

A Data-Driven Approach for Leveraging Inline and Offline Data to Determine the Causes of Monoclonal Antibody Productivity Reduction in the Commercial-Scale Cell Culture Process

Abstract
:
1. Introduction
- A data-driven approach is proposed to leverage both the inline and offline data to identify the causes of mAb productivity reduction.
- The features of inline data are extracted using the FDA method and then fused with offline data to obtain an extended feature matrix to represent the cell culture process. The causes of mAb productivity are analyzed via the ccPCA method based on the extended feature matrix.
- A commercial-scale case study is provided to verify the effectiveness of the proposed approach.
2. Materials and Methods
2.1. Preliminaries
2.1.1. kNN
2.1.2. FDA
2.1.3. ccPCA
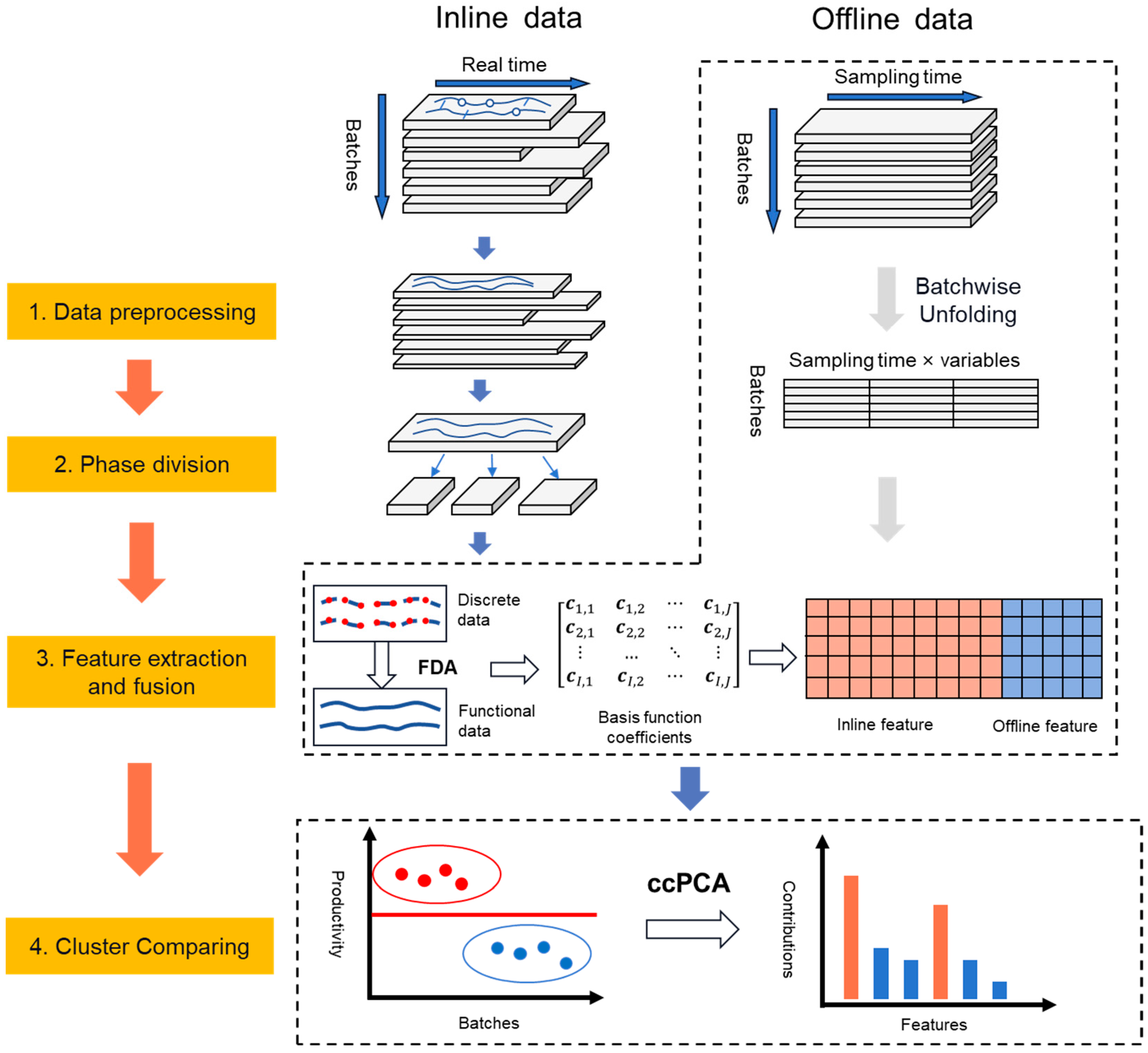
2.2. Proposed Approach
2.2.1. Data Preprocessing
- Missing data caused by network failures: Network failures disconnect the equipment and the real-time database; thus, the values during network downtime are not recorded. The amount of missing data depends on how long it takes to solve network failures.
