

Investigation of Stabilized Amorphous Solid Dispersions to Improve Oral Olaparib Absorption



Abstract
:1. Introduction
2. Materials and Methods
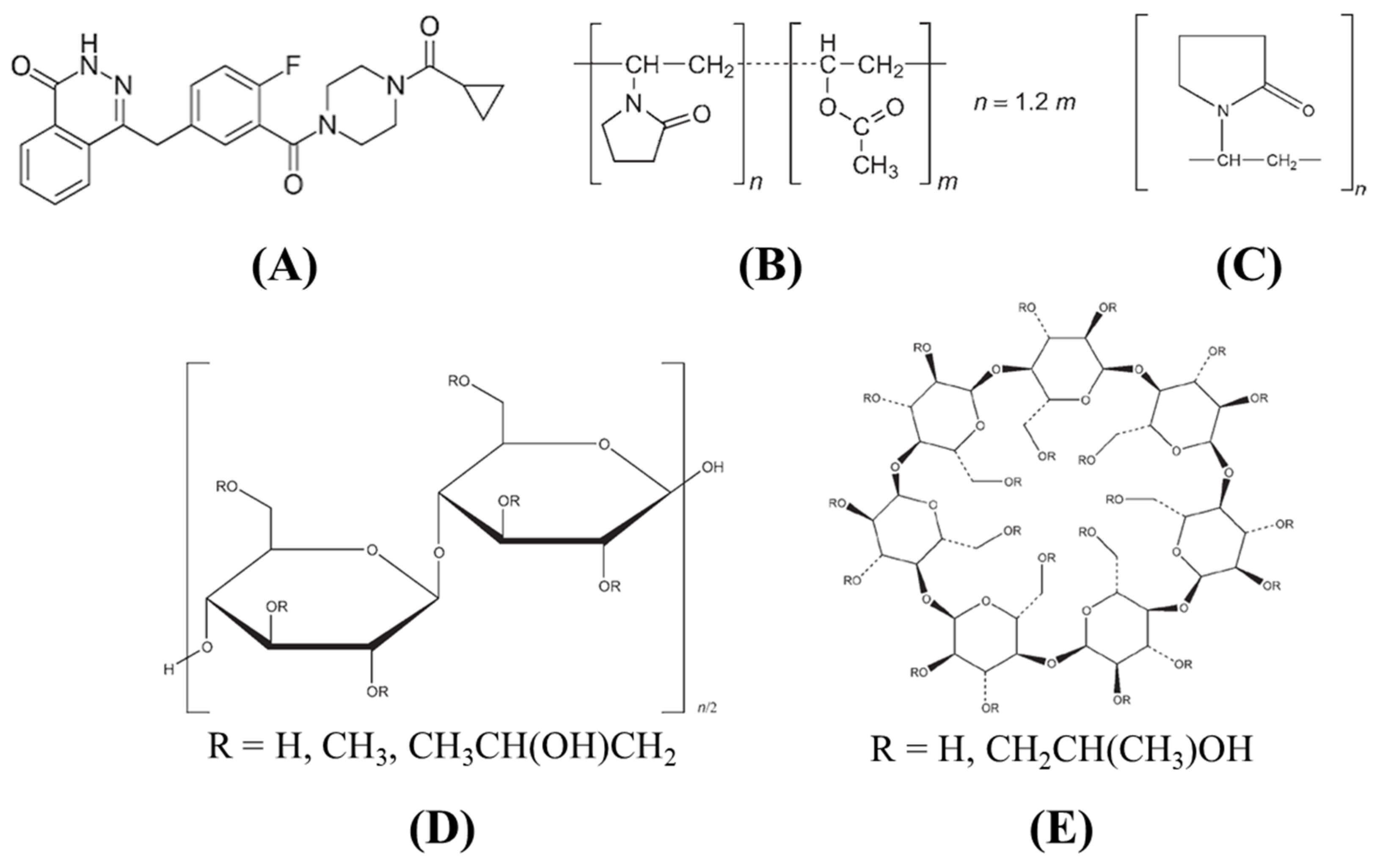
2.1. Materials
2.2. HPLC Analysis Condition
2.3. Drug Solubility Test
2.4. Screening of Polymers
2.5. Preparation of Solid Dispersion
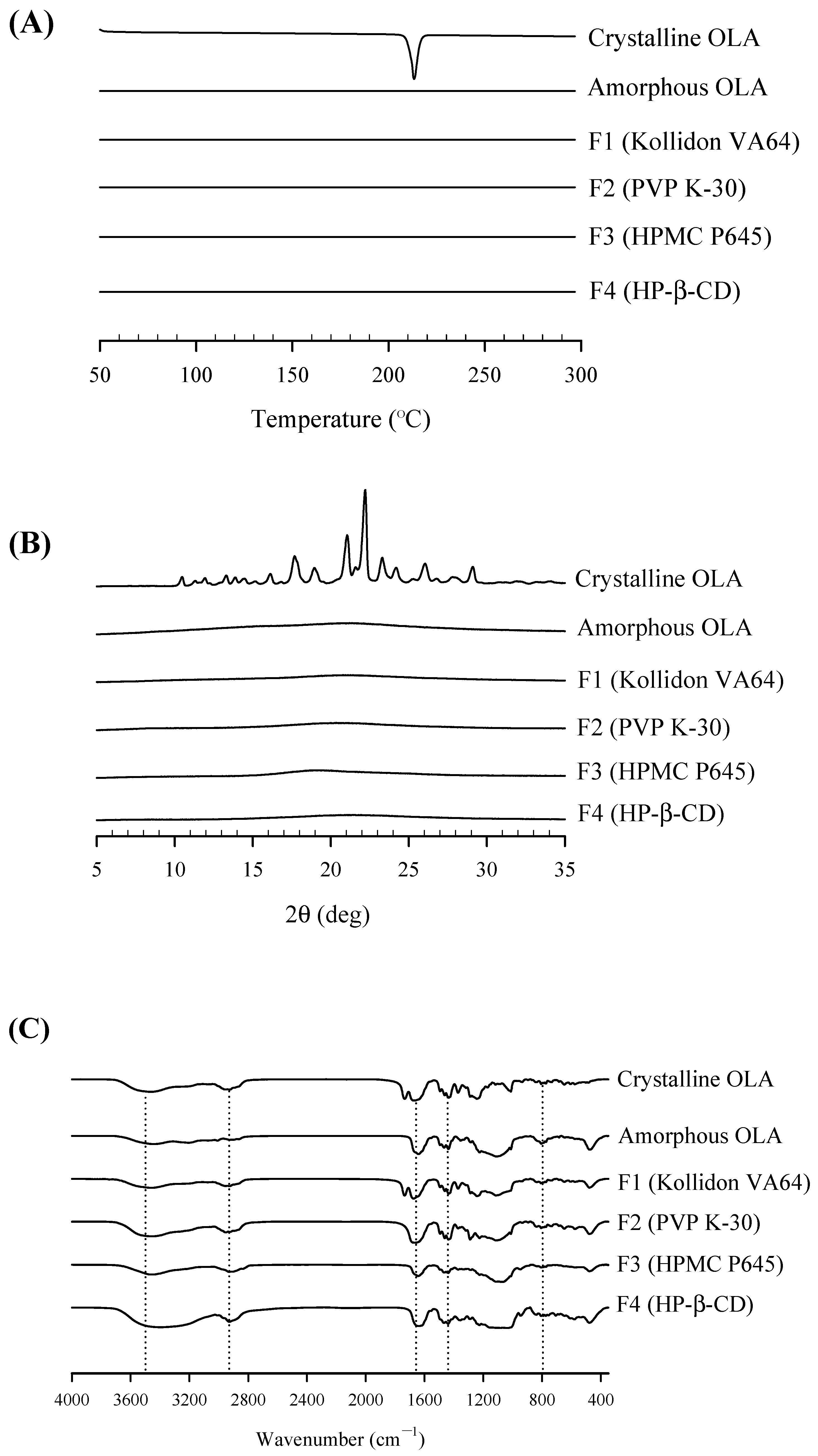
2.6. Physicochemical Characterization of OLA-SD
2.6.1. Surface and Morphological Features
2.6.2. Thermal Properties
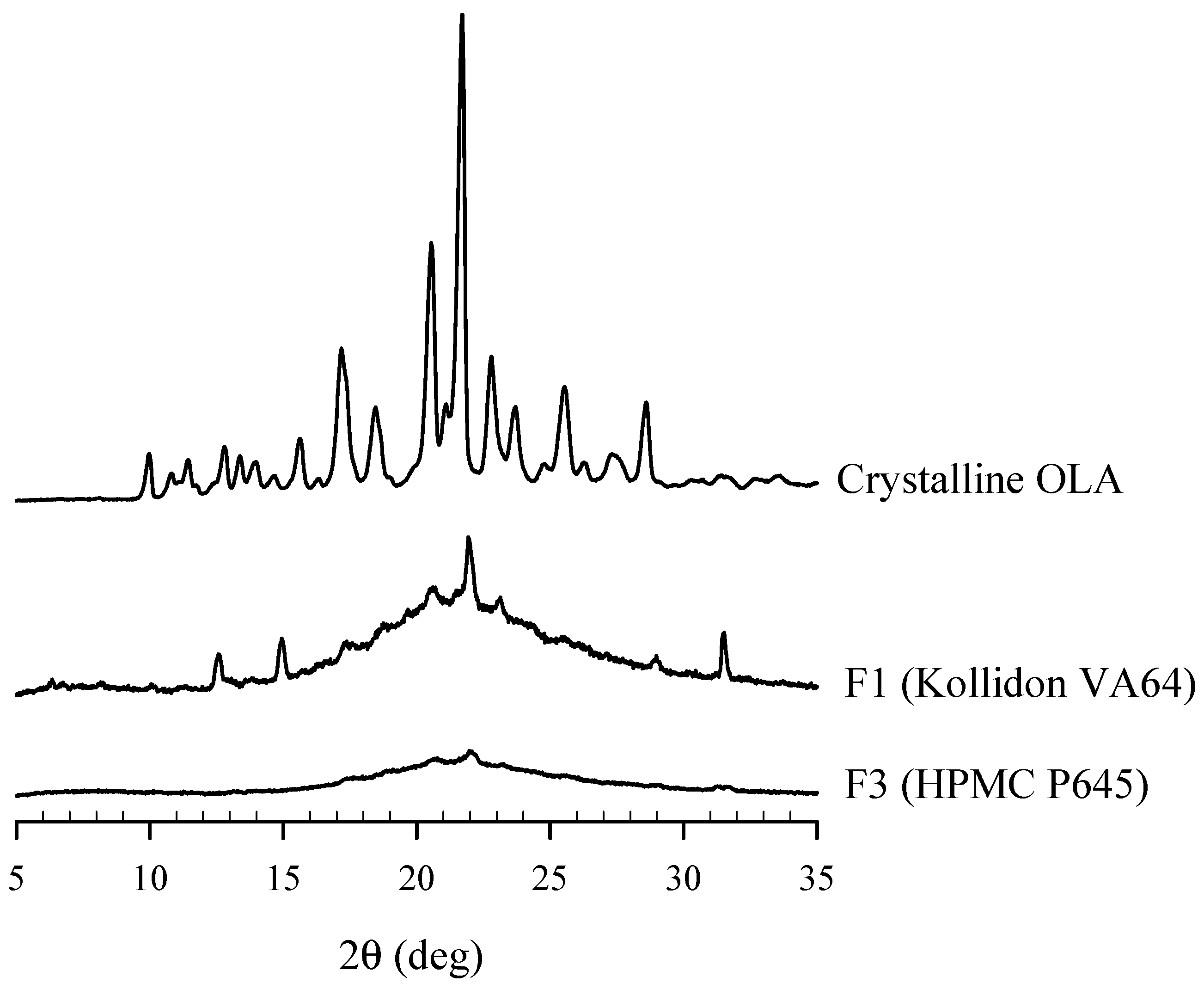
2.6.3. Crystallinity State
2.6.4. Fourier Transform Infrared Spectroscopy Analysis
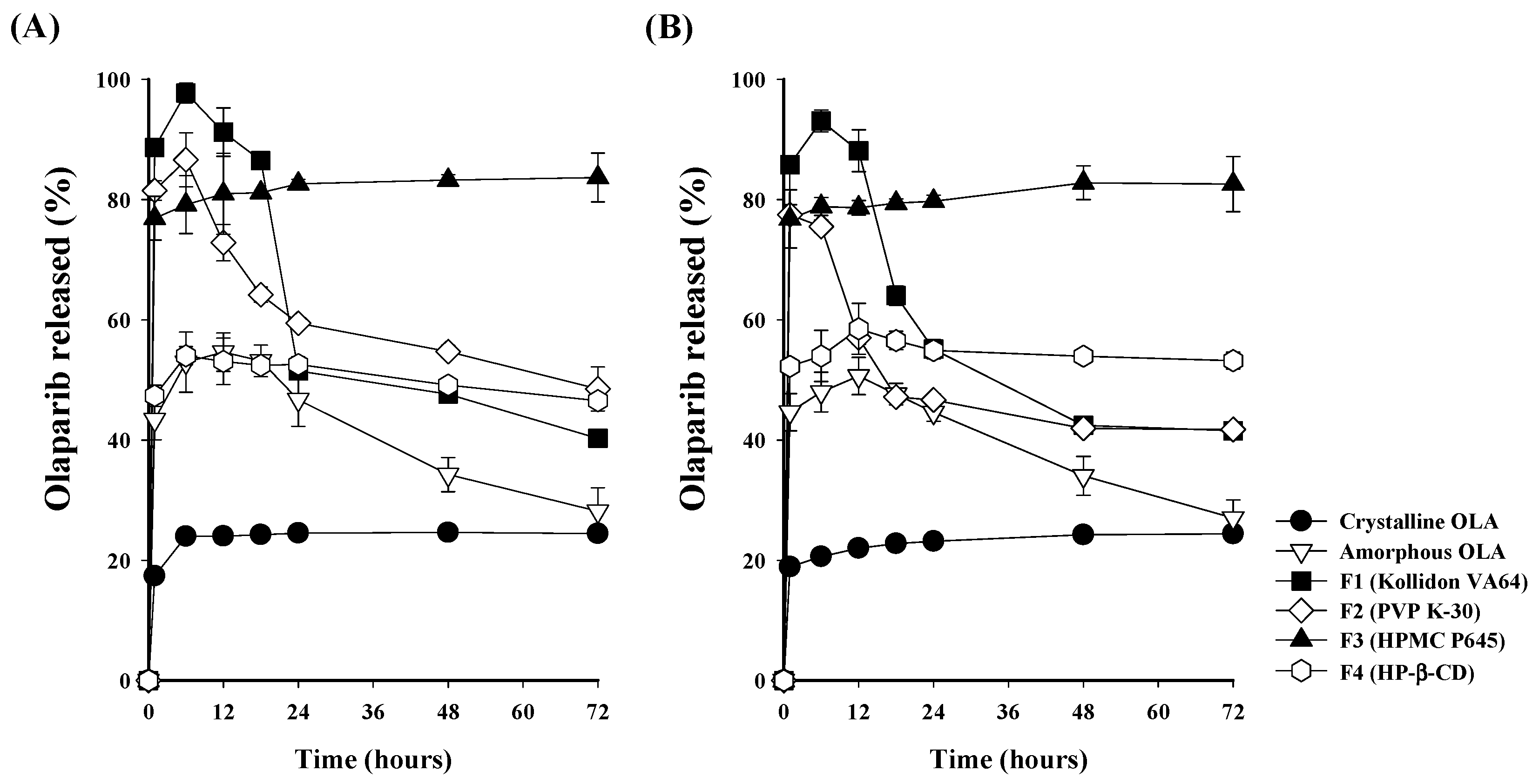
2.7. In Vitro OLA-SD Release
2.8. Morphological Changes in OLA-SD Dispersed in Aqueous Solution
2.9. In Vivo Pharmacokinetic Study
2.10. Statistical Analyses
3. Results
3.1. Solubility of OLA
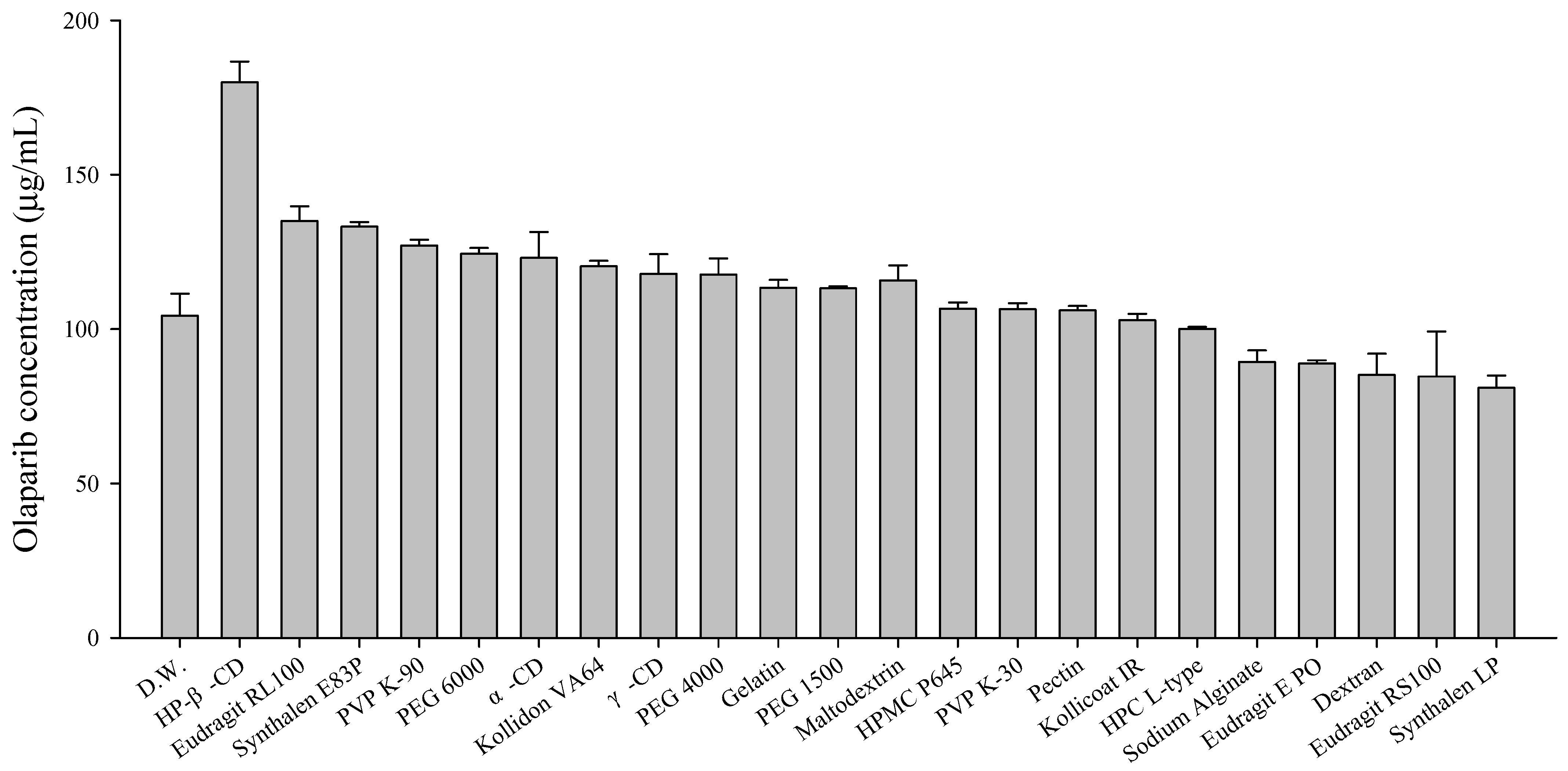
3.2. Selection of Polymers for the Solid Dispersion
3.3. Physicochemical Characterization of OLA-SD
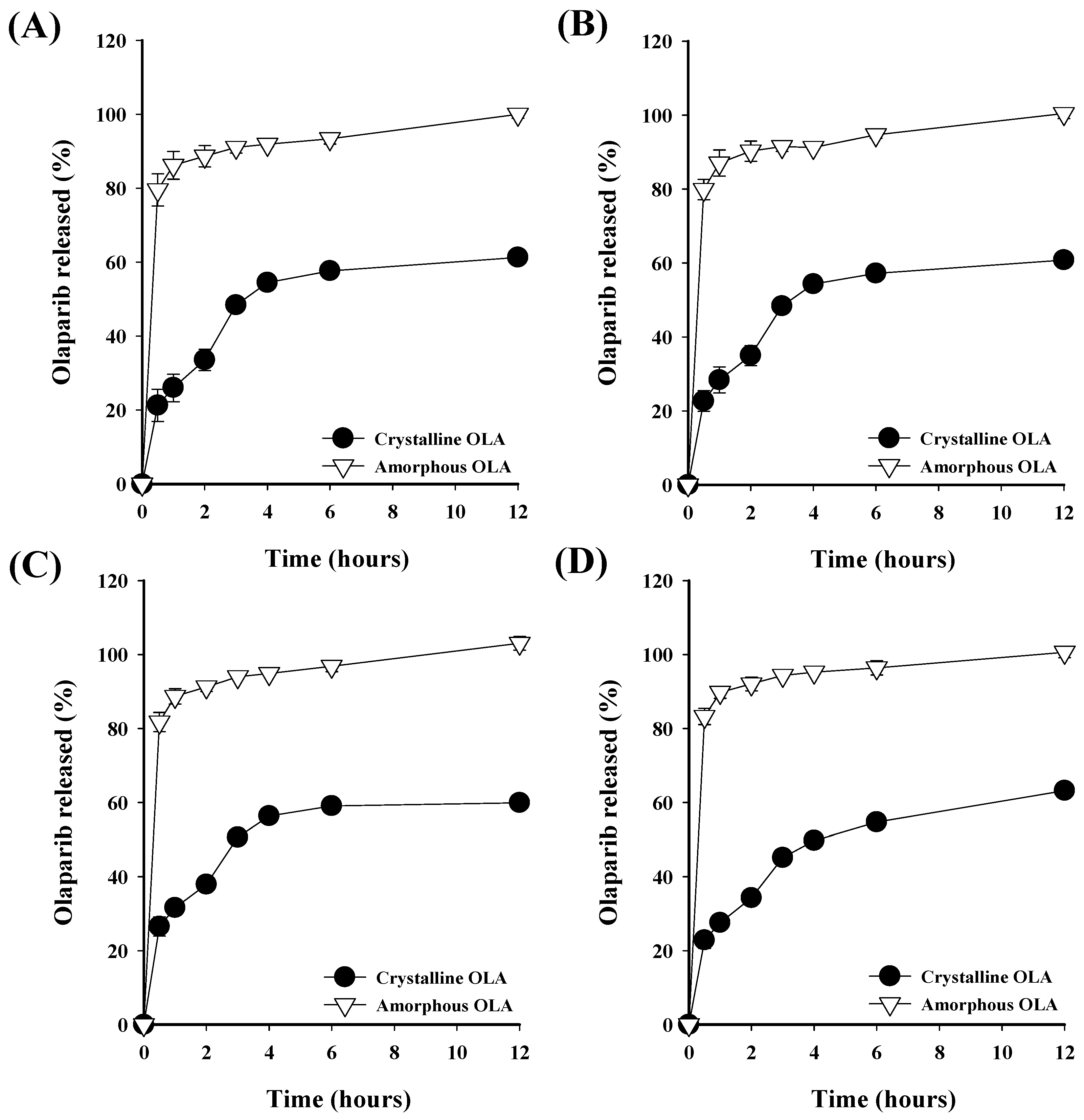
3.4. Dissolution Profile of Prepared OLA-SDs
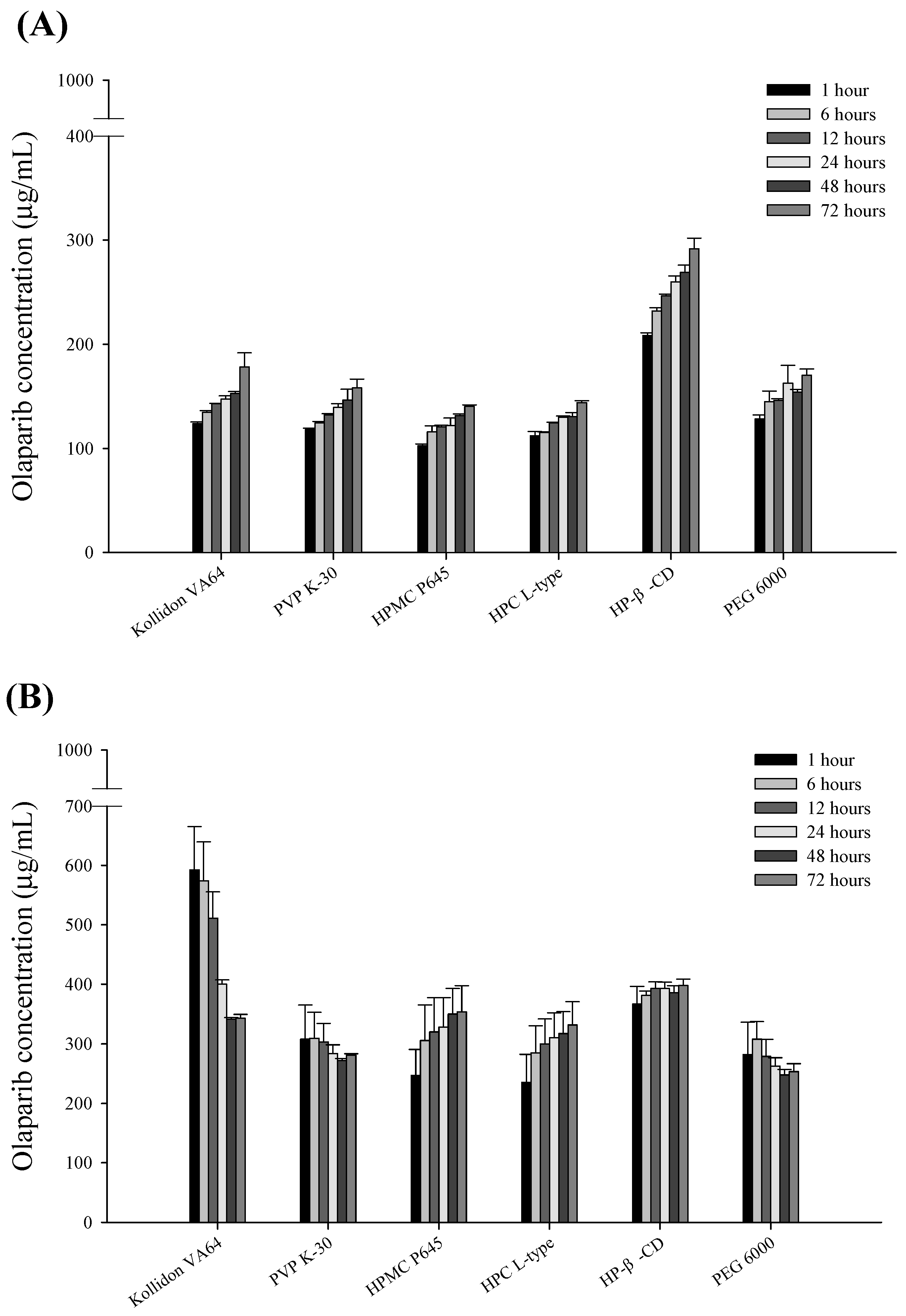
3.5. Comparison of the Characteristics of OLA-SDs Dispersed in Aqueous Solution
3.6. In Vivo Pharmacokinetic Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chatterjee, I.; Roy, D.; Panda, G. A scalable and eco-friendly total synthesis of poly (ADP-ribose) polymerase inhibitor Olaparib. Green Chem. 2023, 25, 9097–9102. [Google Scholar] [CrossRef]
- Freire Boullosa, L.; Van Loenhout, J.; Flieswasser, T.; Hermans, C.; Merlin, C.; Lau, H.W.; Marcq, E.; Verschuuren, M.; De Vos, W.H.; Lardon, F. Auranofin synergizes with the PARP inhibitor olaparib to induce ROS-mediated cell death in mutant p53 cancers. Antioxidants 2023, 12, 667. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.-A.; Dhillon, S. Olaparib tablet: A review in ovarian cancer maintenance therapy. Target. Oncol. 2018, 13, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Hirlekar, B.U.; Nuthi, A.; Singh, K.D.; Murty, U.S.; Dixit, V.A. An overview of compound properties, multiparameter optimization, and computational drug design methods for PARP-1 inhibitor drugs. Eur. J. Med. Chem. 2023, 252, 115300. [Google Scholar] [CrossRef] [PubMed]
- Orleni, M.; Canil, G.; Posocco, B.; Gagno, S.; Toffoli, G. Bioanalytical Methods for Poly (ADP-Ribose) Polymerase Inhibitor Quantification: A Review for Therapeutic Drug Monitoring. Ther. Drug Monit. 2023, 45, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Rocha, B.; de Morais, L.A.; Viana, M.C.; Carneiro, G. Promising strategies for improving oral bioavailability of poor water-soluble drugs. Expert Opin. Drug Discov. 2023, 18, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.; Pyo, Y.-C.; Kim, D.-H.; Lee, S.-E.; Kim, J.-K.; Park, J.-S. Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef]
