
Biological Evaluations and Computer-Aided Approaches of Janus Kinases 2 and 3 Inhibitors for Cancer Treatment: A Review
 ,
,  ,
,  , , , ,
, , , ,
Abstract
1. Introduction
2. The JAK2 Target
3. The JAK3 Target
4. JAK2 Inhibitors
5. Natural Derived JAK2 Inhibitors
6. JAK3 Inhibitors
7. Natural Derived JAK3 Inhibitors
8. Dual JAK2/3 Inhibitors
9. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Bose, S.; Banerjee, S.; Mondal, A.; Chakraborty, U.; Pumarol, J.; Croley, C.R.; Bishayee, A. Targeting the JAK/STAT Signaling Pathway Using Phytocompounds for Cancer Prevention and Therapy. Cells 2020, 11, 1451. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef]
- Leonard, W.J.; O’Shea, J.J. Jaks and STATs: Biological implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef] [PubMed]
- Ihle, J.N.; Witthuhn, B.A.; Quelle, F.W.; Yamamoto, K.; Silvennoinen, O. Signaling through the hematopoietic cytokine receptors. Annu. Rev. Immunol. 1995, 13, 369–398. [Google Scholar] [CrossRef] [PubMed]
- Hammarén, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Haan, C.; Kreis, S.; Margue, C.; Behrmann, I. Jaks and cytokine receptors–an intimate relationship. Biochem. Pharmacol. 2006, 72, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Pölläniemi, A.; Virtanen, A.; Silvennoinen, O.; Haikarainen, T. Development of an enzyme-coupled activity assay for Janus kinase 2 inhibitor screening. SLAS Discov. 2023, 28, 180–187. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef]
- Li, Y.; Guo, F.; Chen, T.; Zhang, L.; Qin, Y. Anthraquinone derivative C10 inhibits proliferation and cell cycle progression in colon cancer cells via the Jak2/Stat3 signaling pathway. Toxicol. Appl. Pharmacol. 2021, 418, 115481. [Google Scholar] [CrossRef]
- Pardanani, A.; Gotlib, J.R.; Jamieson, C.; Cortes, J.E.; Talpaz, M.; Stone, R.M.; Silverman, M.H.; Gilliland, D.G.; Shorr, J.; Tefferi, A. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J. Clin. Oncol. 2011, 9, 789–796. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef]
- Plimack, E.R.; Lorusso, P.M.; McCoon, P.; Tang, W.; Krebs, A.D.; Curt, G.; Eckhardt, S.G. AZD1480: A phase I study of a novel JAK2 inhibitor in solid tumors. Oncol. 2013, 18, 819–820. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Vaddi, K.; Liu, P.; Manshouri, T.; Li, J.; Scherle, P.A.; Caulder, E.; Wen, X.; Li, Y.; Waeltz, P.; et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: Therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010, 115, 3109–3117. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S. JAK2 inhibitors for myeloproliferative neoplasms: What is next? Blood 2017, 130, 115–125. [Google Scholar] [CrossRef]
- Telliez, J.B.; Dowty, M.E.; Wang, L.; Jussif, J.; Lin, T.; Li, L.; Moy, E.; Balbo, P.; Li, W.; Zhao, Y.; et al. Discovery of a JAK3-selective inhibitor: Functional differentiation of JAK3-selective inhibition over pan-JAK or JAK1-selective inhibition. ACS Chem. Biol. 2016, 11, 3442–3451. [Google Scholar] [CrossRef]
- Forster, M.; Liang, X.J.; Schröder, M.; Gerstenecker, S.; Chaikuad, A.; Knapp, S.; Laufer, S.; Gehringer, M. Discovery of a Novel Class of Covalent Dual Inhibitors Targeting the Protein Kinases BMX and BTK. Int. J. Mol. Sci. 2020, 21, 9269. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Marín, H.A.; Tosti, A. Evaluating the Therapeutic Potential of Ritlecitinib for the Treatment of Alopecia Areata. Drug Des. Dev. Ther. 2022, 16, 363–374. [Google Scholar] [CrossRef]
