

Effect of Interindividual Variability in Metabolic Clearance and Relative Bioavailability on Rifampicin Exposure in Tuberculosis Patients with and without HIV Co-Infection: Does Formulation Quality Matter?

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Study
2.2. Population Pharmacokinetic Models
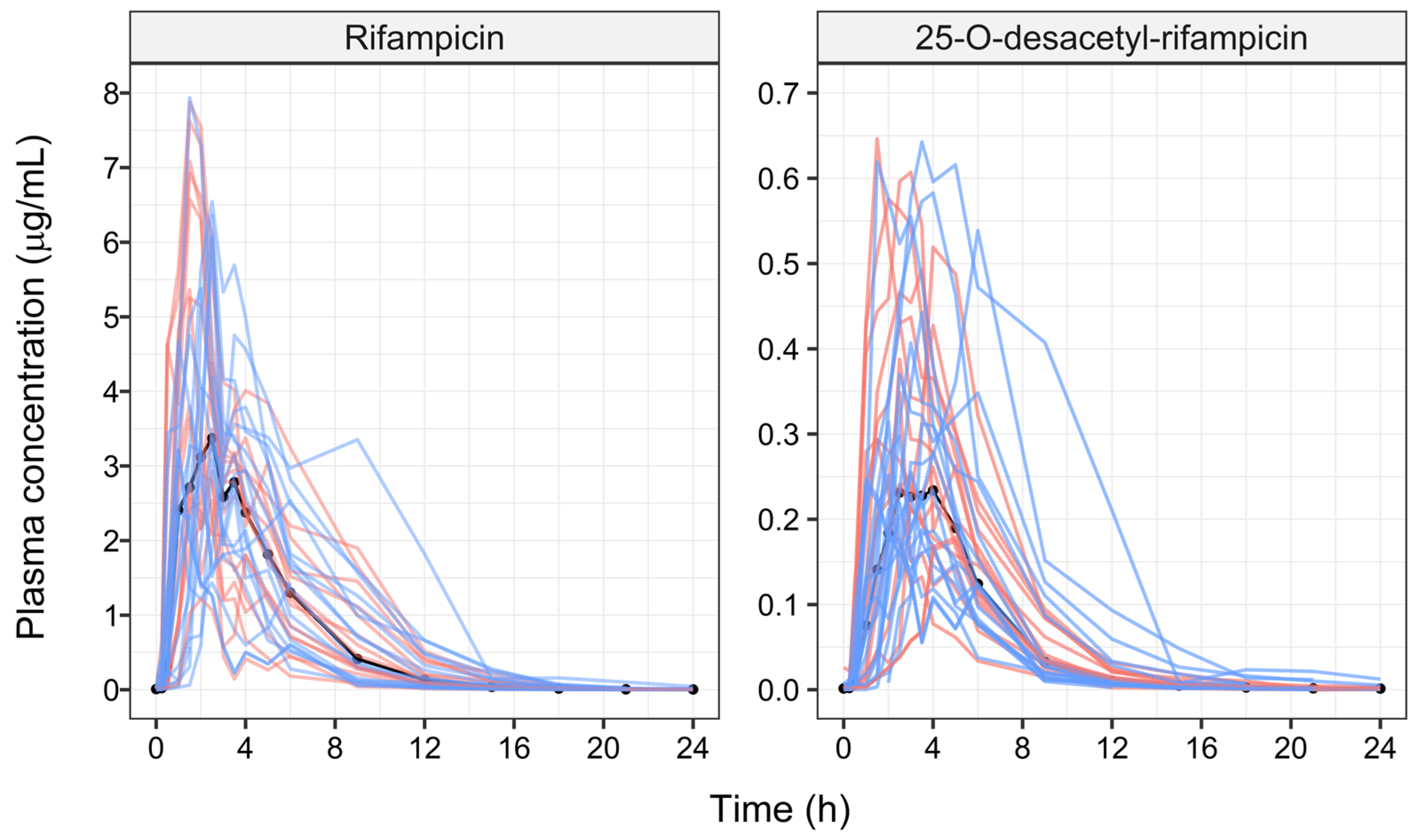
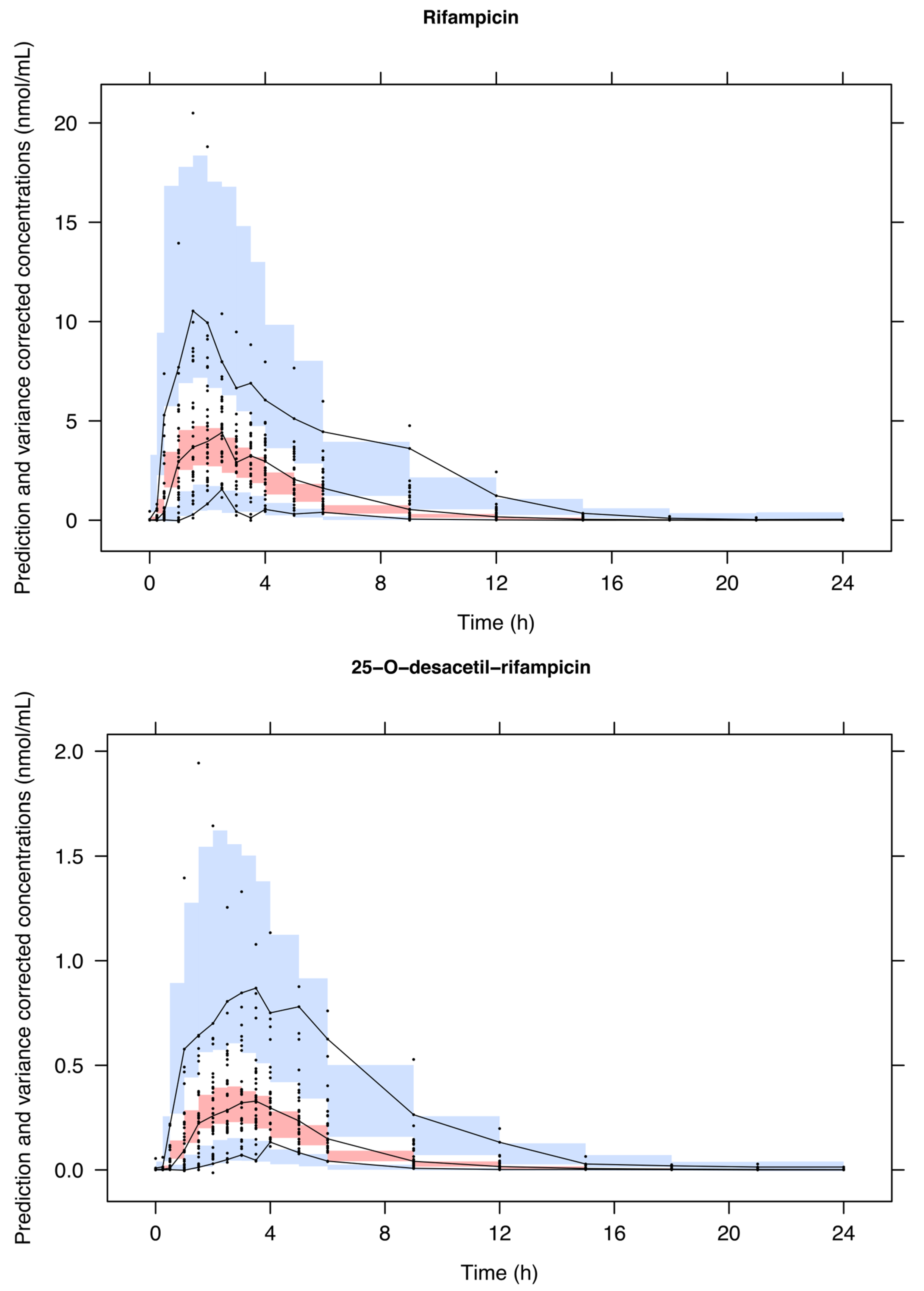
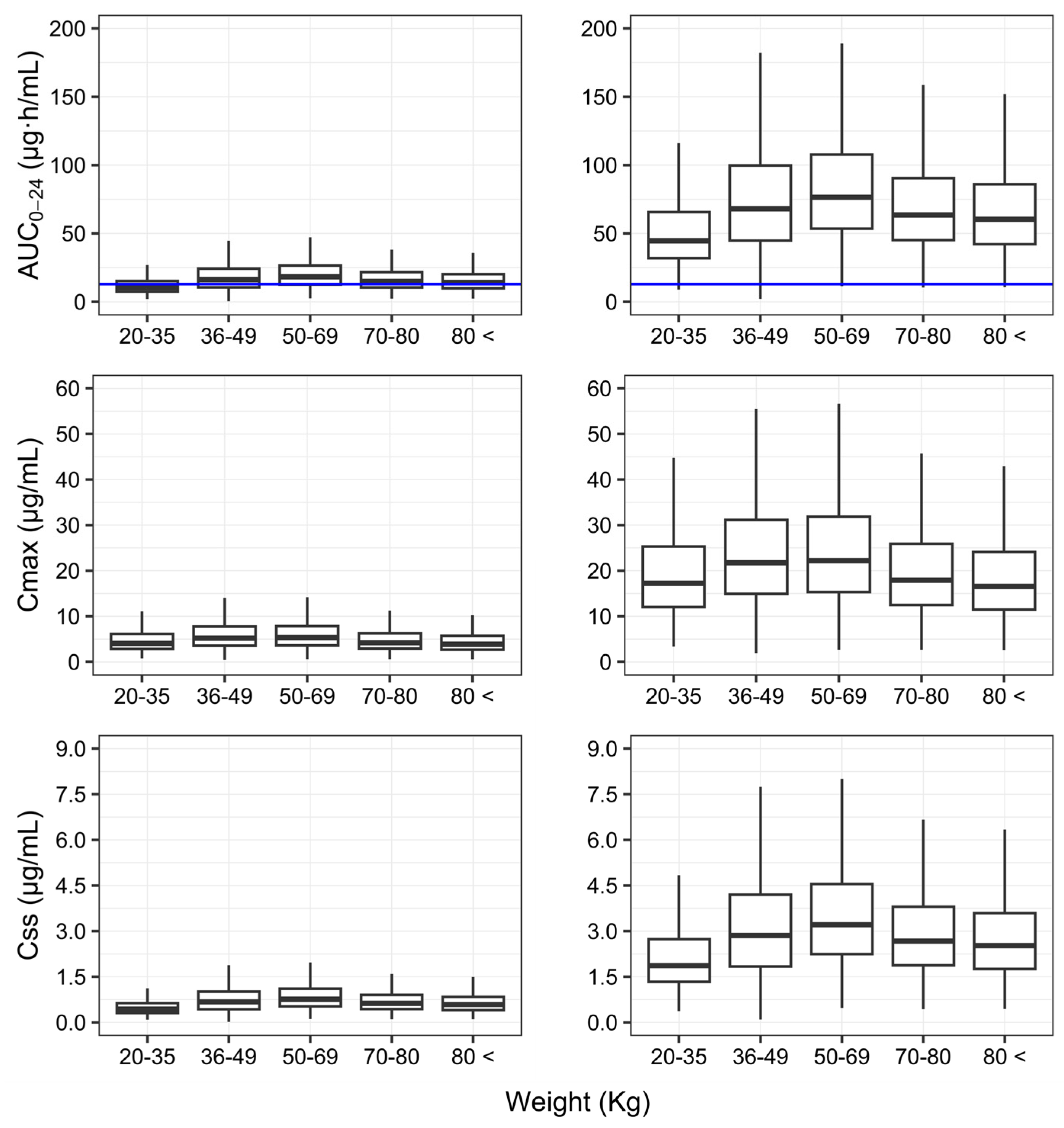
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Global Tuberculosis Report 2023; World Health Organization: Geneva, Switzerland, 2023.
- Gayoso, R.; Dalcolmo, M.; Braga, J.U.; Barreira, D. Predictors of mortality in multidrug-resistant tuberculosis patients from Brazilian reference centers, 2005 to 2012. Braz. J. Infect. Dis. 2018, 22, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Ejeh, F.E.; Undiandeye, A.; Okon, K.; Moshood, K.H. Prevalence of rifampicin resistance tuberculosis among HIV/TB coinfected patients in Benue State, Nigeria. Pan Afr. Med. J. 2021, 38, 203. [Google Scholar] [CrossRef] [PubMed]
- Romaino, S.M.N.; Naing, N.N.; Mat Zuki, M.J. Factors associated with tuberculosis treatment success among tuberculosis and human immunodeficiency virus co-infected patients in Kelantan. Med. J. Malaysia 2022, 77, 696–703. [Google Scholar] [PubMed]
- Ministério da Saúde. Manual de Recomendações Para o Controle da Tuberculose no Brasil; Ministério da Saúde, Secretaria de Vigilância em Saúde, Departamento de Vigilância Epidemiológica: Brasília, Brasil, 2019; pp. 1–288.
