
Drug Nanocrystals in Oral Absorption: Factors That Influence Pharmacokinetics


Abstract
:1. Introduction
2. Influential Factors for Drug Nanocrystals in Oral Absorption
2.1. Particle Size and Morphology
2.2. Dissolution Rate
2.3. Crystalline State
2.4. Stabilizing Agents
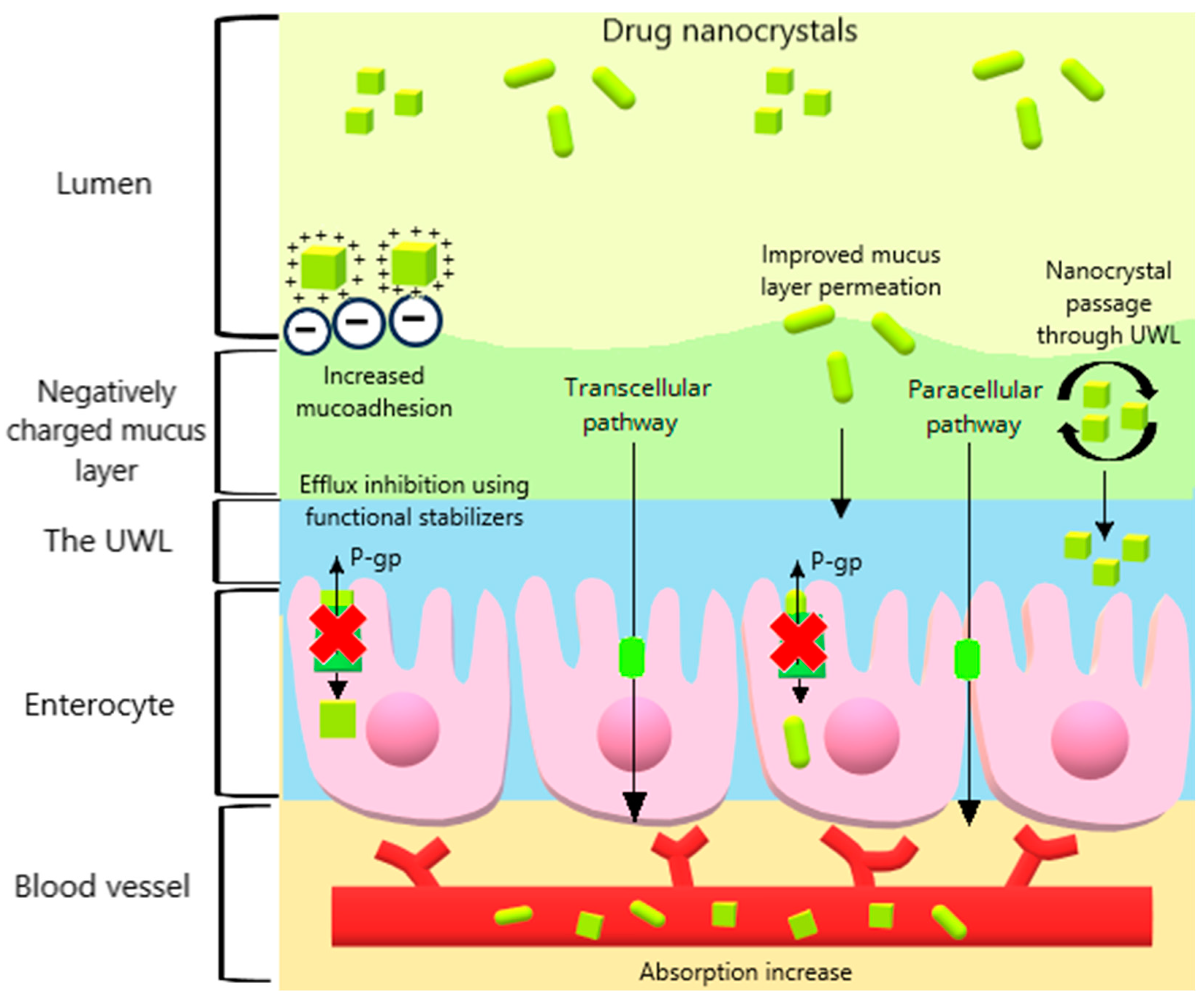
2.5. Surface Charge
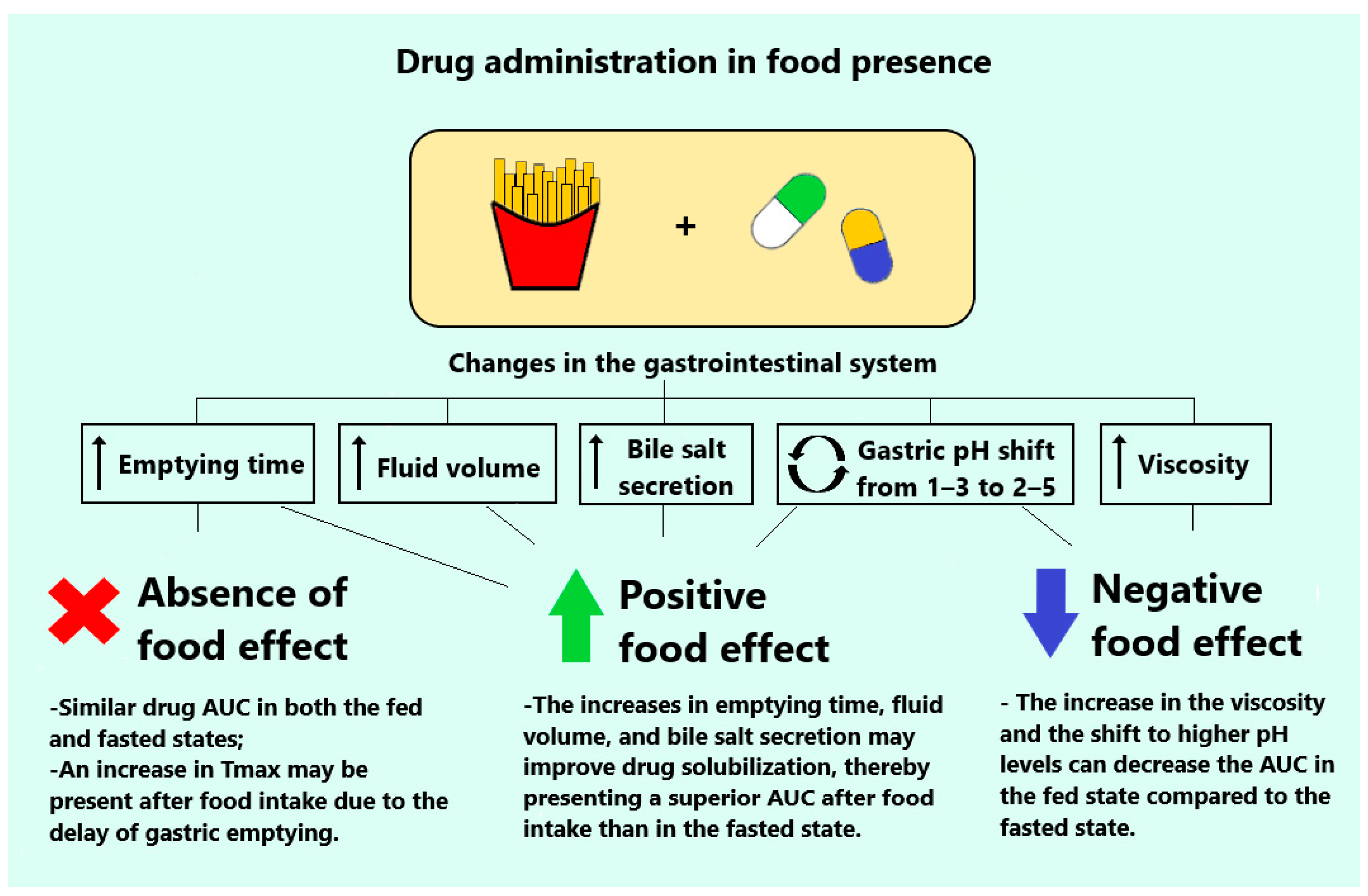
3. Pharmacokinetics Particularities
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in Oral Drug Delivery. Front. Pharmacol. 2021, 12, 618411. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, J.; Steenekamp, J.; Steyn, D.; Hamman, J. The Role of Functional Excipients in Solid Oral Dosage Forms to Overcome Poor Drug Dissolution and Bioavailability. Pharmaceutics 2020, 12, 393. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Bae, Y.H. Bile Acid Transporter-Mediated Oral Drug Delivery. J. Control. Release 2020, 327, 100–116. [Google Scholar] [CrossRef]
- Dahan, A.; Hoffman, A. Rationalizing the Selection of Oral Lipid Based Drug Delivery Systems by an in Vitro Dynamic Lipolysis Model for Improved Oral Bioavailability of Poorly Water Soluble Drugs. J. Control. Release 2008, 129, 1–10. [Google Scholar] [CrossRef]
- Koziolek, M.; Grimm, M.; Schneider, F.; Jedamzik, P.; Sager, M.; Kühn, J.-P.; Siegmund, W.; Weitschies, W. Navigating the Human Gastrointestinal Tract for Oral Drug Delivery: Uncharted Waters and New Frontiers. Adv. Drug Deliv. Rev. 2016, 101, 75–88. [Google Scholar] [CrossRef]
- Cheng, L.; Wong, H. Food Effects on Oral Drug Absorption: Application of Physiologically-Based Pharmacokinetic Modeling as a Predictive Tool. Pharmaceutics 2020, 12, 672. [Google Scholar] [CrossRef]
- O’Shea, J.P.; Holm, R.; O’Driscoll, C.M.; Griffin, B.T. Food for Thought: Formulating Away the Food Effect—A PEARRL Review. J. Pharm. Pharmacol. 2019, 71, 510–535. [Google Scholar] [CrossRef] [PubMed]
- Rangaraj, N.; Sampathi, S.; Junnuthula, V.; Kolimi, P.; Mandati, P.; Narala, S.; Nyavanandi, D.; Dyawanapelly, S. Fast-Fed Variability: Insights into Drug Delivery, Molecular Manifestations, and Regulatory Aspects. Pharmaceutics 2022, 14, 1807. [Google Scholar] [CrossRef]
- Liu, L.; Tian, C.; Dong, B.; Xia, M.; Cai, Y.; Hu, R.; Chu, X. Models to Evaluate the Barrier Properties of Mucus during Drug Diffusion. Int. J. Pharm. 2021, 599, 120415. [Google Scholar] [CrossRef]
- Tian, Z.; Mai, Y.; Meng, T.; Ma, S.; Gou, G.; Yang, J. Nanocrystals for Improving Oral Bioavailability of Drugs: Intestinal Transport Mechanisms and Influencing Factors. AAPS PharmSciTech 2021, 22, 179. [Google Scholar] [CrossRef]
- Sugano, K. Possible Reduction of Effective Thickness of Intestinal Unstirred Water Layer by Particle Drifting Effect. Int. J. Pharm. 2010, 387, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Elmeliegy, M.; Vourvahis, M.; Guo, C.; Wang, D.D. Effect of P-Glycoprotein (P-Gp) Inducers on Exposure of P-Gp Substrates: Review of Clinical Drug–Drug Interaction Studies. Clin. Pharmacokinet. 2020, 59, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Yamazaki, M. Role of P-Glycoprotein in Pharmacokinetics. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef] [PubMed]
- Sanches, B.M.A.; Ferreira, E.I. Is Prodrug Design an Approach to Increase Water Solubility? Int. J. Pharm. 2019, 568, 118498. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Hirlekar, R. Multicomponent Cyclodextrin System for Improvement of Solubility and Dissolution Rate of Poorly Water Soluble Drug. Asian J. Pharm. Sci. 2019, 14, 104–115. [Google Scholar] [CrossRef]
- Sathisaran, I.; Dalvi, S. Engineering Cocrystals of Poorly Water-Soluble Drugs to Enhance Dissolution in Aqueous Medium. Pharmaceutics 2018, 10, 108. [Google Scholar] [CrossRef]
- Karagianni, A.; Kachrimanis, K.; Nikolakakis, I. Co-Amorphous Solid Dispersions for Solubility and Absorption Improvement of Drugs: Composition, Preparation, Characterization and Formulations for Oral Delivery. Pharmaceutics 2018, 10, 98. [Google Scholar] [CrossRef]