- Jiang, J.K.; Ghoreschi, K.; Deflorian, F.; Chen, Z.; Perreira, M.; Pesu, M.; Smith, J.; Nguyen, D.T.; Liu, E.H.; Leister, W.; et al. Examining the chirality, conformation and selective kinase inhibition of 3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile (CP-690,550). J. Med. Chem. 2008, 51, 8012–8018. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Chen, C.; Huan, X.; Huang, H.; Wang, M.; Zhang, J.; Yan, Y.; Liu, J.; Zhang, T.; Zhang, D. Design, synthesis, and pharmacological evaluation of 4- or 6-phenyl-pyrimidine derivatives as novel and selective Janus kinase 3 inhibitors. Eur. J. Med. Chem. 2020, 191, 112148. [Google Scholar] [CrossRef]
- Su, W.; Chen, Z.; Liu, M.; He, R.; Liu, C.; Li, R.; Gao, M.; Zheng, M.; Tu, Z.; Zhang, Z.; et al. Design, synthesis and structure-activity relationship studies of pyrido[2,3-d]pyrimidin-7-ones as potent Janus Kinase 3 (JAK3) covalent inhibitors. Bioorg. Med. Chem. Lett. 2022, 64, 128680. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Diao, Y.; Li, W.; Luo, Y.; Yang, T.; Zhao, Y.; Qi, T.; Xu, F.; Ma, X.; Ge, H.; et al. Design, synthesis and structure-activity relationship study of aminopyridine derivatives as novel inhibitors of Janus kinase 2. Bioorg. Med. Chem. Lett. 2019, 29, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.M.; Li, L.; Ho, Y.; Lin, A.; Li, L.; Stein, A.; Forman, S.; Perrotti, D.; Jove, R.; Bhatia, R. Role of altered growth factor receptor-mediated JAK2 signaling in growth and maintenance of human acute myeloid leukemia stem cells. Blood 2014, 123, 2826–2837. [Google Scholar] [CrossRef]
- Dosil, M.; Wang, S.; Lemischka, I.R. Mitogenic signalling and substrate specificity of the Flk2/Flt3 receptor tyrosine kinase in fibroblasts and interleukin 3-dependent hematopoietic cells. Mol. Cell. Biol. 1993, 13, 6572–6585. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef]
- Alicea-Velázquez, N.L.; Boggon, T.J. The use of structural biology in Janus kinase targeted drug discovery. Curr. Drug Targets 2011, 12, 546–555. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Janus kinases in immune cell signaling. Immunol. Rev. 2009, 228, 273–287. [Google Scholar] [CrossRef]
- Virtanen, A.T.; Haikarainen, T.; Sampathkumar, P.; Palmroth, M.; Liukkonen, S.; Liu, J.; Nekhotiaeva, N.; Hubbard, S.R.; Silvennoinen, O. Identification of Novel Small Molecule Ligands for JAK2 Pseudokinase Domain. Pharmaceuticals 2023, 16, 75. [Google Scholar] [CrossRef]
- Loh, C.Y.; Arya, A.; Naema, A.F.; Wong, W.F.; Sethi, G.; Looi, C.Y. Signal Transducer and Activator of Transcription (STATs) Proteins in Cancer and Inflammation: Functions and Therapeutic Implication. Front. Oncol. 2019, 9, 48. [Google Scholar] [CrossRef]
- Doheny, D.; Sirkisoon, S.; Carpenter, R.L.; Aguayo, N.R.; Regua, A.T.; Anguelov, M.; Manore, S.G.; Arrigo, A.; Jalboush, S.A.; Wong, G.L.; et al. Combined inhibition of JAK2-STAT3 and SMO-GLI1/tGLI1 pathways suppresses breast cancer stem cells, tumor growth, and metastasis. Oncogene 2020, 39, 6589–6605. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef]
- Raivola, J.; Haikarainen, T.; Silvennoinen, O. Characterization of JAK1 Pseudokinase Domain in Cytokine Signaling. Cancers 2020, 12, 78. [Google Scholar] [CrossRef] [PubMed]
- Raivola, J.; Hammarén, H.M.; Virtanen, A.T.; Bulleeraz, V.; Ward, A.C.; Silvennoinen, O. Hyperactivation of Oncogenic JAK3 Mutants Depend on ATP Binding to the Pseudokinase Domain. Front. Oncol. 2018, 8, 560. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Zhao, R.; Xing, S.; Li, Z.; Fu, X.; Li, Q.; Krantz, S.B.; Zhao, Z.J. Identification of an acquired JAK2 mutation in polycythemia vera. J. Biol. Chem. 2005, 280, 22788–22792. [Google Scholar] [CrossRef]