- TB CARE I. International Standards for Tuberculosis Care; International standards for Tuberculosis Care; University of California: San Francisco, CA, USA, 2014; pp. 1–92. [Google Scholar]
- Alsultan, A.; Peloquin, C.A. Therapeutic drug monitoring in the treatment of tuberculosis: An update. Drugs 2014, 74, 839–854. [Google Scholar] [CrossRef] [PubMed]
- Kimerling, M.E.; Phillips, P.; Patterson, P.; Hall, M.; Robinson, C.A.; Dunlap, N.E. Low serum antimycobacterial drug levels in non-HIV-infected tuberculosis patients. Chest 1998, 113, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.B.; Shantaveerapa, H.; Byrd Jr, R.P.; Morton, S.E.; Fountain, F.; Roy, T.M. Utility of rifampin blood levels in the treatment and follow-up of active pulmonary tuberculosis in patients who were slow to respond to routine directly observed therapy. Chest 2001, 120, 1520–1524. [Google Scholar] [CrossRef] [PubMed]
- Sahai, J.; Gallicano, K.; Swick, L.; Tailor, S.; Garber, G.; Seguin, I.; Oliveras, L.; Walker, S.; Rachlis, A.; Cameron, W. Reduced plasma concentrations of antituberculosis drugs in patients with HIV infection. Ann. Intern. Med. 1997, 127, 289. [Google Scholar] [CrossRef] [PubMed]
- Strydom, N.; Gupta, S.V.; Fox, W.S.; Via, L.E.; Bang, H.; Lee, M.; Eum, S.; Shim, T.; Iii, C.E.B.; Zimmerman, M.; et al. Tuberculosis drugs’ sistribution and emergence of resistance in patient’s lung lesions: A mechanistic model and tool for regimen and dose optimization. PLoS Med. 2019, 16, e1002773. [Google Scholar] [CrossRef] [PubMed]
- van Crevel, R.; Alisjahbana, B.; de Lange WC, M.; Borst, F.; Danusantoso, H.; van der Meer JW, M.; Burger, D.; Nelwan RH, H. Low Plasma concentrations of rifampicin in tuberculosis patients in Indonesia. Int. J. Tuberc. Lung Dis. 2002, 6, 497–502. [Google Scholar] [CrossRef]
- Milán-Segovia, R.C.; Ramírez, A.M.D.; Cook, H.J.; Aquino, M.M.; Pérez, M.V.; Brundage, R.C.; Moreno, S.R. Population pharmacokinetics of rifampicin in Mexican patients with tuberculosis. J. Clin. Pharm. Ther. 2013, 38, 56–61. [Google Scholar] [CrossRef]
- Schipani, A.; Pertinez, H.; Mlota, R.; Molyneux, E.; Lopez, N.; Dzinjalamala, F.K.; van Oosterhout, J.J.; Ward, S.A.; Khoo, S.; Davies, G. A simultaneous population pharmacokinetic analysis of rifampicin in Malawian adults and children. Br. J. Clin. Pharmacol. 2016, 81, 679–687. [Google Scholar] [CrossRef]
- Wilkins, J.J.; Savic, R.M.; Karlsson, M.O.; Langdon, G.; McIlleron, H.; Pillai, G.; Smith, P.J.; Simonsson, U.S.H. Population pharmacokinetics of rifampin in pulmonary tuberculosis patients, including a semimechanistic model to describe variable absorption. Antimicrob. Agents Chemother. 2008, 52, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Daskapan, A.; Idrus, L.R.; Postma, M.J.; Wilffert, B.; Kosterink, J.G.W.; Stienstra, Y.; Touw, D.J.; Andersen, A.B.; Bekker, A.; Denti, P.; et al. A systematic review on the effect of HIV infection on the pharmacokinetics of first-line tuberculosis drugs. Clin. Pharmacokinet. 2019, 58, 747–766. [Google Scholar] [CrossRef]
- Seng, K.-Y.; Hee, K.-H.; Soon, G.-H.; Chew, N.; Khoo, S.H.; Lee, L.S.-U. Population pharmacokinetics of rifampicin and 25-desacetyl-rifampicin in healthy Asian adults. J. Antimicrob. Chemother. 2015, 70, 3298–3306. [Google Scholar] [CrossRef]
- Nishimura, T.; Kohno, H.; Nagai, H.; Maruoka, D.; Koike, Y.; Kobayashi, M.; Atsuda, K. The population pharmacokinetics of rifampicin in Japanese pulmonary tuberculosis patients. Drug Res. 2020, 70, 199–205. [Google Scholar] [CrossRef]