- Data spikes caused by sensor failures: Data spikes, although not often seen, manifest as a sudden drop or rise in normal data that fall out of the reasonable ranges (e.g., the pH drops to 0 or the temperature rises to 100 °C during the culture process), and can be easily identified by simple rules.
- Variables with little information: There are many sensors equipped in both bioreactors and their accessories, but the variables not directly linked to the bioreactors may not reflect the dynamic characteristics of the cell culture process. Furthermore, some process variables are stationary across the whole batch, and their variances are zero or very low. Such variables are considered to have little contribution to the data analysis of the cell culture process.
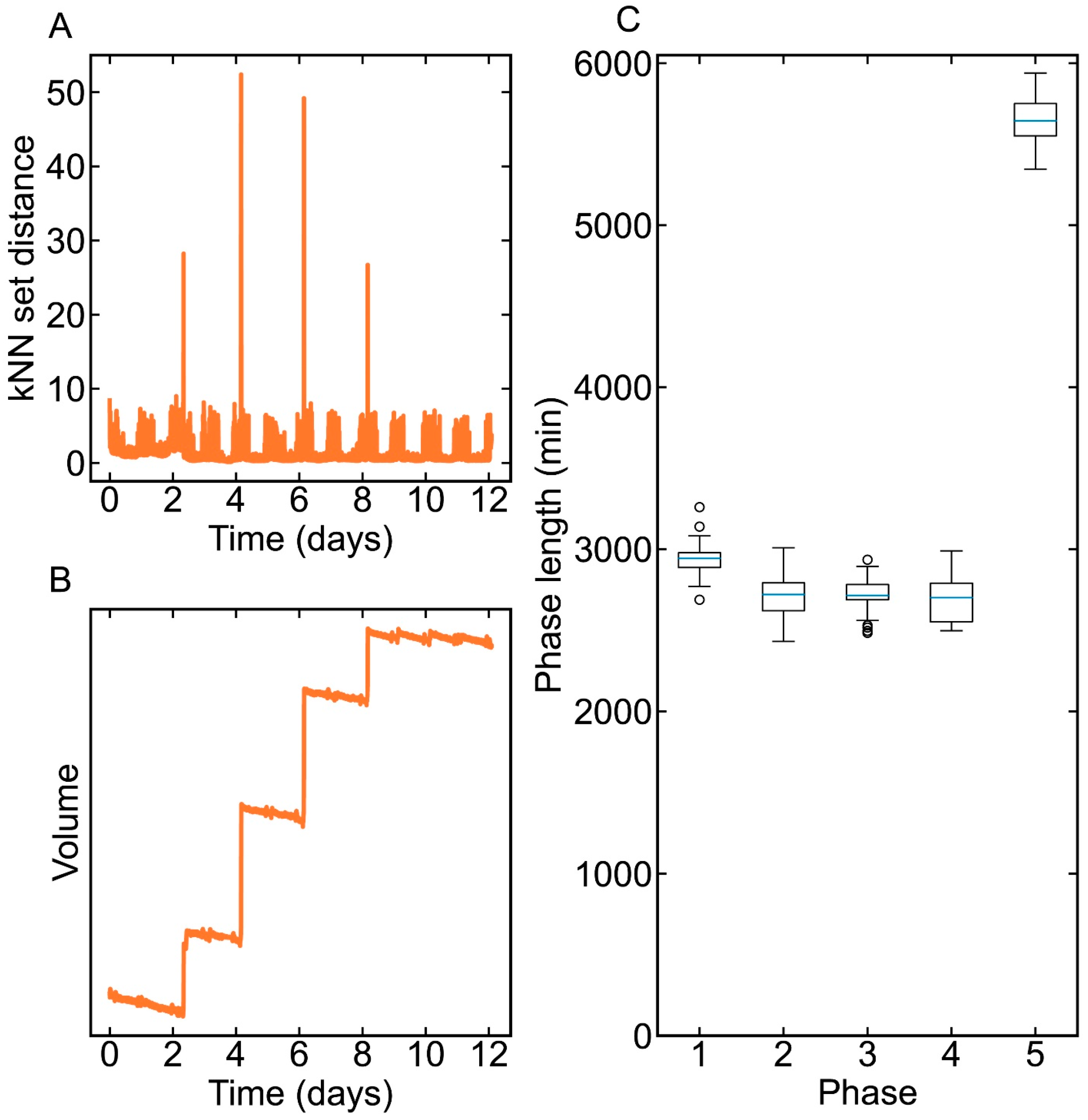
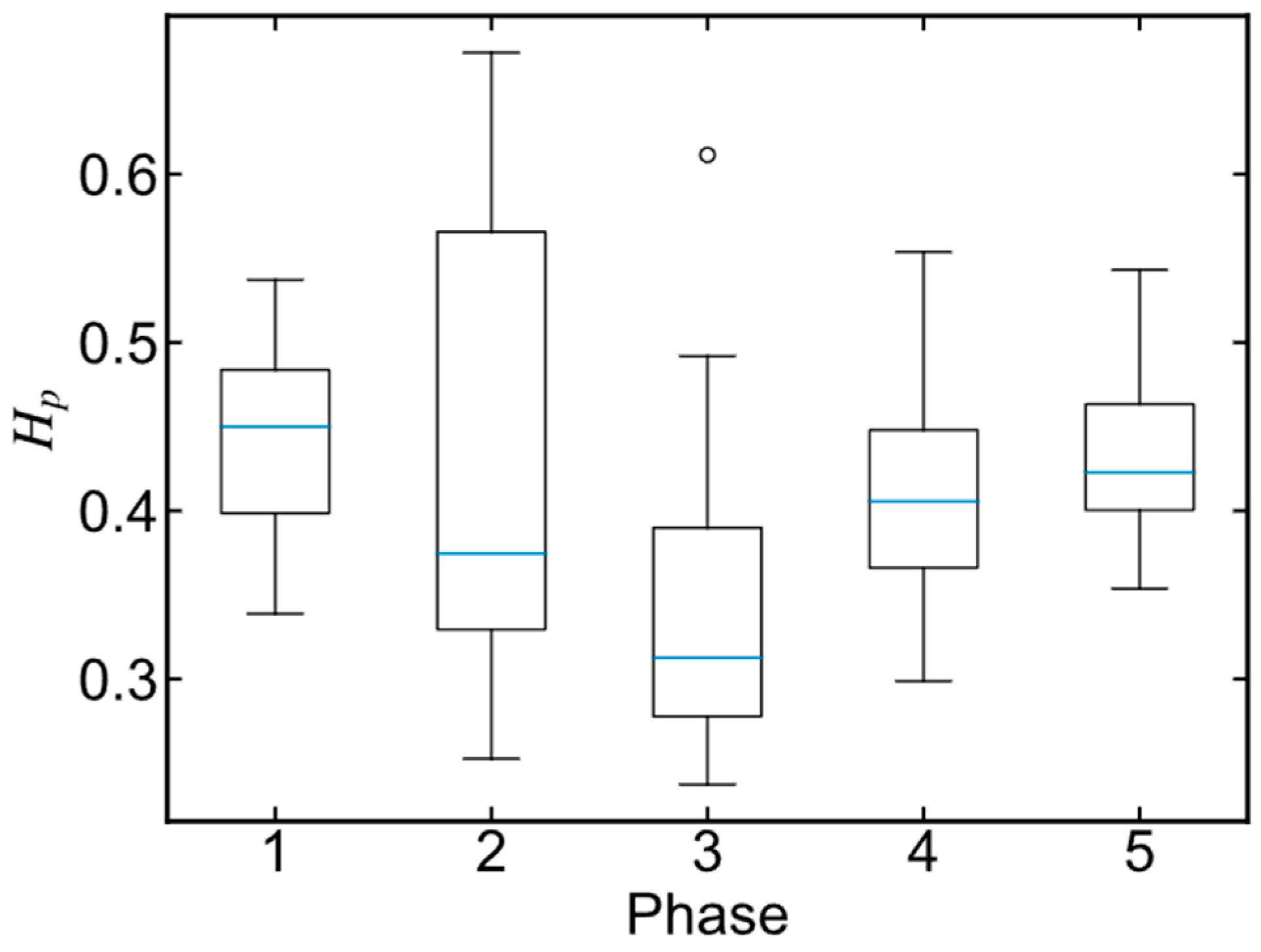
2.2.2. Phase Division
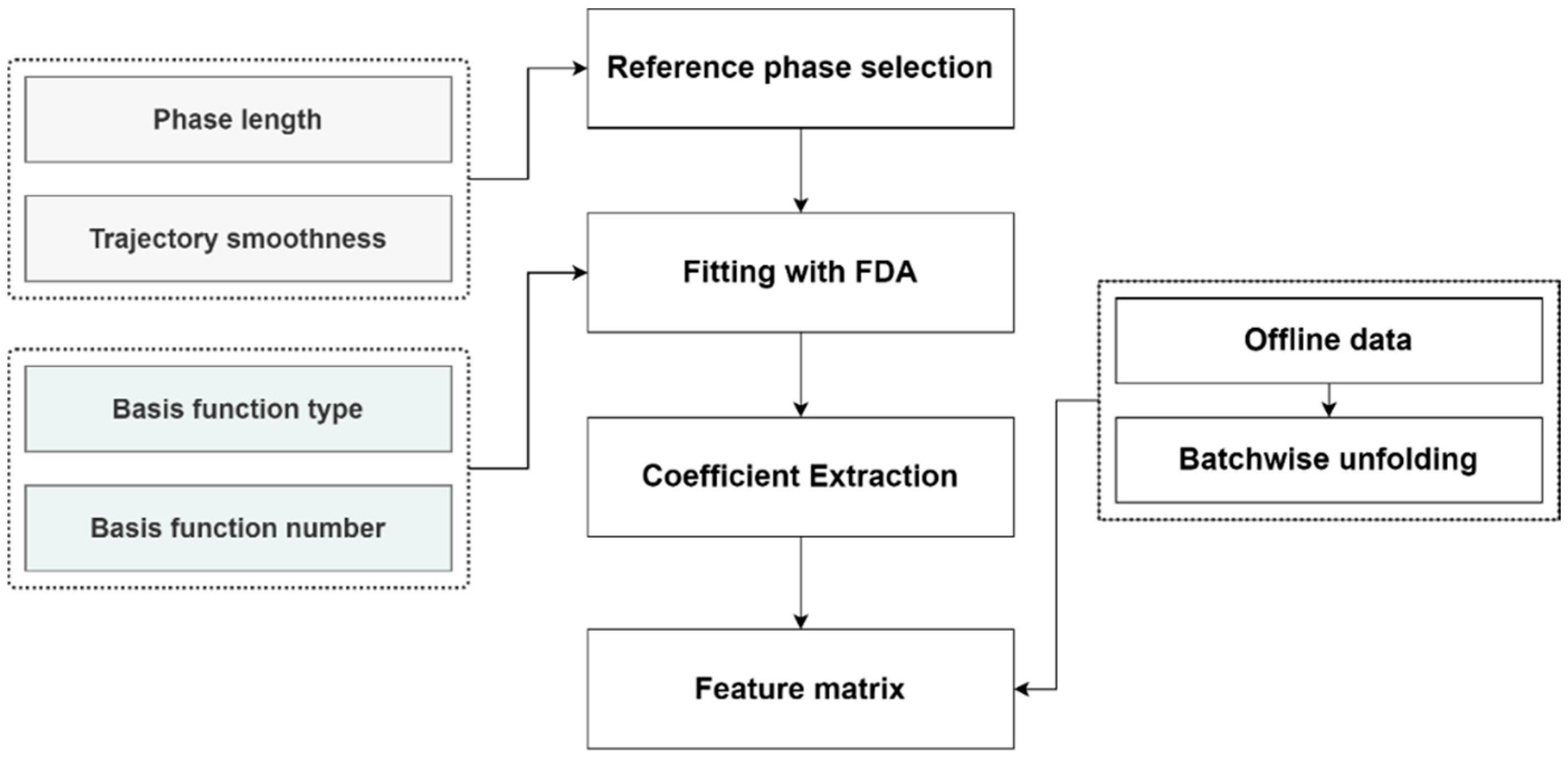