- Lee, S.-M.; Lee, J.-G.; Yun, T.-H.; Cho, J.-H.; Kim, K.-S. Enhanced Stability and Improved Oral Absorption of Enzalutamide with Self-Nanoemulsifying Drug Delivery System. Int. J. Mol. Sci. 2024, 25, 1197. [Google Scholar] [CrossRef] [PubMed]
- Gala, U.H.; Miller, D.A.; Williams III, R.O. Harnessing the therapeutic potential of anticancer drugs through amorphous solid dispersions. Biochim. Biophys. Acta (BBA) Rev. Cancer 2020, 1873, 188319. [Google Scholar] [CrossRef]
- Okada, K.; Ono, T.; Hayashi, Y.; Kumada, S.; Onuki, Y. Use of Time-Domain NMR for 1H T1 Relaxation Measurement and Fitting Analysis in Homogeneity Evaluation of Amorphous Solid Dispersion. J. Pharm. Sci. 2024, 113, 680–687. [Google Scholar] [CrossRef]
- Saha, S.K.; Joshi, A.; Singh, R.; Jana, S.; Dubey, K. An investigation into solubility and dissolution improvement of alectinib hydrochloride as a third-generation amorphous solid dispersion. J. Drug Deliv. Sci. Technol. 2023, 81, 104259. [Google Scholar] [CrossRef]
- Huang, R.; Han, J.; Wang, R.; Zhao, X.; Qiao, H.; Chen, L.; Li, W.; Di, L.; Zhang, W.; Li, J. Surfactant-free solid dispersion of BCS class IV drug in an amorphous chitosan oligosaccharide matrix for concomitant dissolution in vitro-permeability increase. Eur. J. Pharm. Sci. 2019, 130, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Panizzon, G.P.; Giacomini Bueno, F.; Ueda-Nakamura, T.; Nakamura, C.V.; Dias Filho, B.P. Manufacturing different types of solid dispersions of BCS class iv polyphenol (daidzein) by spray drying: Formulation and bioavailability. Pharmaceutics 2019, 11, 492. [Google Scholar] [CrossRef] [PubMed]
- Beig, A.; Fine-Shamir, N.; Lindley, D.; Miller, J.M.; Dahan, A. Advantageous solubility-permeability interplay when using amorphous solid dispersion (ASD) formulation for the BCS class IV P-gp substrate rifaximin: Simultaneous increase of both the solubility and the permeability. AAPS J. 2017, 19, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Dai, W.-G. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm. Sin. B 2014, 4, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Vo, C.L.-N.; Park, C.; Lee, B.-J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Han, W.; Shu, Y.; Li, M.; Sun, Y.; Sui, X.; Liu, B.; Tian, B.; Liu, Y.; Fu, Q. Liquid–liquid phase separation drug aggregate: Merit for oral delivery of amorphous solid dispersions. J. Control. Release 2023, 353, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Ali, M.; Baboota, S.; Ahuja, A.; Kumar, A.; Ali, J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS PharmSciTech 2010, 11, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Hu, Y.; Liu, L.; Su, L.; Li, N.; Yu, J.; Tang, B.; Yang, Z. Physical stability of amorphous solid dispersions: A physicochemical perspective with thermodynamic, kinetic and environmental aspects. Pharm. Res. 2018, 35, 125. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Kim, S.-B.; Choi, S.-H.; Nguyen, T.-T.-L.; Ahn, S.-H.; Moon, K.-S.; Cho, K.-H.; Sim, T.-Y.; Heo, E.-J.; Kim, S.T. Development and Evaluation of Self-Microemulsifying Drug Delivery System for Improving Oral Absorption of Poorly Water-Soluble Olaparib. Pharmaceutics 2023, 15, 1669. [Google Scholar] [CrossRef]