- Haneef, J.; Ali, S.; Chadha, R. Emerging Multi-Drug Eutectics: Opportunities and Challenges. AAPS PharmSciTech 2021, 22, 66. [Google Scholar] [CrossRef]
- Mohammad, I.S.; Hu, H.; Yin, L.; He, W. Drug Nanocrystals: Fabrication Methods and Promising Therapeutic Applications. Int. J. Pharm. 2019, 562, 187–202. [Google Scholar] [CrossRef]
- McGuckin, M.B.; Wang, J.; Ghanma, R.; Qin, N.; Palma, S.D.; Donnelly, R.F.; Paredes, A.J. Nanocrystals as a Master Key to Deliver Hydrophobic Drugs via Multiple Administration Routes. J. Control. Release 2022, 345, 334–353. [Google Scholar] [CrossRef]
- Bilgili, E.; Guner, G. Mechanistic Modeling of Wet Stirred Media Milling for Production of Drug Nanosuspensions. AAPS PharmSciTech 2021, 22, 2. [Google Scholar] [CrossRef]
- Soni, G.; Kale, K.; Shetty, S.; Gupta, M.K.; Yadav, K.S. Quality by Design (QbD) Approach in Processing Polymeric Nanoparticles Loading Anticancer Drugs by High Pressure Homogenizer. Heliyon 2020, 6, e03846. [Google Scholar] [CrossRef] [PubMed]
- Macedo, L.d.O.; Barbosa, E.J.; Löbenberg, R.; Bou-Chacra, N.A. Anti-Inflammatory Drug Nanocrystals: State of Art and Regulatory Perspective. Eur. J. Pharm. Sci. 2021, 158, 105654. [Google Scholar] [CrossRef] [PubMed]
- Chary, P.S.; Shaikh, S.; Bhavana, V.; Rajana, N.; Vasave, R.; Mehra, N.K. Emerging Role of Nanocrystals in Pharmaceutical Applications: A Review of Regulatory Aspects and Drug Development Process. Appl. Mater. Today 2024, 40, 102334. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, W.; Lu, Y. What Is the Future for Nanocrystal-Based Drug-Delivery Systems? Ther. Deliv. 2020, 11, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Z.; Zhang, H.; Gao, J.; Zheng, A. Progress in the Development of Stabilization Strategies for Nanocrystal Preparations. Drug Deliv. 2021, 28, 19–36. [Google Scholar] [CrossRef]
- Arti, S.; Bharti, M.; Kumar, V.; Saruchi; Rehani, V.; Dhiman, J. Drug Nanocrystals as Nanocarrier-Based Drug Delivery Systems. In Industrial Applications of Nanocrystals; Mallakpour, S., Hussain, C.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 179–203. [Google Scholar]
- Joseph, E.; Singhvi, G. Multifunctional Nanocrystals for Cancer Therapy: A Potential Nanocarrier. In Nanomaterials for Drug Delivery and Therapy; Grumezescu, A.M., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; pp. 91–116. ISBN 978-0-12-816505-8. [Google Scholar]
- Chiou, W.L.; Jeong, H.Y.; Chung, S.M.; Wu, T.C. Evaluation of Using Dog as an Animal Model to Study the Fraction of Oral Dose Absorbed of 43 Drugs in Humans. Pharm. Res. 2000, 17, 135–140. [Google Scholar] [CrossRef]
- Arndt, M.; Chokshi, H.; Tang, K.; Parrott, N.J.; Reppas, C.; Dressman, J.B. Dissolution Media Simulating the Proximal Canine Gastrointestinal Tract in the Fasted State. Eur. J. Pharm. Biopharm. 2013, 84, 633–641. [Google Scholar] [CrossRef]
- Martinez, M.N.; Papich, M.G.; Fahmy, R. Impact of Gastrointestinal Differences in Veterinary Species on the Oral Drug Solubility, in Vivo Dissolution, and Formulation of Veterinary Therapeutics. ADMET DMPK 2022, 10, 1–25. [Google Scholar] [CrossRef]
- Papich, M.G.; Martinez, M.N. Applying Biopharmaceutical Classification System (BCS) Criteria to Predict Oral Absorption of Drugs in Dogs: Challenges and Pitfalls. AAPS J. 2015, 17, 948–964. [Google Scholar] [CrossRef]
- Xu, X.; Chen, G.; Li, Y.; Wang, J.; Yin, J.; Ren, L. Enhanced Dissolution and Oral Bioavailability of Cinacalcet Hydrochlorde Nanocrystals with No Food Effect. Nanotechnology 2019, 30, 055102. [Google Scholar] [CrossRef] [PubMed]