2.2.3. Feature Extraction and Fusion
2.2.4. Cluster Comparing
3. Results and Discussion
3.1. The Cell Culture Process
3.1.1. Process Description
3.1.2. Process Data
3.2. Data Preprocessing Result
3.3. Phase Division Result
3.4. Feature Extraction and Fusion Result
3.4.1. Reference Phase Selection
3.4.2. FDA Fitting Result
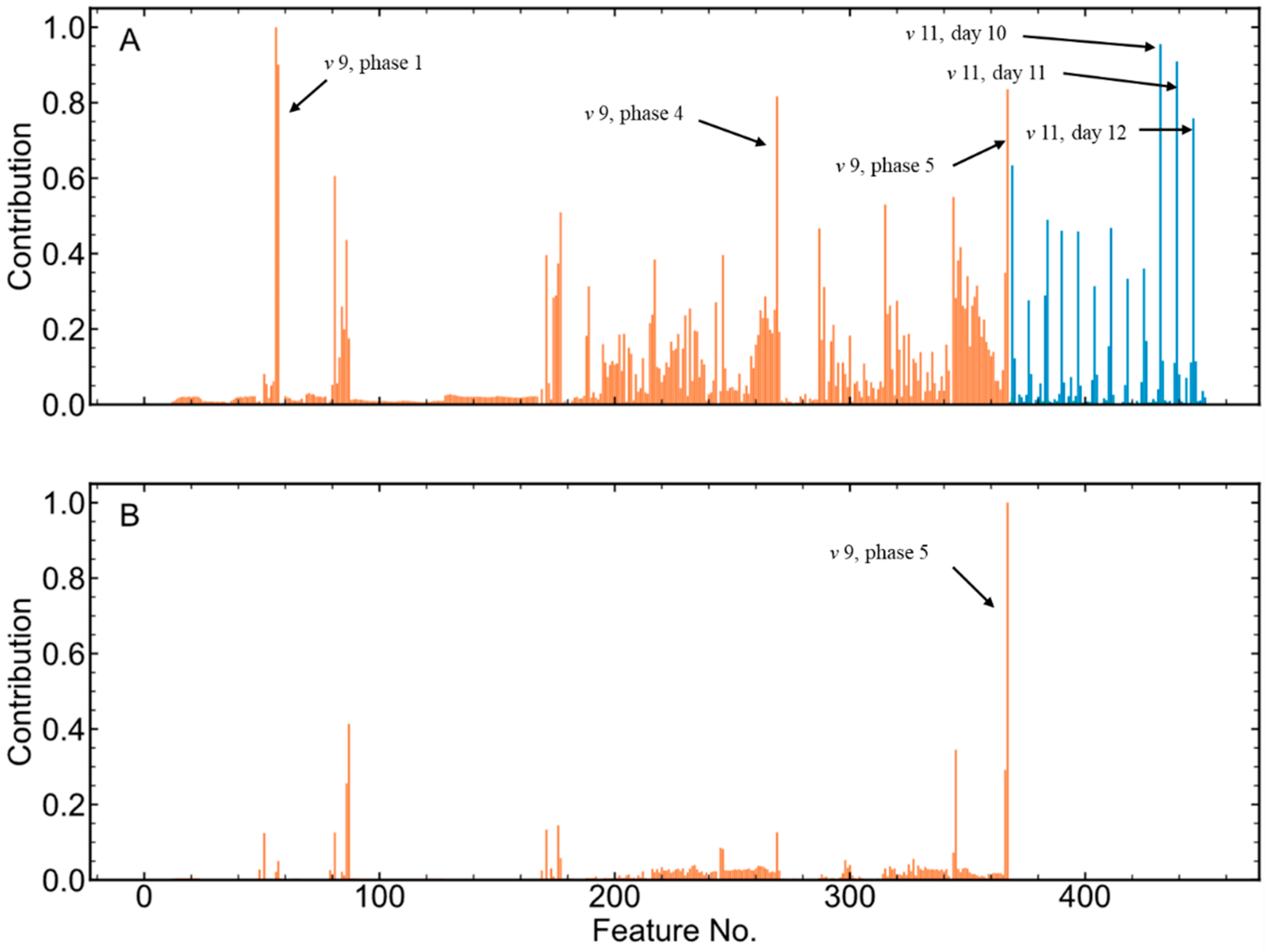
3.5. Cluster Comparing Result
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walsh, G.; Walsh, E. Biopharmaceutical benchmarks 2022. Nat. Biotechnol. 2022, 40, 1722–1760. [Google Scholar] [CrossRef]
- Urquhart, L. Top companies and drugs by sales in 2022. Nat. Rev. Drug Discov. 2023, 22, 260. [Google Scholar] [CrossRef]
- Mordor Intelligence. Biopharmaceutical Industry Size & Share Analysis—Growth Trends & Forecasts (2024–2029). Available online: https://www.mordorintelligence.com/industry-reports/global-biopharmaceuticals-market-industry (accessed on 2 March 2024).