- Pathade, A.D.; Kommineni, N.; Bulbake, U.; Thummar, M.M.; Samanthula, G.; Khan, W. Preparation and comparison of oral bioavailability for different nano-formulations of olaparib. AAPS PharmSciTech 2019, 20, 276. [Google Scholar] [CrossRef]
- Yasu, T.; Nishijima, R.; Ikuta, R.; Shirota, M.; Iwase, H. Development of a simple high-performance liquid chromatography-ultraviolet detection method for olaparib in patients with ovarian cancer. Drug Discov. Ther. 2023, 17, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Turunen, E.; Mannila, J.; Laitinen, R.; Riikonen, J.; Lehto, V.-P.; Järvinen, T.; Ketolainen, J.; Järvinen, K.; Jarho, P. Fast-dissolving sublingual solid dispersion and cyclodextrin complex increase the absorption of perphenazine in rabbits. J. Pharm. Pharmacol. 2011, 63, 19–25. [Google Scholar] [CrossRef]
- Pignatello, R.; Ferro, M.; De Guidi, G.; Salemi, G.; Vandelli, M.A.; Guccione, S.; Geppi, M.; Forte, C.; Puglisi, G. Preparation, characterisation and photosensitivity studies of solid dispersions of diflunisal and Eudragit RS100® and RL100®. Int. J. Pharm. 2001, 218, 27–42. [Google Scholar] [CrossRef]
- Liu, H.; Wang, P.; Zhang, X.; Shen, F.; Gogos, C.G. Effects of extrusion process parameters on the dissolution behavior of indomethacin in Eudragit® E PO solid dispersions. Int. J. Pharm. 2010, 383, 161–169. [Google Scholar] [CrossRef] [PubMed]
- LaFountaine, J.S.; Prasad, L.K.; Miller, D.A.; McGinity, J.W.; Williams III, R.O. Mucoadhesive amorphous solid dispersions for sustained release of poorly water soluble drugs. Eur. J. Pharm. Biopharm. 2017, 113, 157–167. [Google Scholar] [CrossRef]
- Kyaw Oo, M.; Mandal, U.K.; Chatterjee, B. Polymeric behavior evaluation of PVP K30-poloxamer binary carrier for solid dispersed nisoldipine by experimental design. Pharm. Dev. Technol. 2017, 22, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Szafraniec-Szczęsny, J.; Antosik-Rogóż, A.; Kurek, M.; Gawlak, K.; Górska, A.; Peralta, S.; Knapik-Kowalczuk, J.; Kramarczyk, D.; Paluch, M.; Jachowicz, R. How does the addition of Kollidon® VA64 inhibit the recrystallization and improve ezetimibe dissolution from amorphous solid dispersions? Pharmaceutics 2021, 13, 147. [Google Scholar] [CrossRef]
- Janssens, S.; Anné, M.; Rombaut, P.; Van den Mooter, G. Spray drying from complex solvent systems broadens the applicability of Kollicoat IR as a carrier in the formulation of solid dispersions. Eur. J. Pharm. Sci. 2009, 37, 241–248. [Google Scholar] [CrossRef]
- Verheyen, S.; Blaton, N.; Kinget, R.; Van den Mooter, G. Mechanism of increased dissolution of diazepam and temazepam from polyethylene glycol 6000 solid dispersions. Int. J. Pharm. 2002, 249, 45–58. [Google Scholar] [CrossRef]
