, J.L.; Boot, H.; Smits, P.H.; Rosing, H.; Mandigers, C.M.; Soesan, M.; et al. Upfront Genotyping of DPYD*2A to Individualize Fluoropyrimidine Therapy: A Safety and Cost Analysis. J. Clin. Oncol. 2016, 34, 227–234. [Google Scholar] [CrossRef]
- Sharma, B.B.; Rai, K.; Blunt, H.; Zhao, W.; Tosteson, T.D.; Brooks, G.A. Pathogenic DPYD Variants and Treatment-Related Mortality in Patients Receiving Fluoropyrimidine Chemotherapy: A Systematic Review and Meta-Analysis. Oncologist 2021, 26, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Meulendijks, D.; Henricks, L.M.; Sonke, G.S.; Deenen, M.J.; Froehlich, T.K.; Amstutz, U.; Largiadèr, C.R.; Jennings, B.A.; Marinaki, A.M.; Sanderson, J.D.; et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: A systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015, 16, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Whirl-Carrillo, M.; Huddart, R.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Whaley, R.; Klein, T.E. An Evidence-Based Framework for Evaluating Pharmacogenomics Knowledge for Personalized Medicine. Clin. Pharmacol. Ther. 2021, 110, 563–572. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef]
- European Public Assessment Report (EPAR) for Capecitabine. 16 February 2012. Available online: https://www.ema.europa.eu/en/documents/assessment-report/capecitabine-teva-epar-public-assessment-report_en.pdf (accessed on 1 March 2024).
- Food and Drug Administration (FDA) Table of Pharmacogenetic Associations. 26 October 2022. Available online: https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations (accessed on 1 March 2024).
- Amstutz, U.; Henricks, L.M.; Offer, S.M.; Barbarino, J.; Schellens, J.H.M.; Swen, J.J.; Klein, T.E.; McLeod, H.L.; Caudle, K.E.; Diasio, R.B.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin. Pharmacol. Ther. 2018, 103, 210–216. [Google Scholar] [CrossRef]
- Lunenburg, C.A.T.C.; van der Wouden, C.H.; Nijenhuis, M.; Crommentuijn-van Rhenen, M.H.; de Boer-Veger, N.J.; Buunk, A.M.; Houwink, E.J.F.; Mulder, H.; Rongen, G.A.; van Schaik, R.H.N.; et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur. J. Hum. Genet. 2020, 28, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, R.M.; Kirkham, J.J.; Mason, J.R.; Bird, K.A.; Williamson, P.R.; Nunn, A.J.; Turner, M.A.; Smyth, R.L.; Pirmohamed, M. Development and inter-rater reliability of the Liverpool adverse drug reaction causality assessment tool. PLoS ONE 2011, 6, e28096. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE). 2017. Available online: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcae_v5_quick_reference_5x7.pdf (accessed on 1 March 2024).
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Solé, X.; Guinó, E.; Valls, J.; Iniesta, R.; Moreno, V. SNPStats: A web tool for the analysis of association studies. Bioinformatics 2006, 22, 1928–1929. [Google Scholar] [CrossRef]
- Freeman, B.; Smith, N.; Curtis, C.; Huckett, L.; Mill, J.; Craig, I.W. DNA from buccal swabs recruited by mail: Evaluation of storage effects on long-term stability and suitability for multiplex polymerase chain reaction genotyping. Behav. Genet. 2003, 33, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Martín, A.; Hernández, A.F.; Martínez-González, L.J.; González-Alzaga, B.; Rodríguez-Barranco, M.; López-Flores, I.; Aguilar-Garduno, C.; Lacasana, M. Polymorphisms of pesticide-metabolizing genes in children living in intensive farming communities. Chemosphere 2015, 139, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Deenen, M.J.; Tol, J.; Burylo, A.M.; Doodeman, V.D.; de Boer, A.; Vincent, A.; Guchelaar, H.J.; Smits, P.H.; Beijnen, J.H.; Punt, C.J.; et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin. Cancer Res. 2011, 17, 3455–3468. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.W.; Guchelaar, H.J.; Boven, E. The role of pharmacogenetics in capecitabine efficacy and toxicity. Cancer Treat. Rev. 2016, 50, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Seck, K.; Riemer, S.; Kates, R.; Ullrich, T.; Lutz, V.; Harbeck, N.; Schmitt, M.; Kiechle, M.; Diasio, R.; Gross, E. Analysis of the DPYD gene implicated in 5-fluorouracil catabolism in a cohort of Caucasian individuals. Clin. Cancer Res. 2005, 11, 5886–5892. [Google Scholar] [CrossRef] [PubMed]
- Madi, A.; Fisher, D.; Maughan, T.S.; Colley, J.P.; Meade, A.M.; Maynard, J.; Humphreys, V.; Wasan, H.; Adams, R.A.; Idziaszczyk, S.; et al. Pharmacogenetic analyses of 2183 patients with advanced colorectal cancer; potential role for common dihydropyrimidine dehydrogenase variants in toxicity to chemotherapy. Eur. J. Cancer 2018, 102, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kuilenburg, A.B.P.V.; Meijer, J.; Tanck, M.W.T.; Dobritzsch, D.; Zoetekouw, L.; Dekkers, L.L.; Roelofsen, J.; Meinsma, R.; Wymenga, M.; Kulik, W.; et al. Phenotypic and clinical implications of variants in the dihydropyrimidine dehydrogenase gene. Biochim. Biophys. Acta 2016, 1862, 754–762. [Google Scholar] [CrossRef]
- Lee, A.M.; Shi, Q.; Pavey, E.; Alberts, S.R.; Sargent, D.J.; Sinicrope, F.A.; Berenberg, J.L.; Goldberg, R.M.; Diasio, R.B. DPYD variants as predictors of 5-fluorouracil toxicity in adjuvant colon cancer treatment (NCCTG N0147). J. Natl. Cancer Inst. 2014, 106, dju298. [Google Scholar] [CrossRef]
- Díaz-Villamarín, X.; Fernández-Varón, E.; Rojas Romero, M.C.; Callejas-Rubio, J.L.; Cabeza-Barrera, J.; Rodríguez-Nogales, A.; Gálvez, J.; Morón, R. Azathioprine dose tailoring based on pharmacogenetic information: Insights of clinical implementation. Biomed. Pharmacother. 2023, 168, 115706. [Google Scholar] [CrossRef]
- Fragoulakis, V.; Roncato, R.; Bignucolo, A.; Patrinos, G.P.; Toffoli, G.; Cecchin, E.; Mitropoulou, C. Cost-utility analysis and cross-country comparison of pharmacogenomics-guided treatment in colorectal cancer patients participating in the U-PGx PREPARE study. Pharmacol. Res. 2023, 197, 106949. [Google Scholar] [CrossRef]
- Koufaki, M.I.; Fragoulakis, V.; Díaz-Villamarín, X.; Karamperis, K.; Vozikis, A.; Swen, J.J.; Dávila-Fajardo, C.L.; Vasileiou, K.Z.; Patrinos, G.P.; Mitropoulou, C. Economic evaluation of pharmacogenomic-guided antiplatelet treatment in Spanish patients suffering from acute coronary syndrome participating in the U-PGx PREPARE study. Hum. Genom. 2023, 17, 51. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).