- Klingler, F.; Raab, N.; Zeh, N.; Otte, K. Next Generation Cell Engineering Using microRNAs. In Cell Culture Engineering and Technology: In Appreciation to Professor Mohamed Al-Rubeai; Pörtner, R., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 69–92. [Google Scholar]
- Hu, W. Cell Culture Bioprocess Engineering, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2020; p. 465. [Google Scholar]
- Carrara, S.C.; Ulitzka, M.; Grzeschik, J.; Kornmann, H.; Hock, B.; Kolmar, H. From cell line development to the formulated drug product: The art of manufacturing therapeutic monoclonal antibodies. Int. J. Pharm. 2021, 594, 120164. [Google Scholar] [CrossRef]
- McHugh, K.P.; Xu, J.; Aron, K.L.; Borys, M.C.; Li, Z.J. Effective temperature shift strategy development and scale confirmation for simultaneous optimization of protein productivity and quality in Chinese hamster ovary cells. Biotechnol. Prog. 2020, 36, e2959. [Google Scholar] [CrossRef]
- Wohlenberg, O.J.; Kortmann, C.; Meyer, K.V.; Schellenberg, J.; Dahlmann, K.; Bahnemann, J.; Scheper, T.; Solle, D. Optimization of a mAb production process with regard to robustness and product quality using quality by design principles. Eng. Life Sci. 2022, 22, 484–494. [Google Scholar] [CrossRef]
- Wang, C.; Wang, J.; Chen, M.; Fan, L.; Zhao, L.; Tan, W. Ultra-low carbon dioxide partial pressure improves the galactosylation of a monoclonal antibody produced in Chinese hamster ovary cells in a bioreactor. Biotechnol. Lett. 2018, 40, 1201–1208. [Google Scholar] [CrossRef]
- Reuveny, S.; Velez, D.; Macmillan, J.D.; Miller, L. Factors affecting cell growth and monoclonal antibody production in stirred reactors. J. Immunol. Methods 1986, 86, 53–59. [Google Scholar] [CrossRef]
- Das, T.K.; Narhi, L.O.; Sreedhara, A.; Menzen, T.; Grapentin, C.; Chou, D.K.; Antochshuk, V.; Filipe, V. Stress Factors in mAb Drug Substance Production Processes: Critical Assessment of Impact on Product Quality and Control Strategy. J. Pharm. Sci. USA 2020, 109, 116–133. [Google Scholar] [CrossRef]
- Fouladiha, H.; Marashi, S.; Torkashvand, F.; Mahboudi, F.; Lewis, N.E.; Vaziri, B. A metabolic network-based approach for developing feeding strategies for CHO cells to increase monoclonal antibody production. Bioprocess Biosyst. Eng. 2020, 43, 1381–1389. [Google Scholar] [CrossRef]
- Domján, J.; Fricska, A.; Madarász, L.; Gyürkés, M.; Köte, Á.; Farkas, A.; Vass, P.; Fehér, C.; Horváth, B.; Könczöl, K.; et al. Raman-based dynamic feeding strategies using real-time glucose concentration monitoring system during adalimumab producing CHO cell cultivation. Biotechnol. Prog. 2020, 36, e3052. [Google Scholar] [CrossRef]
- Mun, M.; Khoo, S.; Do Minh, A.; Dvornicky, J.; Trexler-Schmidt, M.; Kao, Y.; Laird, M.W. Air sparging for prevention of antibody disulfide bond reduction in harvested CHO cell culture fluid. Biotechnol. Bioeng. 2015, 112, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Dong, X.; Wan, Y.; Gao, D.; Chen, Z.; Zhang, Y.; Zheng, Z.; Chen, K.; Jiao, J.; Sun, Y.; et al. Development of an in-line Raman analytical method for commercial-scale CHO cell culture process monitoring: Influence of measurement channels and batch number on model performance. Biotechnol. J. 2024, 19, 2300395. [Google Scholar] [CrossRef] [PubMed]
- Kornecki, M.; Strube, J. Process Analytical Technology for Advanced Process Control in Biologics Manufacturing with the Aid of Macroscopic Kinetic Modeling. Bioengineering 2018, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.V.; Raudenbush, K.; Papoutsakis, E.T.; Ierapetritou, M. Cell-culture process optimization via model-based predictions of metabolism and protein glycosylation. Biotechnol. Adv. 2023, 67, 108179. [Google Scholar] [CrossRef] [PubMed]