- Benavent, C.; Torrado-Salmerón, C.; Torrado-Santiago, S. Development of a solid dispersion of nystatin with maltodextrin as a carrier agent: Improvements in antifungal efficacy against Candida spp. biofilm infections. Pharmaceuticals 2021, 14, 397. [Google Scholar] [CrossRef] [PubMed]
- Varshosaz, J.; Ahmadi, F.; Emami, J.; Tavakoli, N.; Minaiyan, M.; Mahzouni, P.; Dorkoosh, F. Colon delivery of budesonide using solid dispersion in dextran for the treatment and secondary prevention of ulcerative colitis in rat. Int. J. Prev. Med. 2010, 1, 115. [Google Scholar] [PubMed]
- Fathanah, A.; Setyawan, D.; Sari, R. Improving solubility and dissolution of meloxicam by solid dispersion using hydroxypropyl methylcellulose 2910 3 cps and nicotinamide. J. Basic Clin. Physiol. Pharmacol. 2019, 30. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Ahmad, S.; Tarabokija, J.; Parker, N.; Bilgili, E. Spray-dried amorphous solid dispersions of griseofulvin in HPC/soluplus/SDS: Elucidating the multifaceted impact of SDS as a minor component. Pharmaceutics 2020, 12, 197. [Google Scholar] [CrossRef] [PubMed]
- Borba, P.A.A.; Pinotti, M.; de Campos, C.E.M.; Pezzini, B.R.; Stulzer, H.K. Sodium alginate as a potential carrier in solid dispersion formulations to enhance dissolution rate and apparent water solubility of BCS II drugs. Carbohydr. Polym. 2016, 137, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Pas, T.; Vergauwen, B.; Van den Mooter, G. Exploring the feasibility of the use of biopolymers as a carrier in the formulation of amorphous solid dispersions–Part I: Gelatin. Int. J. Pharm. 2018, 535, 47–58. [Google Scholar] [CrossRef]
- Kumar, S.; Gupta, S.K. Mango peel pectin as a carrier for solid dispersions. Polim. Med. 2013, 43, 231–233. [Google Scholar] [PubMed]
- Sosnik, A.; Seremeta, K.P. Advantages and challenges of the spray-drying technology for the production of pure drug particles and drug-loaded polymeric carriers. Adv. Colloid Interface Sci. 2015, 223, 40–54. [Google Scholar] [CrossRef]
- Sandhu, H.; Shah, N.; Chokshi, H.; Malick, A.W. Overview of amorphous solid dispersion technologies. In Amorphous Solid Dispersions: Theory and Practice; Springer: Berlin/Heidelberg, Germany, 2014; pp. 91–122. [Google Scholar]
- Choi, M.-J.; Woo, M.R.; Choi, H.-G.; Jin, S.G. Effects of polymers on the drug solubility and dissolution enhancement of poorly water-soluble rivaroxaban. Int. J. Mol. Sci. 2022, 23, 9491. [Google Scholar] [CrossRef]
- Kim, D.S.; Choi, J.S.; Kim, D.W.; Kim, K.S.; Seo, Y.G.; Cho, K.H.; Kim, J.O.; Yong, C.S.; Youn, Y.S.; Lim, S.-J. Comparison of solvent⿿ wetted and kneaded l-sulpiride loaded solid dispersions: Powder characterization and in vivo evaluation. Int. J. Pharm. 2016, 511, 351–358. [Google Scholar] [CrossRef]