- Smythe, W.; Khandelwal, A.; Merle, C.; Rustomjee, R.; Gninafon, M.; Lo, M.B.; Sow, O.B.; Olliaro, P.L.; Lienhardt, C.; Horton, J.; et al. A semimechanistic pharmacokinetic-enzyme turnover model for rifampin autoinduction in adult tuberculosis patients. Antimicrob. Agents Chemother. 2012, 56, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Svensson, R.J.; Aarnoutse, R.E.; Diacon, A.H.; Dawson, R.; Gillespie, S.H.; Boeree, M.J.; Simonsson, U.S.H. A population pharmacokinetic model incorporating saturable pharmacokinetics and autoinduction for high rifampicin doses. Clin. Pharmacol. Ther. 2018, 103, 674–683. [Google Scholar] [CrossRef]
- Egelund, E.; Alsultan, A.; Peloquin, C. Optimizing the clinical pharmacology of tuberculosis medications. Clin. Pharmacol. Ther. 2015, 98, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Clewe, O.; Karlsson, M.O.; Simonsson, U.S.H. Evaluation of optimized bronchoalveolar lavage sampling designs for characterization of pulmonary drug distribution. J. Pharmacokinet. Pharmacodyn. 2015, 42, 699–708. [Google Scholar] [CrossRef]
- McCune, J.S.; Reynolds, K.S. Developing and using therapeutics for emerging infections. Clin. Pharmacol. Ther. 2015, 98, 346–351. [Google Scholar] [CrossRef]
- Verbeeck, R.K.; Günther, G.; Kibuule, D.; Hunter, C.; Rennie, T.W. Optimizing treatment outcome of first-line anti-tuberculosis drugs: The role of therapeutic drug monitoring. Eur. J. Clin. Pharmacol. 2016, 72, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Vinks, A.A. Therapeutic optimization as part of the precision medicine paradigm. Clin. Pharmacol. Ther. 2016, 99, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Weld, E.D.; Dooley, K.E. State-of-the-art review of HIV-TB coinfection in special populations. Clin. Pharmacol. Ther. 2018, 104, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Muliaditan, M.; Della Pasqua, O. How long will treatment guidelines for TB continue to overlook variability in drug exposure? J. Antimicrob. Chemother. 2019, 74, 3274–3280. [Google Scholar] [CrossRef] [PubMed]
- Abulfathi, A.A.; Decloedt, E.H.; Svensson, E.M.; Diacon, A.H.; Donald, P.; Reuter, H. Clinical pharmacokinetics and pharmacodynamics of rifampicin in human tuberculosis. Clin. Pharmacokinet. 2019, 58, 1103–1129. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Panchagnula, R. Implication of biopharmaceutics and pharmacokinetics of rifampicin in variable bioavailability from solid oral dosage forms. Biopharm. Drug Dispos. 2005, 26, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Bart, G.; Jaber, M.; Giang, L.M.; Brundage, R.C.; Korthuis, P.T. Findings from a pilot study of buprenorphine population pharmacokinetics: A potential effect of HIV on buprenorphine bioavailability. Drug Alcohol Depend. 2022, 241, 109696. [Google Scholar] [CrossRef] [PubMed]
- Jamis-Dow, C.A.; Katki, A.G.; Collins, J.M.; Klecker, R.W. Rifampin and rifabutin and their metabolism by human liver esterases. Xenobiotica 1997, 27, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Aristoff, P.A.; Garcia, G.A.; Kirchhoff, P.D.; Hollis Showalter, H.D. Rifamycins—Obstacles and Opportunities. Tuberculosis 2010, 90, 94–118. [Google Scholar] [CrossRef]
- Song, S.H.; Chang, H.E.; Jun, S.H.; Park, K.U.; Lee, J.H.; Lee, E.-M.; Song, Y.-H.; Song, J. Relationship between CES2 genetic variations and rifampicin metabolism. J. Antimicrob. Chemother. 2013, 68, 1281–1284. [Google Scholar] [CrossRef]