- Karakucuk, A.; Teksin, Z.S.; Eroglu, H.; Celebi, N. Evaluation of Improved Oral Bioavailability of Ritonavir Nanosuspension. Eur. J. Pharm. Sci. 2019, 131, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Wei, M.; Li, W.; Guo, M.; Guo, C.; Ma, M.; Wang, Y.; Yang, Z.; Li, M.; Fu, Q.; et al. Impacts of Particle Shapes on the Oral Delivery of Drug Nanocrystals: Mucus Permeation, Transepithelial Transport and Bioavailability. J. Control. Release 2019, 307, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, W.; Xiong, S.; Luo, J.; Li, Y.; Zhao, Y.; Wang, Q.; Zhang, Z.; Chen, X.; Chen, T. Highly Stabilized Nanocrystals Delivering Ginkgolide B in Protecting against the Parkinson’s Disease. Int. J. Pharm. 2020, 577, 119053. [Google Scholar] [CrossRef] [PubMed]
- Imono, M.; Uchiyama, H.; Yoshida, S.; Miyazaki, S.; Tamura, N.; Tsutsumimoto, H.; Kadota, K.; Tozuka, Y. The Elucidation of Key Factors for Oral Absorption Enhancement of Nanocrystal Formulations: In Vitro—In Vivo Correlation of Nanocrystals. Eur. J. Pharm. Biopharm. 2020, 146, 84–92. [Google Scholar] [CrossRef]
- Kala, S.G.; Chinni, S. Development and Characterization of Venetoclax Nanocrystals for Oral Bioavailability Enhancement. AAPS PharmSciTech 2021, 22, 92. [Google Scholar] [CrossRef]
- Zhu, Y.; Fu, Y.; Zhang, A.; Wang, X.; Zhao, Z.; Zhang, Y.; Yin, T.; Gou, J.; Wang, Y.; He, H.; et al. Rod-Shaped Nintedanib Nanocrystals Improved Oral Bioavailability through Multiple Intestinal Absorption Pathways. Eur. J. Pharm. Sci. 2022, 168, 106047. [Google Scholar] [CrossRef]
- Shaikh, F.; Patel, M.; Patel, V.; Patel, A.; Shinde, G.; Shelke, S.; Pathan, I. Formulation and Optimization of Cilnidipine Loaded Nanosuspension for the Enhancement of Solubility, Dissolution and Bioavailability. J. Drug Deliv. Sci. Technol. 2022, 69, 103066. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Ao, W.; Li, Y.; Xiao, Y.; Bai, M. Fabrication of an Aprepitant Nanosuspension Using Hydroxypropyl Chitosan to Increase the Bioavailability. Biochem. Biophys. Res. Commun. 2022, 631, 72–77. [Google Scholar] [CrossRef]
- Bakhaidar, R.B.; Naveen, N.R.; Basim, P.; Murshid, S.S.; Kurakula, M.; Alamoudi, A.J.; Bukhary, D.M.; Jali, A.M.; Majrashi, M.A.; Alshehri, S.; et al. Response Surface Methodology (RSM) Powered Formulation Development, Optimization and Evaluation of Thiolated Based Mucoadhesive Nanocrystals for Local Delivery of Simvastatin. Polymers 2022, 14, 5184. [Google Scholar] [CrossRef]
- Jin, G.; Ngo, H.V.; Wang, J.; Cui, J.-H.; Cao, Q.-R.; Park, C.; Jung, M.; Lee, B.-J. Design and Evaluation of in Vivo Bioavailability in Beagle Dogs of Bilayer Tablet Consisting of Immediate Release Nanosuspension and Sustained Release Layers of Rebamipide. Int. J. Pharm. 2022, 619, 121718. [Google Scholar] [CrossRef] [PubMed]
- Sampathi, S.; Prajapati, S.; Junnuthula, V.; Dyawanapelly, S. Pharmacokinetics and Anti-Diabetic Studies of Gliclazide Nanosuspension. Pharmaceutics 2022, 14, 1947. [Google Scholar] [CrossRef]
- Sun, W.; Gao, J.; Fan, R.; Zhang, T.; Tian, Y.; Wang, Z.; Zhang, H.; Zheng, A. The Effect of Particle Size on the Absorption of Cyclosporin A Nanosuspensions. Int. J. Nanomed. 2022, 17, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, X.; Chen, G.; Zhang, Y.; Zhang, H.; Zhang, W. Effect of Particle Size on the Oral Absorption of Isoliquiritigenin Nanocrystals. Braz. J. Pharm. Sci. 2022, 58, e201186. [Google Scholar] [CrossRef]
- Chougule, M.; Sirvi, A.; Saini, V.; Kashyap, M.; Sangamwar, A.T. Enhanced Biopharmaceutical Performance of Brick Dust Molecule Nilotinib via Stabilized Amorphous Nanosuspension Using a Facile Acid–Base Neutralization Approach. Drug Deliv. Transl. Res. 2023, 13, 2503–2519. [Google Scholar] [CrossRef]
- Bhairam, M.; Pandey, R.K.; Shukla, S.S.; Gidwani, B. Preparation, Optimization, and Evaluation of Dolutegravir Nanosuspension: In Vitro and In Vivo Characterization. J. Pharm. Innov. 2023, 18, 1798–1811. [Google Scholar] [CrossRef]
- Sarwar, A.R.; Iqbal, F.M.; Jamil, M.A.; Abbas, K. Nanocrystals of Mangiferin Using Design Expert: Preparation, Characterization, and Pharmacokinetic Evaluation. Molecules 2023, 28, 5918. [Google Scholar] [CrossRef]