- Choi, M.-J.; Woo, M.R.; Baek, K.; Park, J.H.; Joung, S.; Choi, Y.S.; Choi, H.-G.; Jin, S.G. Enhanced oral bioavailability of Rivaroxaban-Loaded microspheres by optimizing the polymer and surfactant based on molecular interaction mechanisms. Mol. Pharm. 2023, 20, 4153–4164. [Google Scholar] [CrossRef]
- Park, J.-B.; Kang, C.-Y.; Kang, W.-S.; Choi, H.-G.; Han, H.-K.; Lee, B.-J. New investigation of distribution imaging and content uniformity of very low dose drugs using hot-melt extrusion method. Int. J. Pharm. 2013, 458, 245–253. [Google Scholar] [CrossRef]
- Nepal, P.R.; Han, H.-K.; Choi, H.-K. Enhancement of solubility and dissolution of Coenzyme Q10 using solid dispersion formulation. Int. J. Pharm. 2010, 383, 147–153. [Google Scholar] [CrossRef]
- Ha, E.-S.; Baek, I.-h.; Cho, W.; Hwang, S.-J.; Kim, M.-S. Preparation and evaluation of solid dispersion of atorvastatin calcium with Soluplus® by spray drying technique. Chem. Pharm. Bull. 2014, 62, 545–551. [Google Scholar] [CrossRef]
- Duret, C.; Wauthoz, N.; Sebti, T.; Vanderbist, F.; Amighi, K. Solid dispersions of itraconazole for inhalation with enhanced dissolution, solubility and dispersion properties. Int. J. Pharm. 2012, 428, 103–113. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Pereira, J.; Edgar, K.J.; Taylor, L.S. Influence of additives on the properties of nanodroplets formed in highly supersaturated aqueous solutions of ritonavir. Mol. Pharm. 2013, 10, 3392–3403. [Google Scholar] [CrossRef]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef]
- Araujo-Fernandez, A.S.; Uribe-Villarreal, J.C.; Perez-Chauca, E.; Alva-Plasencia, P.M.; Caballero-Aquiño, O.E.; Ganoza-Yupanqui, M.L. Validation of a UV spectrophotometric method to quantify losartan potassium in tablets from the dissolution test at pH 1.2, 4.5 and 6.8. J. Pharm. Pharmacogn. Res. 2022, 10, 310–317. [Google Scholar] [CrossRef]
- Murdande, S.B.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Solubility advantage of amorphous pharmaceuticals: II. Application of quantitative thermodynamic relationships for prediction of solubility enhancement in structurally diverse insoluble pharmaceuticals. Pharm. Res. 2010, 27, 2704–2714. [Google Scholar] [CrossRef]
- Song, J.; Kawakami, K. Nucleation During Storage Impeded Supersaturation in the Dissolution Process of Amorphous Celecoxib. Mol. Pharm. 2023, 20, 4050–4057. [Google Scholar] [CrossRef]
- Feng, X.; Ye, X.; Park, J.-B.; Lu, W.; Morott, J.; Beissner, B.; Lian, Z.J.; Pinto, E.; Bi, V.; Porter, S. Evaluation of the recrystallization kinetics of hot-melt extruded polymeric solid dispersions using an improved Avrami equation. Drug Dev. Ind. Pharm. 2015, 41, 1479–1487. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X. Preparation and characterization of the inclusion complex of Ofloxacin with β-CD and HP-β-CD. J. Incl. Phenom. Macrocycl. Chem. 2011, 69, 173–179. [Google Scholar] [CrossRef]