- Muda, M.R.; Harun, S.N.; Syed Sulaiman, S.A.; Sheikh Ghadzi, S.M. Population pharmacokinetics analyses of rifampicin in adult and children populations: A systematic review. Br. J. Clin. Pharmacol. 2022, 88, 3132–3152. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, E.G.; Schinkel, A.H.; Relling, M.V.; Schuetz, J.D. P-glycoprotein: A major determinant of rifampicin-inducible expression of cytochrome P4503A in mice and humans. Proc. Natl. Acad. Sci. USA 1996, 93, 4001–4005. [Google Scholar] [CrossRef] [PubMed]
- Chigutsa, E.; Visser, M.E.; Swart, E.C.; Denti, P.; Pushpakom, S.; Egan, D.; Holford, N.H.G.; Smith, P.J.; Maartens, G.; Owen, A.; et al. The SLCO1B1 Rs4149032 polymorphism is highly prevalent in South Africans and is associated with reduced rifampin concentrations: Dosing implications. Antimicrob. Agents Chemother. 2011, 55, 4122–4127. [Google Scholar] [CrossRef] [PubMed]
- Weiner, M.; Peloquin, C.; Burman, W.; Luo, C.-C.; Engle, M.; Prihoda, T.J.; Kenzie, W.R.M.; Bliven-Sizemore, E.; Johnson, J.L.; Vernon, A. Effects of tuberculosis, race, and human gene SLCO1B1 polymorphisms on rifampin concentrations. Antimicrob. Agents Chemother. 2010, 54, 4192–4200. [Google Scholar] [CrossRef] [PubMed]
- Sundell, J.; Bienvenu, E.; Äbelö, A.; Ashton, M. Effect of efavirenz-based ART on the pharmacokinetics of rifampicin and its primary metabolite in patients coinfected with TB and HIV. J. Antimicrob. Chemother. 2021, 76, 2950–2957. [Google Scholar] [CrossRef] [PubMed]
- Medellin-Garibay, S.E.; Huerta-García, A.P.; Rodríguez-Báez, A.S.; Magaña-Aquino, M.; Ortiz-Álvarez, A.; Portales-Pérez, D.P.; del Carmen Milán-Segovia, R.; Romano-Moreno, S. A population approach of rifampicin pharmacogenetics and pharmacokinetics in Mexican patients with tuberculosis. Tuberculosis 2020, 124, 101982. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, G.E.; Brooks, M.B.; Coit, J.M.; Pertinez, H.; Vargas Vásquez, D.; Sánchez Garavito, E.; Calderón, R.I.; Jiménez, J.; Tintaya, K.; Peloquin, C.A.; et al. Efficacy and safety of high-dose rifampin in pulmonary tuberculosis. A randomized controlled trial. Am. J. Respir. Crit. Care Med. 2018, 198, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Nardotto, G.H.B.; Bollela, V.R.; Rocha, A.; Della Pasqua, O.; Lanchote, V.L. No implication of HIV coinfection on the plasma exposure to rifampicin, pyrazinamide, and ethambutol in tuberculosis patients. Clin. Transl. Sci. 2022, 15, 514–523. [Google Scholar] [CrossRef] [PubMed]
- van Beek, S.W.; ter Heine, R.; Alffenaar, J.-W.C.; Magis-Escurra, C.; Aarnoutse, R.E.; Svensson, E.M. A model-informed method for the purpose of precision dosing of isoniazid in pulmonary tuberculosis. Clin. Pharmacokinet. 2021, 60, 943–953. [Google Scholar] [CrossRef]
- Shen, J.; Boeckmann, A.; Vick, A. Implementation of dose superimposition to introduce multiple doses for a mathematical absorption model (Transit compartment model). J. Pharmacokinet. Pharmacodyn. 2012, 39, 251–262. [Google Scholar] [CrossRef]
- Savic, R.M.; Jonker, D.M.; Kerbusch, T.; Karlsson, M.O. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 2007, 34, 711–726. [Google Scholar] [CrossRef]
- Carlsson, K.C.; Savić, R.M.; Hooker, A.C.; Karlsson, M.O. Modeling subpopulations with the $MIXTURE subroutine in NONMEM: Finding the individual probability of belonging to a subpopulation for the use in model analysis and improved decision making. AAPS J. 2009, 11, 148–154. [Google Scholar] [CrossRef]