- Chary, S.S.; Bhikshapathi, D.V.R.N.; Vamsi, N.M.; Kumar, J.P. Optimizing Entrectinib Nanosuspension: Quality by Design for Enhanced Oral Bioavailability and Minimized Fast-Fed Variability. Bionanoscience 2024, 1–19. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, F.; Shen, C.; Shen, B.; Yuan, H. Quercetin Nanocrystals for Bioavailability Enhancement: Impact of Different Functional Stabilizers on In Vitro/In Vivo Drug Performances. Pharm. Dev. Technol. 2024, 29, 551–558. [Google Scholar] [CrossRef]
- Malamatari, M.; Taylor, K.M.G.; Malamataris, S.; Douroumis, D.; Kachrimanis, K. Pharmaceutical Nanocrystals: Production by Wet Milling and Applications. Drug Discov. Today 2018, 23, 534–547. [Google Scholar] [CrossRef]
- Vinchhi, P.; Patel, J.K.; Patel, M.M. High-Pressure Homogenization Techniques for Nanoparticles. In Emerging Technologies for Nanoparticle Manufacturing; Patel, J.K., Pathak, Y.V., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 263–285. [Google Scholar]
- Sinha, B.; Müller, R.H.; Möschwitzer, J.P. Bottom-up Approaches for Preparing Drug Nanocrystals: Formulations and Factors Affecting Particle Size. Int. J. Pharm. 2013, 453, 126–141. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Llanos-González, E.; Alcain, F.J. The Use of Coenzyme Q10 in Cardiovascular Diseases. Antioxidants 2021, 10, 755. [Google Scholar] [CrossRef]
- Sun, J.; Wang, F.; Sui, Y.; She, Z.; Zhai, W.; Wang, C.; Deng, Y. Effect of Particle Size on Solubility, Dissolution Rate, and Oral Bioavailability: Evaluation Using Coenzyme Q10 as Naked Nanocrystals. Int. J. Nanomed. 2012, 7, 5733–5744. [Google Scholar] [CrossRef]
- Uhlemann, J.; Diedam, H.; Hoheisel, W.; Schikarski, T.; Peukert, W. Modeling and Simulation of Process Technology for Nanoparticulate Drug Formulations—A Particle Technology Perspective. Pharmaceutics 2020, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.C. The Stokes-Einstein Law for Diffusion in Solution. Proc. R. Soc. Lond. A 1924, 106, 724–749. [Google Scholar] [CrossRef]
- Yu, M.; Wang, J.; Yang, Y.; Zhu, C.; Su, Q.; Guo, S.; Sun, J.; Gan, Y.; Shi, X.; Gao, H. Rotation-Facilitated Rapid Transport of Nanorods in Mucosal Tissues. Nano Lett. 2016, 16, 7176–7182. [Google Scholar] [CrossRef] [PubMed]
- Noyes, A.A.; Whitney, W.R. The Rate of Solution of Solid Substances in Their Own Solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef]
- Junyaprasert, V.B.; Morakul, B. Nanocrystals for Enhancement of Oral Bioavailability of Poorly Water-Soluble Drugs. Asian J. Pharm. Sci. 2015, 10, 13–23. [Google Scholar] [CrossRef]
- Gao, L.; Liu, G.; Ma, J.; Wang, X.; Zhou, L.; Li, X.; Wang, F. Application of Drug Nanocrystal Technologies on Oral Drug Delivery of Poorly Soluble Drugs. Pharm. Res. 2013, 30, 307–324. [Google Scholar] [CrossRef]
- Rangaraj, N.; Pailla, S.R.; Chowta, P.; Sampathi, S. Fabrication of Ibrutinib Nanosuspension by Quality by Design Approach: Intended for Enhanced Oral Bioavailability and Diminished Fast Fed Variability. AAPS PharmSciTech 2019, 20, 326. [Google Scholar] [CrossRef]
- Mou, D.; Chen, H.; Wan, J.; Xu, H.; Yang, X. Potent Dried Drug Nanosuspensions for Oral Bioavailability Enhancement of Poorly Soluble Drugs with PH-Dependent Solubility. Int. J. Pharm. 2011, 413, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Thombre, A.G.; Caldwell, W.B.; Friesen, D.T.; McCray, S.B.; Sutton, S.C. Solid Nanocrystalline Dispersions of Ziprasidone with Enhanced Bioavailability in the Fasted State. Mol. Pharm. 2012, 9, 3526–3534. [Google Scholar] [CrossRef]
- Reddy, M.R.; Gubbiyappa, K.S. Formulation Development, Optimization, and Characterization of Entrectinib-Loaded Supersaturable Self-Nanoemulsifying Drug Delivery Systems. Bionanoscience 2023, 13, 521–540. [Google Scholar] [CrossRef]
- Feldman, M.; Barnett, C. Fasting Gastric PH and Its Relationship to True Hypochlorhydria in Humans. Dig. Dis. Sci. 1991, 36, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, G.; Ma, J.; Wang, X.; Zhou, L.; Li, X. Drug Nanocrystals: In Vivo Performances. J. Control. Release 2012, 160, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA). Chemistry Review: Norvir (Ritonavir) Tablets. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022417s000_ChemR.pdf (accessed on 14 August 2024).