- Farzan, P.; Ierapetritou, M.G. Integrated modeling to capture the interaction of physiology and fluid dynamics in biopharmaceutical bioreactors. Comput. Chem. Eng. 2017, 97, 271–282. [Google Scholar] [CrossRef]
- Bayer, B.; Duerkop, M.; Pörtner, R.; Möller, J. Comparison of mechanistic and hybrid modeling approaches for characterization of a CHO cultivation process: Requirements, pitfalls and solution paths. Biotechnol. J. 2023, 18, 2200381. [Google Scholar] [CrossRef] [PubMed]
- Manapragada, C.; Pham, T.D.; Rajan, N.; Aickelin, U. Pharmaceutical process optimisation: Decision support under high uncertainty. Comput. Chem. Eng. 2023, 170, 108100. [Google Scholar] [CrossRef]
- Park, S.; Kim, S.; Park, C.; Kim, J.; Lee, D. Data-driven prediction models for forecasting multistep ahead profiles of mammalian cell culture toward bioprocess digital twins. Biotechnol. Bioeng. 2023, 120, 2494–2508. [Google Scholar] [CrossRef]
- Appl, C.; Moser, A.; Baganz, F.; Hass, V.C. Digital Twins for Bioprocess Control Strategy Development and Realisation. In Digital Twins: Applications to the Design and Optimization of Bioprocesses; Herwig, C., Pörtner, R., Möller, J., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 63–94. [Google Scholar] [CrossRef]
- Sokolov, M.; von Stosch, M.; Narayanan, H.; Feidl, F.; Butté, A. Hybrid modeling—A key enabler towards realizing digital twins in biopharma? Curr. Opin. Chem. Eng. 2021, 34, 100715. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, O.; Sampat, C.; Bhalode, P.; Ramachandran, R.; Ierapetritou, M. Digital Twins in Pharmaceutical and Biopharmaceutical Manufacturing: A Literature Review. Processes 2020, 8, 1088. [Google Scholar] [CrossRef]
- Gargalo, C.L.; Udugama, I.; Pontius, K.; Lopez, P.C.; Nielsen, R.F.; Hasanzadeh, A.; Mansouri, S.S.; Bayer, C.; Junicke, H.; Gernaey, K.V. Towards smart biomanufacturing: A perspective on recent developments in industrial measurement and monitoring technologies for bio-based production processes. J. Ind. Microbiol. Biotechnol. 2020, 47, 947–964. [Google Scholar] [CrossRef] [PubMed]
- Goldrick, S.; Sandner, V.; Cheeks, M.; Turner, R.; Farid, S.S.; McCreath, G.; Glassey, J. Multivariate Data Analysis Methodology to Solve Data Challenges Related to Scale-Up Model Validation and Missing Data on a Micro-Bioreactor System. Biotechnol. J. 2020, 15, 1800684. [Google Scholar] [CrossRef]
- Sokolov, M.; Morbidelli, M.; Butté, A.; Souquet, J.; Broly, H. Sequential Multivariate Cell Culture Modeling at Multiple Scales Supports Systematic Shaping of a Monoclonal Antibody toward a Quality Target. Biotechnol. J. 2018, 13, 1700461. [Google Scholar] [CrossRef]
- Le, H.; Kabbur, S.; Pollastrini, L.; Sun, Z.; Mills, K.; Johnson, K.; Karypis, G.; Hu, W. Multivariate analysis of cell culture bioprocess data—Lactate consumption as process indicator. J. Biotechnol. 2012, 162, 210–223. [Google Scholar] [CrossRef]
- Bayrak, E.S.; Wang, T.; Tulsyan, A.; Coufal, M.; Undey, C. Product Attribute Forecast: Adaptive Model Selection Using Real-Time Machine Learning. IFAC-PapersOnLine 2018, 51, 121–125. [Google Scholar] [CrossRef]
- Jin, Y.; Qin, S.J.; Huang, Q.; Saucedo, V.; Li, Z.; Meier, A.; Kundu, S.; Lehr, B.; Charaniya, S. Classification and Diagnosis of Bioprocess Cell Growth Productions Using Early-Stage Data. Ind. Eng. Chem. Res. 2019, 58, 13469–13480. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, C.; Wang, S.; Wang, F. Pseudo Time-Slice Construction Using a Variable Moving Window k Nearest Neighbor Rule for Sequential Uneven Phase Division and Batch Process Monitoring. Ind. Eng. Chem. Res. 2017, 56, 728–740. [Google Scholar] [CrossRef]