- Kim, J.S.; Choi, Y.J.; Woo, M.R.; Cheon, S.; Ji, S.H.; Im, D.; ud Din, F.; Kim, J.O.; Youn, Y.S.; Oh, K.T. New potential application of hydroxypropyl-β-cyclodextrin in solid self-nanoemulsifying drug delivery system and solid dispersion. Carbohydr. Polym. 2021, 271, 118433. [Google Scholar] [CrossRef]
- Nair, A.R.; Lakshman, Y.D.; Anand, V.S.K.; Sree, K.N.; Bhat, K.; Dengale, S.J. Overview of extensively employed polymeric carriers in solid dispersion technology. AAPS PharmSciTech 2020, 21, 309. [Google Scholar] [CrossRef]
- Wilson, V.R.; Lou, X.; Osterling, D.J.; Stolarik, D.F.; Jenkins, G.J.; Nichols, B.L.; Dong, Y.; Edgar, K.J.; Zhang, G.G.; Taylor, L.S. Amorphous solid dispersions of enzalutamide and novel polysaccharide derivatives: Investigation of relationships between polymer structure and performance. Sci. Rep. 2020, 10, 18535. [Google Scholar] [CrossRef]
- Chavan, R.B.; Rathi, S.; Jyothi, V.G.S.; Shastri, N.R. Cellulose based polymers in development of amorphous solid dispersions. Asian J. Pharm. Sci. 2019, 14, 248–264. [Google Scholar] [CrossRef]
- Trivino, A.; Gumireddy, A.; Meng, F.; Prasad, D.; Chauhan, H. Drug–polymer miscibility, interactions, and precipitation inhibition studies for the development of amorphous solid dispersions for the poorly soluble anticancer drug flutamide. Drug Dev. Ind. Pharm. 2019, 45, 1277–1291. [Google Scholar] [CrossRef]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous solid dispersions: An update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int. J. Pharm. 2020, 586, 119560. [Google Scholar] [CrossRef]
- Goyal, M.; Tulsyan, G.; Kanabar, D.D.; Chavan, T.; Muth, A.; Gupta, V. Poly vinyl pyrrolidone (PVP) based inhaled delivery carriers for olaparib for non-small cell lung cancer (NSCLC) treatment. J. Drug Deliv. Sci. Technol. 2023, 87, 104767. [Google Scholar] [CrossRef]
- Zaini, E.; Evert, A.; Octavia, M.D. Increase in Dissolution Rate of Simvastatin by Amorphous Solid Dispersion System with Hydroxypropylmethylcellulose Polymer. Orient. J. Chem. 2017, 33, 2080. [Google Scholar] [CrossRef]
- Liu, P.; Zhou, J.-y.; Chang, J.-h.; Liu, X.-g.; Xue, H.-f.; Wang, R.-x.; Li, Z.-s.; Li, C.-s.; Wang, J.; Liu, C.-z. Soluplus-mediated diosgenin amorphous solid dispersion with high solubility and high stability: Development, characterization and oral bioavailability. In Drug Design, Development and Therapy; Taylor & Francis: Abingdon, UK, 2020; Volume 14, pp. 2959–2975. [Google Scholar]
- Alali, A.S.; Kalam, M.A.; Ahmed, M.M.; Aboudzadeh, M.A.; Alhudaithi, S.S.; Anwer, M.K.; Fatima, F.; Iqbal, M. Nanocrystallization improves the solubilization and cytotoxic effect of a poly (Adp-ribose)-polymerase-i inhibitor. Polymers 2022, 14, 4827. [Google Scholar] [CrossRef]
- Anwer, M.K.; Ali, E.A.; Iqbal, M.; Ahmed, M.M.; Aldawsari, M.F.; Saqr, A.A.; Alalaiwe, A.; Soliman, G.A. Development of chitosan-coated PLGA-based nanoparticles for improved oral olaparib delivery: In vitro characterization, and in vivo pharmacokinetic studies. Processes 2022, 10, 1329. [Google Scholar] [CrossRef]