- Food and Drug Administration (FDA). Product Quality Review: Entrectinib Capsules. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212725Orig1s000,%20212726Orig1s000ChemR.pdf (accessed on 14 August 2024).
- Wang, Y.; Tan, X.; Fan, X.; Zhao, L.; Wang, S.; He, H.; Yin, T.; Zhang, Y.; Tang, X.; Jian, L.; et al. Current Strategies for Oral Delivery of BCS IV Drug Nanocrystals: Challenges, Solutions and Future Trends. Expert. Opin. Drug Deliv. 2021, 18, 1211–1228. [Google Scholar] [CrossRef]
- Arora, S.; Pansari, A.; Kilford, P.; Jamei, M.; Gardner, I.; Turner, D.B. Biopharmaceutic In Vitro In Vivo Extrapolation (IVIV_E) Informed Physiologically-Based Pharmacokinetic Model of Ritonavir Norvir Tablet Absorption in Humans Under Fasted and Fed State Conditions. Mol. Pharm. 2020, 17, 2329–2344. [Google Scholar] [CrossRef]
- Ng, J.; Klein, C.; Chui, Y.; Awni, W.; Morris, J.; Podsadecki, T.; Cui, Y.; Bernstein, B.; Kim, D. The Effect of Food on Ritonavir Bioavailability Following Administration of Ritonavir 100 Mg Film-Coated Tablet in Healthy Adult Subjects. J. Int. AIDS Soc. 2008, 11, P247. [Google Scholar] [CrossRef]
- Vermunt, M.A.C.; de Weger, V.A.; Janssen, J.M.; Lopez-Yurda, M.I.; Keessen, M.; Thijssen, B.; Rosing, H.; Huitema, A.D.R.; Beijnen, J.H.; Marchetti, S. Effect of Food on the Pharmacokinetics of the Oral. Docetaxel Tablet Formulation ModraDoc006 Combined with Ritonavir (ModraDoc006/r) in Patients with Advanced Solid Tumours. Drugs R&D 2021, 21, 103–111. [Google Scholar] [CrossRef]
- DeSesso, J.M.; Jacobson, C.F. Anatomical and Physiological Parameters Affecting Gastrointestinal Absorption in Humans and Rats. Food Chem. Toxicol. 2001, 39, 209–228. [Google Scholar] [CrossRef]
- Cao, X.; Gibbs, S.T.; Fang, L.; Miller, H.A.; Landowski, C.P.; Shin, H.-C.; Lennernas, H.; Zhong, Y.; Amidon, G.L.; Yu, L.X.; et al. Why Is It Challenging to Predict Intestinal Drug Absorption and Oral Bioavailability in Human Using Rat Model. Pharm. Res. 2006, 23, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Femia, R.A.; Goyette, R.E. The Science of Megestrol Acetate Delivery. BioDrugs 2005, 19, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Park, S.R.; Hwang, J.G.; Jeong, S.I.; Choi, Y.-S.; Min, H.J.; Kim, H.Y.; Choi, B.-H.; Park, M.K. Comparison of the Pharmacokinetic Characteristics and Bioequivalence between Two Nanosuspension Formulations of Megestrol Acetate in Healthy Korean Male Subjects. Transl. Clin. Pharmacol. 2024, 32, 63–72. [Google Scholar] [CrossRef]
- Koziolek, M.; Grimm, M.; Bollmann, T.; Schäfer, K.J.; Blattner, S.M.; Lotz, R.; Boeck, G.; Weitschies, W. Characterization of the GI Transit Conditions in Beagle Dogs with a Telemetric Motility Capsule. Eur. J. Pharm. Biopharm. 2019, 136, 221–230. [Google Scholar] [CrossRef]
- Apley, M.; Crist, B.; Gonzalez, M.; Hunter, R.P.; Martinez, M.N.; Modric, S.; Papich, M.G.; Parr, A.F.; Riviere, J.E.; Marques, M.R.C. Solubility Criteria for Veterinary Drugs—Workshop Report. Pharm. Forum 2013, 39, 1–9. [Google Scholar]
- Jang, K.; Yoon, S.; Kim, S.-E.; Cho, J.-Y.; Yoon, S.H.; Lim, K.S.; Yu, K.-S.; Jang, I.-J.; Lee, H. Novel Nanocrystal Formulation of Megestrol Acetate Has Improved Bioavailability Compared with the Conventional Micronized Formulation in the Fasting State. Drug Des. Devel Ther. 2014, 8, 851–858. [Google Scholar] [CrossRef]
- Xiong, S.; Liu, W.; Li, D.; Chen, X.; Liu, F.; Yuan, D.; Pan, H.; Wang, Q.; Fang, S.; Chen, T. Oral Delivery of Puerarin Nanocrystals To Improve Brain Accumulation and Anti-Parkinsonian Efficacy. Mol. Pharm. 2019, 16, 1444–1455. [Google Scholar] [CrossRef]