- Yin, Y.; Chen, C.J.; Yu, R.N.; Shu, L.; Zhang, T.T.; Zhang, D.Y. Discovery of novel selective Janus kinase 2 (JAK2) inhibitors bearing a 1H-pyrazolo[3,4-d]pyrimidin-4-amino scaffold. Bioorg. Chem. 2019, 27, 1562–1576. [Google Scholar] [CrossRef]
- Xiong, H.; Du, W.; Zhang, Y.J.; Hong, J.; Su, W.Y.; Tang, J.T.; Wang, Y.C.; Lu, R.; Fang, J.Y. Trichostatin A, a histone deacetylase inhibitor, suppresses JAK2/STAT3 signaling via inducing the promoter-associated histone acetylation of SOCS1 and SOCS3 in human colorectal cancer cells. Mol. Carcinog. 2012, 51, 174–184. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, F.; Li, G.; Li, G.; Yang, X.; Liu, L.; Zhang, R.; Zhang, B.; Feng, Y. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018, 18, 25. [Google Scholar] [CrossRef]
- Leroy, E.; Constantinescu, S.N. Rethinking JAK2 inhibition: Towards novel strategies of more specific and versatile janus kinase inhibition. Leukemia 2017, 31, 1023–1038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Gadina, M.; Schreiber, R.D. Cytokine signaling in 2002: New surprises in the Jak/Stat pathway. Cell 2002, 109, S121–S131. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Shi, W.; Zhao, D.; Wang, Q.; Chang, X.; He, X.; Wang, X.; Gao, Y.; Lu, P.; Zhang, X.; et al. Design and synthesis of boron-containing diphenylpyrimidines as potent BTK and JAK3 dual inhibitors. Bioorg. Med. Chem. 2020, 28, 115236. [Google Scholar] [CrossRef] [PubMed]
- Sanachai, K.; Mahalapbutr, P.; Hengphasatporn, K.; Shigeta, Y.; Seetaha, S.; Tabtimmai, L.; Langer, T.; Wolschann, P.; Kittikool, T.; Yotphan, S.; et al. Pharmacophore-Based Virtual Screening and Experimental Validation of Pyrazolone-Derived Inhibitors toward Janus Kinases. ACS Omega 2022, 7, 33548–33559. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Rassidakis, G.Z.; Lin, Q.; Atwell, C.; Medeiros, L.J.; Amin, H.M. Jak3 activation is significantly associated with ALK expression in anaplastic large cell lymphoma. Hum. Pathol. 2005, 36, 939–944. [Google Scholar] [CrossRef]
- Gee, K.; Kozlowski, M.; Kryworuchko, M.; Diaz-Mitoma, F.; Kumar, A. Differential effect of IL-4 and IL-13 on CD44 expression in the Burkitt’s lymphoma B cell line BL30/B95-8 and in Epstein-Barr virus (EBV) transformed human B cells: Loss of IL-13 receptors on Burkitt’s lymphoma B cells. Cell. Immunol. 2001, 211, 131–142. [Google Scholar] [CrossRef]
- Yared, M.A.; Khoury, J.D.; Medeiros, L.J.; Rassidakis, G.Z.; Lai, R. Activation status of the JAK/STAT3 pathway in mantle cell lymphoma. Arch. Pathol. Lab. Med. 2005, 129, 990–996. [Google Scholar] [CrossRef]
- Malamut, G.; El Machhour, R.; Montcuquet, N.; Martin-Lannerée, S.; Dusanter-Fourt, I.; Verkarre, V.; Mention, J.J.; Rahmi, G.; Kiyono, H.; Butz, E.A.; et al. IL-15 triggers an antiapoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease-associated inflammation and lymphomagenesis. J. Clin. Investig. 2010, 120, 2131–2143. [Google Scholar] [CrossRef]
- Forster, M.; Gehringer, M.; Laufer, S.A. Recent advances in JAK3 inhibition: Isoform selectivity by covalent cysteine targeting. Bioorg. Med. Chem. Lett. 2017, 27, 4229–4237. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.A.; El-Kady, D.S.; Abd-Rabou, A.A.; Tantawy, M.A.; AbdElhalim, M.M.; Elazabawy, S.R.; Abdallah, A.E.M.; Elmegeed, G.A. Synthesis of novel hybrid hetero-steroids: Molecular docking study augmented anti-proliferative properties against cancerous cells. Steroids 2020, 54, 108527. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, T.; Xu, L.; Dong, Y.; Luo, Y.; Wang, C.; Han, Y.; Chen, K.; Qin, M.; Liu, Y.; et al. Discovery of 4-piperazinyl-2-aminopyrimidine derivatives as dual inhibitors of JAK2 and FLT3. Eur. J. Med. Chem. 2019, 181, 111590. [Google Scholar] [CrossRef] [PubMed]