- Proost, J.H. Combined proportional and additive residual error models in population pharmacokinetic modelling. Eur. J. Pharm. Sci. 2017, 109, S78–S82. [Google Scholar]
- Mould, D.R.; Upton, R.N. Basic concepts in population modeling, simulation, and model-based drug development—Part 2: Introduction to pharmacokinetic modeling methods. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e38. [Google Scholar] [CrossRef]
- Bauer, R.J. NONMEM Tutorial Part II: Estimation methods and advanced examples. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 538–556. [Google Scholar] [CrossRef] [PubMed]
- Duffull, S.B.; Wright, D.F.B.; Winter, H.R. Interpreting population pharmacokinetic-pharmacodynamic analyses—A clinical viewpoint. Br. J. Clin. Pharmacol. 2011, 71, 807–814. [Google Scholar] [CrossRef]
- Nguyen, T.H.T.; Mouksassi, M.-S.; Holford, N.; Al-Huniti, N.; Freedman, I.; Hooker, A.C.; John, J.; Karlsson, M.O.; Mould, D.R.; Ruixo, J.J.P.; et al. Model evaluation of continuous data pharmacometric models: Metrics and graphics. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 87–109. [Google Scholar] [CrossRef]
- Bergstrand, M.; Hooker, A.C.; Wallin, J.E.; Karlsson, M.O. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011, 13, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Beal, S.L.; Sheiner, L.B. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J. Pharmacokinet. Pharmacodyn. 2001, 28, 171–192. [Google Scholar] [CrossRef]
- Comets, E.; Brendel, K.; Mentré, F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effects models: The Npde add-on package for R. Comput. Methods Programs Biomed. 2008, 90, 154–166. [Google Scholar] [CrossRef]
- Ribbing, J.; Niclas Jonsson, E. Power, selection bias and predictive performance of the population pharmacokinetic covariate model. J. Pharmacokinet. Pharmacodyn. 2004, 31, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Lindbom, L.; Pihlgren, P.; Jonsson, N. PsN-Toolkit—A Collection of computer intensive statistical methods for non-linear mixed effects modeling using NONMEM. Comput. Methods Programs Biomed. 2005, 79, 241–257. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Italy, 2022. [Google Scholar]
- Pasipanodya, J.G.; McIlleron, H.; Burger, A.; Wash, P.A.; Smith, P.; Gumbo, T. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J. Infect. Dis. 2013, 208, 1464–1473. [Google Scholar] [CrossRef]
- Acocella, G. Clinical pharmacokinetics of rifampicin. Clin. Pharmacokinet. 1978, 3, 108–127. [Google Scholar] [CrossRef]
- Loos, U.; Musch, E.; Jensen, J.C.; Mikus, G.; Schwabe, H.K.; Eichelbaum, M. Pharmacokinetics of oral and intravenous rifampicin during chronic administration. Klin. Wochenschr. 1985, 63, 1205–1211. [Google Scholar] [CrossRef]
- Jing, Y.; Zhu, L.Q.; Yang, J.W.; Huang, S.P.; Wang, Q.; Zhang, J. Population pharmacokinetics of rifampicin in Chinese patients with pulmonary tuberculosis. J. Clin. Pharmacol. 2016, 56, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Marsot, A.; Ménard, A.; Dupouey, J.; Muziotti, C.; Guilhaumou, R.; Blin, O. Population pharmacokinetics of rifampicin in adult patients with osteoarticular infections: Interaction with fusidic acid. Br. J. Clin. Pharmacol. 2017, 83, 1039–1047. [Google Scholar] [CrossRef]