- García-Herrero, V.; Torrado, C.; García-Rodríguez, J.J.; López-Sánchez, A.; Torrado, S.; Torrado-Santiago, S. Improvement of the Surface Hydrophilic Properties of Naproxen Particles with Addition of Hydroxypropylmethyl Cellulose and Sodium Dodecyl Sulphate: In Vitro and in Vivo Studies. Int. J. Pharm. 2017, 529, 381–390. [Google Scholar] [CrossRef]
- Narula, A.; Sabra, R.; Li, N. Mechanisms and Extent of Enhanced Passive Permeation by Colloidal Drug Particles. Mol. Pharm. 2022, 19, 3085–3099. [Google Scholar] [CrossRef]
- Ramachandran, G.; Sudheesh, M.S. Role of Permeability on the Biopredictive Dissolution of Amorphous Solid Dispersions. AAPS PharmSciTech 2021, 22, 243. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutical Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Amidon, K.S.; Langguth, P.; Lennernäs, H.; Yu, L.; Amidon, G.L. Bioequivalence of Oral Products and the Biopharmaceutics Classification System: Science, Regulation, and Public Policy. Clin. Pharmacol. Ther. 2011, 90, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Gigliobianco, M.R.; Casadidio, C.; Censi, R.; Di Martino, P. Nanocrystals of Poorly Soluble Drugs: Drug Bioavailability and Physicochemical Stability. Pharmaceutics 2018, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Asif, M.; Kumar, A.; Aggarval, A. Biopharmaceutical Classification System: An Account. Int. J. Pharmtech Res. 2010, 2, 1681–1690. [Google Scholar]
- Peltonen, L.; Strachan, C.J. Degrees of Order: A Comparison of Nanocrystal and Amorphous Solids for Poorly Soluble Drugs. Int. J. Pharm. 2020, 586, 119492. [Google Scholar] [CrossRef]
- Jermain, S.V.; Brough, C.; Williams, R.O. Amorphous Solid Dispersions and Nanocrystal Technologies for Poorly Water-Soluble Drug Delivery—An Update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef]
- Merisko-Liversidge, E.; Liversidge, G.G. Nanosizing for Oral and Parenteral Drug Delivery: A Perspective on Formulating Poorly-Water Soluble Compounds Using Wet Media Milling Technology. Adv. Drug Deliv. Rev. 2011, 63, 427–440. [Google Scholar] [CrossRef]
- Tuomela, A.; Hirvonen, J.; Peltonen, L. Stabilizing Agents for Drug Nanocrystals: Effect on Bioavailability. Pharmaceutics 2016, 8, 16. [Google Scholar] [CrossRef]
- Fu, Q.; Sun, J.; Ai, X.; Zhang, P.; Li, M.; Wang, Y.; Liu, X.; Sun, Y.; Sui, X.; Sun, L.; et al. Nimodipine Nanocrystals for Oral Bioavailability Improvement: Role of Mesenteric Lymph Transport in the Oral Absorption. Int. J. Pharm. 2013, 448, 290–297. [Google Scholar] [CrossRef]
- Yu, Q.; Wang, Z.; Li, P.; Yang, Q. The Effect of Various Absorption Enhancers on Tight Junction in the Human Intestinal Caco-2 Cell Line. Drug Dev. Ind. Pharm. 2013, 39, 587–592. [Google Scholar] [CrossRef]
- Brunner, J.; Ragupathy, S.; Borchard, G. Target Specific Tight Junction Modulators. Adv. Drug Deliv. Rev. 2021, 171, 266–288. [Google Scholar] [CrossRef]
- Luiz, M.T.; Di Filippo, L.D.; Alves, R.C.; Araújo, V.H.S.; Duarte, J.L.; Marchetti, J.M.; Chorilli, M. The Use of TPGS in Drug Delivery Systems to Overcome Biological Barriers. Eur. Polym. J. 2021, 142, 110129. [Google Scholar] [CrossRef]
- Collnot, E.-M.; Baldes, C.; Schaefer, U.F.; Edgar, K.J.; Wempe, M.F.; Lehr, C.-M. Vitamin E TPGS P-Glycoprotein Inhibition Mechanism: Influence on Conformational Flexibility, Intracellular ATP Levels, and Role of Time and Site of Access. Mol. Pharm. 2010, 7, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Macedo, L.d.O.; Morales, I.A.C.; Barbosa, E.J.; Stephano, M.A.; de Araujo, G.L.B.; Bou-Chacra, N.A. Thermal Study, Process Optimization, and Water Solubility Improvement of a Freeze-Dried Artemether Nanosuspension for Malaria Treatment. J. Drug Deliv. Sci. Technol. 2022, 78, 103915. [Google Scholar] [CrossRef]
- Guo, Y.; Luo, J.; Tan, S.; Otieno, B.O.; Zhang, Z. The Applications of Vitamin E TPGS in Drug Delivery. Eur. J. Pharm. Sci. 2013, 49, 175–186. [Google Scholar] [CrossRef]