- Jyothi-Buggana, S.; Paturi, M.C.; Perka, H.; Gade, D.R.; Vvs, R.P. Novel 2,4-disubstituted quinazolines as cytotoxic agents and JAK2 inhibitors: Synthesis, in vitro evaluation and molecular dynamics studies. Comput. Biol. Chem. 2019, 79, 110–118. [Google Scholar] [CrossRef]
- Sinha, I.; Null, K.; Wolter, W.; Suckow, M.A.; King, T.; Pinto, J.T.; Sinha, R. Methylseleninic acid downregulates hypoxia-inducible factor-1α in invasive prostate cancer. Int. J. Cancer 2012, 130, 1430–1439. [Google Scholar] [CrossRef]
- Tarrado-Castellarnau, M.; Cortés, R.; Zanuy, M.; Tarragó-Celada, J.; Polat, I.H.; Hill, R.; Fan, T.W.; Link, W.; Cascante, M. Methylseleninic acid promotes antitumour effects via nuclear FOXO3a translocation through Akt inhibition. Pharmacol. Res. 2015, 102, 218–234. [Google Scholar] [CrossRef]
- Zhang, T.; Zhu, X.; Qiu, J.; Jiang, K.; Zhao, G.; Wu, H.; Deng, G.; Qiu, C. Correction to: Methylseleninic Acid Suppresses Breast Cancer Growth via the JAK2/STAT3 Pathway. Reprod. Sci. 2021, 28, 614. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Diao, Y.; Ge, H.; Xu, F.; Zhu, L.; Zhao, Z.; Li, H. Discovery and optimization of 2-aminopyridine derivatives as novel and selective JAK2 inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127048. [Google Scholar] [CrossRef]
- Li, Y.; Wang, P.; Chen, C.; Ye, T.; Han, Y.; Hou, Y.; Liu, Y.; Gong, P.; Qin, M.; Zhao, Y. Discovery and rational design of 2-aminopyrimidine-based derivatives targeting Janus kinase 2 (JAK2) and FMS-like tyrosine kinase 3 (FLT3). Bioorg. Chem. 2020, 104, 104361. [Google Scholar] [CrossRef]
- Bottiglieri, T. S-Adenosyl-L-methionine (SAMe): From the bench to the bedside—Molecular basis of a pleiotrophic molecule. Am. J. Clin. Nutr. 2002, 76, 1151S–1157S. [Google Scholar] [CrossRef]
- Mahmood, N.; Cheishvili, D.; Arakelian, A.; Tanvir, I.; Khan, H.A.; Pépin, A.S.; Szyf, M.; Rabbani, S.A. Methyl donor S-adenosylmethionine (SAM) supplementation attenuates breast cancer growth, invasion, and metastasis in vivo; therapeutic and chemopreventive applications. Oncotarget 2017, 9, 5169–5183. [Google Scholar] [CrossRef]
- Ma, D.; Shen, B.; Seewoo, V.; Tong, H.; Yang, W.; Cheng, X.; Jin, Z.; Peng, C.; Qiu, W. GADD45β induction by S-adenosylmethionine inhibits hepatocellular carcinoma cell proliferation during acute ischemia-hypoxia. Oncotarget 2016, 7, 37215–37225. [Google Scholar] [CrossRef] [PubMed]
- Parashar, S.; Cheishvili, D.; Arakelian, A.; Hussain, Z.; Tanvir, I.; Khan, H.A.; Szyf, M.; Rabbani, S.A. S-adenosylmethionine blocks osteosarcoma cells proliferation and invasion in vitro and tumor metastasis in vivo: Therapeutic and diagnostic clinical applications. Cancer Med. 2015, 4, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Li, T.W.; Zhang, Q.; Oh, P.; Xia, M.; Chen, H.; Bemanian, S.; Lastra, N.; Circ, M.; Moyer, M.P.; Mato, J.M.; et al. S-Adenosylmethionine and methylthioadenosine inhibit cellular FLICE inhibitory protein expression and induce apoptosis in colon cancer cells. Mol. Pharmacol. 2009, 76, 192–200. [Google Scholar] [CrossRef]
- Liu, Y.; Bi, T.; Yuan, F.; Gao, X.; Jia, G.; Tian, Z. S-adenosylmethionine induces apoptosis and cycle arrest of gallbladder carcinoma cells by suppression of JAK2/STAT3 pathways. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 2507–2515. [Google Scholar] [CrossRef]