- Chirehwa, M.T.; Rustomjee, R.; Mthiyane, T.; Onyebujoh, P.; Smith, P.; McIlleron, H.; Denti, P. Model-based evaluation of higher doses of rifampin using a semimechanistic model incorporating autoinduction and saturation of hepatic extraction. Antimicrob. Agents Chemother. 2015, 60, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Milán-Segovia, R.C.; Domínguez-Ramírez, A.M.; Jung-Cook, H.; Magaña-Aquino, M.; Romero-Méndez, M.C.; Medellín-Garibay, S.E.; Vigna-Pérez, M.; Romano-Moreno, S. Relative bioavailability of rifampicin in a three-drug fixed-dose combination formulation. Int. J. Tuberc. Lung Dis. Off. J. Int. Union Tuberc. Lung Dis. 2010, 14, 1454–1460. [Google Scholar]
- Johnston, A.; Holt, D.W. Substandard drugs: A potential crisis for public health. Br. J. Clin. Pharmacol. 2014, 78, 218–243. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fixed Effects | Random Effects | |
|---|---|---|
| Parameters | Typical Value (RSE%) | CV% (RSE%) |
| CL/F (L/h) | 35.2 (10.1) | 26.9 (10.6) |
| V/F (L) | 108.0 (9.4) | |
| MTT (h) | 1.13 (9.0) | 53.2 (7.8) |
| F | ---- | 46.9 (10.7) |
| Nn | 3 (fix) | |
| Fm | ---- | 38.8 (15.8) |
| CLm/(FFm) (L/h) | 368 (9.8) | |
| Vm/(FFm) (L) | 226 (15.2) | 63.3 (22.0) |
| Correlation CL/F–Fm | 71.5 (2.2) | |
| Residual variability (ε) | ||
| RIF proportional | 45.6 (8.5) | |
| RIF additive (nmol/mL)2 | 1.85 × 10−5 (38.6) | |
| η1 * | 20.4 (37.7) | |
| desRIF proportional | 35.5 (10.1) | |
| desRIF additive (nmol/mL)2 | 3.21 × 10−6 (15.7) | |
| η2 * | 35.1 (28.5) | |
| * η1 and η2 are random deviations of individual i from the variance of ε, which is assumed to be the same for all subjects. RSE: relative standard error. | ||
| Model parameterisation: | ||
| Ka = Ktr = (Nn + 1)/MTT | ||
| Priors Included | Frel * | Median AUC0–24 (25th–75th Percentiles) | AUC0–24 Ratio |
|---|---|---|---|
| Schipani et al., 2016 [14] FDC | 0.678 | 30.83 (10.94–75.77) | 0.620 |
| Seng et al., 2015 [17] FDC | 0.292 | 51.26 (16.87–143.27) | 1.031 |
| Wilkins et al. 2008 [15] FDC | 0.499 | 28.50 (9.91–72.22) | 0.573 |
| Milán-Segovia et al., 2013 [13] Formulation A FDC | 0.497 | 23.28 (9.42–43.11) | 0.468 |
| Milán-Segovia et al., 2013 [13] Reference FDC | 0.232 | 49.74 (20.14–92.12) | 1 |
| Current study, FDC tablets | --- | 19.94 (7.15–25.36) | 0.330 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nardotto, G.H.B.; Svenson, E.M.; Bollela, V.R.; Rocha, A.; Slavov, S.N.; Ximenez, J.P.B.; Della Pasqua, O.; Lanchote, V.L. Effect of Interindividual Variability in Metabolic Clearance and Relative Bioavailability on Rifampicin Exposure in Tuberculosis Patients with and without HIV Co-Infection: Does Formulation Quality Matter? Pharmaceutics 2024, 16, 970. https://doi.org/10.3390/pharmaceutics16080970
Nardotto GHB, Svenson EM, Bollela VR, Rocha A, Slavov SN, Ximenez JPB, Della Pasqua O, Lanchote VL. Effect of Interindividual Variability in Metabolic Clearance and Relative Bioavailability on Rifampicin Exposure in Tuberculosis Patients with and without HIV Co-Infection: Does Formulation Quality Matter? Pharmaceutics. 2024; 16(8):970. https://doi.org/10.3390/pharmaceutics16080970
Chicago/Turabian StyleNardotto, Glauco Henrique Balthazar, Elin M. Svenson, Valdes Roberto Bollela, Adriana Rocha, Svetoslav Nanev Slavov, João Paulo Bianchi Ximenez, Oscar Della Pasqua, and Vera Lucia Lanchote. 2024. "Effect of Interindividual Variability in Metabolic Clearance and Relative Bioavailability on Rifampicin Exposure in Tuberculosis Patients with and without HIV Co-Infection: Does Formulation Quality Matter?" Pharmaceutics 16, no. 8: 970. https://doi.org/10.3390/pharmaceutics16080970