- Huang, Y.-B.; Tsai, Y.-H.; Lee, S.-H.; Chang, J.-S.; Wu, P.-C. Optimization of pH-independent release of nicardipine hydrochloride extended-release matrix tablets using response surface methodology. Int. J. Pharm. 2005, 289, 87–95. [Google Scholar] [CrossRef]
- Guo, Y.; Patel, H.; Saraswat, A.; Mateti, K.V.; Patel, K.; Squillante, E. Screening and optimization of supercritical fluid process for the preparation of albendazole solid dispersion. J. Drug Deliv. Sci. Technol. 2023, 88, 104852. [Google Scholar] [CrossRef]
- Saokham, P.; Muankaew, C.; Jansook, P.; Loftsson, T. Solubility of cyclodextrins and drug/cyclodextrin complexes. Molecules 2018, 23, 1161. [Google Scholar] [CrossRef]
- Ghauri, Z.H.; Islam, A.; Qadir, M.A.; Gull, N.; Haider, B.; Khan, R.U.; Riaz, T. Development and evaluation of pH-sensitive biodegradable ternary blended hydrogel films (chitosan/guar gum/PVP) for drug delivery application. Sci. Rep. 2021, 11, 21255. [Google Scholar] [CrossRef]
- Skrdla, P.J.; Floyd, P.D.; Dell’Orco, P.C. Modeling recrystallization kinetics following the dissolution of amorphous drugs. Mol. Pharm. 2019, 17, 219–228. [Google Scholar] [CrossRef]
- Lee, S.-M.; Lee, J.-G.; Yun, T.-H.; Kim, C.-H.; Cho, J.-H.; Kim, K.-S. The Impact of Polymers on Enzalutamide Solid Self-Nanoemulsifying Drug Delivery System and Improved Bioavailability. Pharmaceutics 2024, 16, 457. [Google Scholar] [CrossRef]
- Miller, J.M.; Beig, A.; Carr, R.A.; Spence, J.K.; Dahan, A. A win–win solution in oral delivery of lipophilic drugs: Supersaturation via amorphous solid dispersions increases apparent solubility without sacrifice of intestinal membrane permeability. Mol. Pharm. 2012, 9, 2009–2016. [Google Scholar] [CrossRef]
- Lee, T.W.-Y.; Boersen, N.A.; Yang, G.; Hui, H.-W. Evaluation of different screening methods to understand the dissolution behaviors of amorphous solid dispersions. Drug Dev. Ind. Pharm. 2014, 40, 1072–1083. [Google Scholar] [CrossRef]
- Dedroog, S.; Huygens, C.; Van den Mooter, G. Chemically identical but physically different: A comparison of spray drying, hot melt extrusion and cryo-milling for the formulation of high drug loaded amorphous solid dispersions of naproxen. Eur. J. Pharm. Biopharm. 2019, 135, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zhang, J.; Zhao, Y.; Zhang, L.; Feng, J.; Zhang, L. Discovery of a potent olaparib–chlorambucil hybrid inhibitor of PARP1 for the treatment of cancer. Front. Pharmacol. 2023, 13, 1054616. [Google Scholar] [CrossRef]