- Speich, B.; Ame, S.M.; Ali, S.M.; Alles, R.; Hattendorf, J.; Utzinger, J.; Albonico, M.; Keiser, J. Efficacy and safety of nitazoxanide, albendazole, and nitazoxanide-albendazole against Trichuris trichiura infection: A randomized controlled trial. PLoS Negl. Trop. Dis. 2012, 6, e1685. [Google Scholar] [CrossRef]
- Stockis, A.; Allemon, A.M.; De Bruyn, S.; Gengler, C. Nitazoxanide pharmacokinetics and tolerability in man using single ascending oral doses. Int. J. Clin. Pharmacol. Ther. 2002, 40, 213–220. [Google Scholar] [CrossRef]
- Di Santo, N.; Ehrisman, J. Research perspective: Potential role of nitazoxanide in ovarian cancer treatment. Old drug, new purpose? Cancers 2013, 5, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Tantawy, M.A.; El-Sherbeeny, N.A.; Helmi, N.; Alazragi, R.; Salem, N.; Elaidy, S.M. Synthetic antiprotozoal thiazolide drug induced apoptosis in colorectal cancer cells: Implications of IL-6/JAK2/STAT3 and p53/caspases-dependent signaling pathways based on molecular docking and in vitro study. Mol. Cell Biochem. 2020, 469, 143–157. [Google Scholar] [CrossRef]
- Nafie, M.S.; Mahgoub, S.; Amer, A.M. Antimicrobial and antiproliferative activities of novel synthesized 6-(quinolin-2-ylthio) pyridine derivatives with molecular docking study as multi-targeted JAK2/STAT3 inhibitors. Chem. Biol. Drug Des. 2021, 97, 553–564. [Google Scholar] [CrossRef]
- Li, W.; Yuan, B.; Zhao, Y.; Lu, T.; Zhang, S.; Ding, Z.; Wang, D.; Zhong, S.; Gao, G.; Yan, M. Transcriptome profiling reveals target in primary myelofibrosis together with structural biology study on novel natural inhibitors regarding JAK2. Aging 2021, 13, 8248–8275. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.F.; Li, Z.C.; Chen, P.P.; Zhou, X. Bufothionine induced the mitochondria-mediated apoptosis in H22 liver tumor and acute liver injury. Chin. Med. 2015, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.S.; Shen, F.X.; Xie, R.F.; Zhou, G.; Feng, Y.M.; Zhou, X. Bufothionine induces autophagy in H22 hepatoma-bearing mice by inhibiting JAK2/STAT3 pathway, a possible anti-cancer mechanism of cinobufacini. J. Ethnopharmacol. 2021, 270, 113848. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Shen, P.; Wang, H.; Qin, L.; Ren, J.; Sun, Q.; Ge, R.; Bian, J.; Zhong, Y.; Li, Z.; et al. Discovery of imidazopyrrolopyridines derivatives as novel and selective inhibitors of JAK2. Eur. J. Med. Chem. 2021, 218, 113394. [Google Scholar] [CrossRef] [PubMed]
- Sanachai, K.; Aiebchun, T.; Mahalapbutr, P.; Seetaha, S.; Tabtimmai, L.; Maitarad, P.; Xenikakis, I.; Geronikaki, A.; Choowongkomon, K.; Rungrotmongkol, T. Discovery of novel JAK2 and EGFR inhibitors from a series of thiazole-based chalcone derivatives. RSC Med. Chem. 2021, 12, 430–438. [Google Scholar] [CrossRef]
- Newton, A.S.; Liosi, M.E.; Henry, S.P.; Deiana, L.; Faver, J.C.; Krimmer, S.G.; Puleo, D.E.; Schlessinger, J.; Jorgensen, W.L. Indoloxytriazines as binding molecules for the JAK2 JH2 pseudokinase domain and its V617F variant. Tetrahedron Lett. 2021, 77, 153248. [Google Scholar] [CrossRef]
- Tantawy, M.A.; Shaheen, S.; Kattan, S.W.; Alelwani, W.; Barnawi, I.O.; Elmgeed, G.A.; Nafie, M.S. Cytotoxicity, in silico predictions and molecular studies for androstane heterocycle compounds revealed potential antitumor agent against lung cancer cells. J. Biomol. Struct. Dyn. 2022, 40, 4352–4365. [Google Scholar] [CrossRef]