- Kou, L.; Sun, R.; Bhutia, Y.D.; Yao, Q.; Chen, R. Emerging Advances in P-Glycoprotein Inhibitory Nanomaterials for Drug Delivery. Expert. Opin. Drug Deliv. 2018, 15, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Halder, J.; Pradhan, D.; Kar, B.; Ghosh, G.; Rath, G. Nanotherapeutics Approaches to Overcome P-Glycoprotein-Mediated Multi-Drug Resistance in Cancer. Nanomedicine 2022, 40, 102494. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, D.A.; Langer, R.; Traverso, G. Mucus Interaction to Improve Gastrointestinal Retention and Pharmacokinetics of Orally Administered Nano-Drug Delivery Systems. J. Nanobiotechnol. 2022, 20, 362. [Google Scholar] [CrossRef]
- Cone, R.A. Barrier Properties of Mucus. Adv. Drug Deliv. Rev. 2009, 61, 75–85. [Google Scholar] [CrossRef]
- Feng, C.; Chen, X.; Zhang, J.; Sun, G.; Cheng, X.; Wang, Z.; Park, H.-J. The Effect of Carboxymethyl-Chitosan Nanoparticles on Proliferation of Keloid Fibroblast. Front. Chem. China 2011, 6, 31–37. [Google Scholar] [CrossRef]
- Tan, S.L.J.; Billa, N. Improved Bioavailability of Poorly Soluble Drugs through Gastrointestinal Muco-Adhesion of Lipid Nanoparticles. Pharmaceutics 2021, 13, 1817. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tu, L.; Cheng, M.; Feng, J.; Jin, Y. Mechanisms for Oral Absorption Enhancement of Drugs by Nanocrystals. J. Drug Deliv. Sci. Technol. 2020, 56, 101607. [Google Scholar] [CrossRef]
- Tian, Z.; Zhao, Y.; Mai, Y.; Qiao, F.; Guo, J.; Dong, L.; Niu, Y.; Gou, G.; Yang, J. Nanocrystals with Different Stabilizers Overcome the Mucus and Epithelial Barriers for Oral Delivery of Multicomponent Bufadienolides. Int. J. Pharm. 2022, 616, 121522. [Google Scholar] [CrossRef] [PubMed]
- Sillman, B.; Bade, A.N.; Dash, P.K.; Bhargavan, B.; Kocher, T.; Mathews, S.; Su, H.; Kanmogne, G.D.; Poluektova, L.Y.; Gorantla, S.; et al. Creation of a Long-Acting Nanoformulated Dolutegravir. Nat. Commun. 2018, 9, 443. [Google Scholar] [CrossRef]
- Jahangir, M.A.; Imam, S.S.; Muheem, A.; Chettupalli, A.; Al-Abbasi, F.A.; Nadeem, M.S.; Kazmi, I.; Afzal, M.; Alshehri, S. Nanocrystals: Characterization Overview, Applications in Drug Delivery, and Their Toxicity Concerns. J. Pharm. Innov. 2022, 17, 237–248. [Google Scholar] [CrossRef]
- Ahmad, A.; Imran, M.; Sharma, N. Precision Nanotoxicology in Drug Development: Current Trends and Challenges in Safety and Toxicity Implications of Customized Multifunctional Nanocarriers for Drug-Delivery Applications. Pharmaceutics 2022, 14, 2463. [Google Scholar] [CrossRef]
- Seok, S.H.; Ha, J.-M.; Kim, T.H.; Kim, G.-W.; Kim, B.H.; Lee, D.W.; Choi, M.-S.; Lee, S.-H.; Kim, J.-Y.; Park, E.-S. Effect of Gastric Residence Time on the Oral Absorption of Rebamipide Sustained-Release Tablets in Beagle Dogs. J. Pharm. Investig. 2021, 51, 759–766. [Google Scholar] [CrossRef]
- Nguyen, T.-T.; Hwang, K.-M.; Kim, S.-H.; Park, E.-S. Development of Novel Bilayer Gastroretentive Tablets Based on Hydrophobic Polymers. Int. J. Pharm. 2020, 574, 118865. [Google Scholar] [CrossRef]
- Lv, Y.; Wu, W.; Corpstein, C.D.; Li, T.; Lu, Y. Biological and Intracellular Fates of Drug Nanocrystals through Different Delivery Routes: Recent Development Enabled by Bioimaging and PK Modeling. Adv. Drug Deliv. Rev. 2022, 188, 114466. [Google Scholar] [CrossRef]
- Nowacek, A.S.; McMillan, J.; Miller, R.; Anderson, A.; Rabinow, B.; Gendelman, H.E. Nanoformulated Antiretroviral Drug Combinations Extend Drug Release and Antiretroviral Responses in HIV-1-Infected Macrophages: Implications for NeuroAIDS Therapeutics. J. Neuroimmune Pharmacol. 2010, 5, 592–601. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Drug Nanocrystal | In Vivo Performance | ||||
|---|---|---|---|---|---|
| Composition | Preparation Method | Particle Size (nm) | Control | Main Observations | References |
| Cinacalcet Soluplus® | Antisolvent precipitation | 244 ± 2 | Commercial product | Increase by 2-fold in Cmax and 1.5-fold in AUC0–t in the fasted state; elimination of food effect | [33] |