- Dai, X.-L.; Pang, B.-W.; Lv, W.-T.; Zhen, J.-F.; Gao, L.; Li, C.-W.; Xiong, J.; Lu, T.-B.; Chen, J.-M. Improving the physicochemical and pharmacokinetic properties of olaparib through cocrystallization strategy. Int. J. Pharm. 2023, 647, 123497. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, H.; He, H.; Sun, L.; Zhang, X.; Wang, Q.; Li, K.; He, Z. Cooperative effect of polyvinylpyrrolidone and HPMC E5 on dissolution and bioavailability of nimodipine solid dispersions and tablets. Asian J. Pharm. Sci. 2019, 14, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Marsac, P.J.; Konno, H.; Rumondor, A.C.; Taylor, L.S. Recrystallization of nifedipine and felodipine from amorphous molecular level solid dispersions containing poly (vinylpyrrolidone) and sorbed water. Pharm. Res. 2008, 25, 647–656. [Google Scholar] [CrossRef]
- Huang, S.; O’Donnell, K.P.; Keen, J.M.; Rickard, M.A.; McGinity, J.W.; Williams, R.O. A new extrudable form of hypromellose: AFFINISOL™ HPMC HME. AAPS PharmSciTech 2016, 17, 106–119. [Google Scholar] [CrossRef]
- Maddineni, S.; Battu, S.K.; Morott, J.; Majumdar, S.; Murthy, S.; Repka, M.A. Influence of process and formulation parameters on dissolution and stability characteristics of Kollidon® VA 64 hot-melt extrudates. AAPS PharmSciTech 2015, 16, 444–454. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation (mg) | Amorphous OLA | F1 | F2 | F3 | F4 |
|---|---|---|---|---|---|
| OLA | 150 | 150 | 150 | 150 | 150 |
| Kollidon VA64 | - | 300 | - | - | - |
| PVP K-30 | - | - | 300 | - | - |
| HPMC P645 | - | - | - | 300 | - |
| HP-β-CD | - | - | - | - | 300 |
| Colloidal silica | - | 75 | 75 | 75 | 75 |
| (Ethanol) | (11,000) | (11,000) | (11,000) | (11,000) | (11,000) |
| (D.W.) | (4000) | (4000) | (4000) | (4000) | (4000) |
| Total | 150 | 525 | 525 | 525 | 525 |
| PK Parameters | Crystalline OLA | Amorphous OLA | F1 (Kollidon VA64) | F3 (HPMC P645) |
|---|---|---|---|---|
| AUC0–24 (ng∙h/mL) | 370.11 ± 63.75 | 471.40 ± 150.17 | 995.38 ± 194.76 | 1551.47 ± 484.09 |
| Cmax (ng/mL) | 187.31 ± 82.55 | 415.24 ± 148.20 | 1459.05 ± 548.86 | 2001.25 ± 734.43 |
| Tmax (h) | 0.63 ± 0.31 | 0.29 ± 0.10 | 0.25 ± 0.00 | 0.29 ± 0.10 |
| Kel (h−1) | 0.19 ± 0.06 | 0.20 ± 0.05 | 0.18 ± 0.04 | 0.21 ± 0.04 |
| T1/2 (h) | 4.18 ± 2.27 | 3.68 ± 1.04 | 4.10 ± 0.84 | 3.36 ± 0.63 |
| MRT (h) | 2.97 ± 0.69 | 4.28 ± 4.52 | 3.28 ± 1.14 | 2.04 ± 0.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, T.; Lee, S.; Yun, S.; Cho, D.; Bang, K.; Kim, K. Investigation of Stabilized Amorphous Solid Dispersions to Improve Oral Olaparib Absorption. Pharmaceutics 2024, 16, 958. https://doi.org/10.3390/pharmaceutics16070958
Yun T, Lee S, Yun S, Cho D, Bang K, Kim K. Investigation of Stabilized Amorphous Solid Dispersions to Improve Oral Olaparib Absorption. Pharmaceutics. 2024; 16(7):958. https://doi.org/10.3390/pharmaceutics16070958
Chicago/Turabian StyleYun, Taehan, Sumin Lee, Seowan Yun, Daeyeong Cho, Kyuho Bang, and Kyeongsoo Kim. 2024. "Investigation of Stabilized Amorphous Solid Dispersions to Improve Oral Olaparib Absorption" Pharmaceutics 16, no. 7: 958. https://doi.org/10.3390/pharmaceutics16070958