- Ramsay, J.; Silverman, B.W. Functional Data Analysis, 3rd ed.; Springer: New York, NY, USA, 2005; pp. 37–57. [Google Scholar]
- Fujiwara, T.; Kwon, O.; Ma, K. Supporting Analysis of Dimensionality Reduction Results with Contrastive Learning. IEEE Trans. Vis. Comput. Graph. 2020, 26, 45–55. [Google Scholar] [CrossRef]
- Nomikos, P.; MacGregor, J.F. Monitoring batch processes using multiway principal component analysis. AIChE J. 1994, 40, 1361–1375. [Google Scholar] [CrossRef]
- Ündey, C.; Williams, B.A.; Çınar, A. Monitoring of batch pharmaceutical fermentations: Data synchronization, landmark alignment, and real-time monitoring. IFAC Proc. Vol. 2002, 35, 271–276. [Google Scholar] [CrossRef]
- Yao, M.; Wang, H.; Xu, W. Batch process monitoring based on functional data analysis and support vector data description. J. Process Control 2014, 24, 1085–1097. [Google Scholar] [CrossRef]
- Zhang, S.; Xie, X.; Qu, H. A data-driven workflow for evaporation performance degradation analysis: A full-scale case study in the herbal medicine manufacturing industry. J. Intell. Manuf. 2023, 34, 651–668. [Google Scholar] [CrossRef]
- Abid, A.; Zhang, M.J.; Bagaria, V.K.; Zou, J. Exploring patterns enriched in a dataset with contrastive principal component analysis. Nat. Commun. 2018, 9, 2134. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Inline Process Variables | Unit | No. | Offline Process Variables | Unit |
|---|---|---|---|---|---|
| 1 | pH 1 | ― | 10 | VCD | 106·mL−1 |
| 2 | pH 2 | ― | 11 | cell viability | % |
| 3 | DO 1 | % | 12 | average diameter of cells | μm |
| 4 | DO 2 | % | 13 | cell agglomeration ratio | % |
| 5 | temperature | °C | 14 | pH | ― |
| 6 | air flow sparger | L·min−1 | 15 | glucose concentration | g·L−1 |
| 7 | air flow overlay | L·min−1 | 16 | lactate concentration | g·L−1 |
| 8 | O2 flow sparger | L·min−1 | |||
| 9 | volume | L |
| Variable No. | Phase 1 | Phase 2 | Phase 3 | Phase 4 | Phase 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Type * | Number | Type | Number | Type | Number | Type | Number | Type | Number | |
| 1 | B | 24 | B | 9 | B | 40 | B | 5 | B | 8 |
| 2 | B | 24 | B | 11 | B | 40 | B | 5 | B | 8 |
| 3 | P | 1 | P | 1 | P | 1 | B | 28 | B | 28 |
| 4 | P | 1 | P | 1 | P | 1 | B | 28 | B | 28 |
| 5 | P | 1 | P | 1 | P | 1 | P | 1 | P | 1 |
| 6 | P | 1 | P | 1 | P | 1 | P | 1 | P | 1 |
| 7 | P | 1 | P | 1 | P | 1 | P | 1 | P | 1 |
| 8 | P | 3 | P | 3 | P | 3 | B | 22 | B | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Chen, H.; Wan, Y.; Wang, H.; Qu, H. A Data-Driven Approach for Leveraging Inline and Offline Data to Determine the Causes of Monoclonal Antibody Productivity Reduction in the Commercial-Scale Cell Culture Process. Pharmaceutics 2024, 16, 1082. https://doi.org/10.3390/pharmaceutics16081082
Zhang S, Chen H, Wan Y, Wang H, Qu H. A Data-Driven Approach for Leveraging Inline and Offline Data to Determine the Causes of Monoclonal Antibody Productivity Reduction in the Commercial-Scale Cell Culture Process. Pharmaceutics. 2024; 16(8):1082. https://doi.org/10.3390/pharmaceutics16081082
Chicago/Turabian StyleZhang, Sheng, Hang Chen, Yuxiang Wan, Haibin Wang, and Haibin Qu. 2024. "A Data-Driven Approach for Leveraging Inline and Offline Data to Determine the Causes of Monoclonal Antibody Productivity Reduction in the Commercial-Scale Cell Culture Process" Pharmaceutics 16, no. 8: 1082. https://doi.org/10.3390/pharmaceutics16081082