| Ritonavir HPMC 3cps + SDS | HPH | 541.8 ± 14.5 | Stabilizer solution Coarse suspension Physical mixture Commercial product | Increase by 2-fold in Cmax and AUC0–8h compared with commercial product in the fed state | [34] |
| Lovastatin P188 | Spherical: WBM Rod: antisolvent precipitation–HPH Flaky: cooling crystallization–HPH | 405.8–417.3 | Drug solution in ethanol and Cremophor® EL | Rod-shaped increased AUC0–24h by 1.8-fold and 1.4-fold compared with other morphologies | [35] |
| Ginkgolide B HPMC E5 | Antisolvent precipitation | 83.5 ± 1.8 | Active compound suspended in HPMC | Increase by 13-fold in Cmax and 5-fold in AUC0–t | [36] |
| Megestrol acetate HPMC + SDS | WBM | 158.0 | Microsuspension Commercial product Coarse nanocrystals | Increase by 2.7-fold in Cmax and 3.6-fold in AUC0–2h compared with microsuspension | [37] |
| Venetoclax PVA | Antisolvent precipitation–HPH | ~340.0 | Drug suspension | Increase by 1.8-fold in Cmax and 2-fold in AUC0–∞ | [38] |
| Nintedanib Na-CMC | Antisolvent precipitation | 370.3 ± 19.3 | Drug suspension in T80 | Increase by 2.5-fold in AUC0–t | [39] |
| Cilnidipine P188 + T80 | Antisolvent precipitation | 280.1 ± 3.7 | Drug suspended in Na-CMC Commercial product | Increase by 3-fold in Cmax and 1.8-fold in AUC0–t compared with the commercial product | [40] |
| Aprepitant HPCS | WBM | 151.0 ± 14.5 | Active compound suspended in Na-CMC Commercial product | Increase by 1.3-fold in Cmax and 1.5-fold in AUC0–t compared with the commercial product | [41] |
| Simvastatin P407 | Antisolvent precipitation | 137.8 | Drug suspension | Increase of 6 h in Tmax, 73 h in MRT, and a decrease by 2-fold in Cmax | [42] |
| Rebamipide P188 + P407 | Antisolvent precipitation | 148.0 ± 2.5 | Commercial product | No significant difference between the tablets | [43] |
| Gliclazide SDS + lecithin | Antisolvent precipitation | 96.5 ± 15.0 | Drug suspended in CMC Commercial product | Increase by 3-fold in Cmax and 1.7-fold in AUC0–t compared with the commercial product | [44] |
| Cyclosporin A HPC + TPGS + SDS | WBM | 280–522 | Commercial microemulsion suspended in water | Smaller nanocrystals increased Cmax and AUC0–48h compared with larger ones | [45] |
| Isoliquiritigenin mPEG-PCL | Antisolvent precipitation | 46–540.0 | Active compound suspension | Nanocrystals of ~300 nm increased 5.8-fold of Cmax and 2.7-fold in AUC0–t | [46] |
| Nilotinib Soluplus® + HPMCAS | Antisolvent precipitation | 130.5 ± 1.2 | Drug suspension in MC | Increase by 1.5-fold in Cmax and 1.5-fold in AUC0–24h | [47] |
| Dolutegravir T80 + T20 + Soluplus® | HSH–antisolvent precipitation | 337.1 ± 0.2 | Drug suspension in MC | Increase by 3-fold in Cmax, 1.9-fold in AUC, and prolonged t1/2 | [48] |
| Mangiferin HPMC + P407 + T80 | HSH–antisolvent precipitation | 100.2 | Active compound suspension | Increase by 1.1-fold in Cmax, 1.2-fold in AUC0–t, prolonged t1/2, and MRT | [49] |
| Entrectinib P188 | Antisolvent precipitation | 83 ± 15 | Drug suspension in HPMC Vehicle | Increase by 4-fold in Cmax and 3-fold in AUC0–24h; elimination of food effect | [50] |
| Quercetin HPMC E15, P188, P407, TPGS or GL | WBM | 193–210 | Active compound suspended in water | Almost 11-fold increase in AUC0–t for HPMC E15 and P188 preparations | [51] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macedo, L.d.O.; Masiero, J.F.; Bou-Chacra, N.A. Drug Nanocrystals in Oral Absorption: Factors That Influence Pharmacokinetics. Pharmaceutics 2024, 16, 1141. https://doi.org/10.3390/pharmaceutics16091141
Macedo LdO, Masiero JF, Bou-Chacra NA. Drug Nanocrystals in Oral Absorption: Factors That Influence Pharmacokinetics. Pharmaceutics. 2024; 16(9):1141. https://doi.org/10.3390/pharmaceutics16091141
Chicago/Turabian StyleMacedo, Luiza de Oliveira, Jéssica Fagionato Masiero, and Nádia Araci Bou-Chacra. 2024. "Drug Nanocrystals in Oral Absorption: Factors That Influence Pharmacokinetics" Pharmaceutics 16, no. 9: 1141. https://doi.org/10.3390/pharmaceutics16091141