- Singh, A.; Mishra, A. Molecular modelling study to discover novel JAK2 signaling pathway inhibitor. J. Biomol. Struct. Dyn. 2023, 41, 5827–5838. [Google Scholar] [CrossRef]
- He, L.; Liu, J.; Zhao, H.L.; Zhang, L.C.; Yu, R.L.; Kang, C.M. De novo design of dual-target JAK2, SMO inhibitors based on deep reinforcement learning, molecular docking and molecular dynamics simulations. Biochem. Biophys. Res. Commun. 2023, 638, 23–27. [Google Scholar] [CrossRef]
- Guo, Y.; Zou, Y.; Chen, Y.; Deng, D.; Zhang, Z.; Liu, K.; Tang, M.; Yang, T.; Fu, S.; Zhang, C.; et al. Design, synthesis and biological evaluation of purine-based derivatives as novel JAK2/BRD4(BD2) dual target inhibitors. Bioorg. Chem. 2023, 132, 106386. [Google Scholar] [CrossRef]
- Diao, Y.; Liu, D.; Ge, H.; Zhang, R.; Jiang, K.; Bao, R.; Zhu, X.; Bi, H.; Liao, W.; Chen, Z.; et al. Macrocyclization of linear molecules by deep learning to facilitate macrocyclic drug candidates discovery. Nat. Commun. 2023, 14, 4552. [Google Scholar] [CrossRef] [PubMed]
- Suriya, U.; Mahalapbutr, P.; Geronikaki, A.; Kartsev, V.; Zubenko, A.; Divaeva, L.; Chekrisheva, V.; Petrou, A.; Oopkaew, L.; Somngam, P.; et al. Discovery of furopyridine-based compounds as novel inhibitors of Janus kinase 2: In silico and in vitro studies. Int. J. Biol. Macromol. 2024, 260 Pt 2, 129308. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Park, J.; Han, E.T.; Park, W.S.; Han, J.H.; Chun, W. Drug Repositioning via Graph Neural Networks: Identifying Novel JAK2 Inhibitors from FDA-Approved Drugs through Molecular Docking and Biological Validation. Molecules 2024, 29, 1363. [Google Scholar] [CrossRef]
- Kubo, I.; Muroi, H.; Himejima, M. Combination effects of antifungal nagilactones against Candida albicans and two other fungi with phenylpropanoids. J. Nat. Prod. 1993, 56, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Yao, S.; Ye, Y.; Yu, Q. 3-Deoxy-2β,16-dihydroxynagilactone E, a natural compound from Podocarpus nagi, preferentially inhibits JAK2/STAT3 signaling by allosterically interacting with the regulatory domain of JAK2 and induces apoptosis of cancer cells. Acta Pharmacol. Sin. 2019, 40, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Niu, J.; Wang, F.; Hu, L.; Yu, Q. A natural compound derivative P-13 inhibits STAT3 signaling by covalently inhibiting Janus kinase 2. Investig. New Drugs 2019, 37, 452–460. [Google Scholar] [CrossRef]
- Jiang, D.; Xu, J.; Liu, S.; Nasser, M.I.; Wei, W.; Mao, T.; Liu, X.; Zou, X.; Li, J.; Li, X. Rosmanol induces breast cancer cells apoptosis by regulating PI3K/AKT and STAT3/JAK2 signaling pathways. Oncol. Lett. 2021, 22, 631. [Google Scholar] [CrossRef]
- Zhong, Y.Y.; Chen, H.P.; Tan, B.Z.; Yu, H.H.; Huang, X.S. Triptolide avoids cisplatin resistance and induces apoptosis via the reactive oxygen species/nuclear factor-κB pathway in SKOV3PT platinum-resistant human ovarian cancer cells. Oncol. Lett. 2013, 6, 1084–1092. [Google Scholar] [CrossRef]
- Wei, Y.M.; Wang, Y.H.; Xue, H.Q.; Luan, Z.H.; Liu, B.W.; Ren, J.H. Triptolide, A potential autophagy modulator. Chin. J. Integr. Med. 2019, 25, 233–240. [Google Scholar] [CrossRef]
- Zhong, Y.; Le, F.; Cheng, J.; Luo, C.; Zhang, X.; Wu, X.; Xu, F.; Zuo, Q.; Tan, B. Triptolide inhibits JAK2/STAT3 signaling and induces lethal autophagy through ROS generation in cisplatin-resistant SKOV3/DDP ovarian cancer cells. Oncol. Rep. 2021, 45, 69. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, C.; Niu, Y.; Xia, J.; Fan, L.; Wu, Y.; Gao, W. Procyanidins mediates antineoplastic effects against non-small cell lung cancer via the JAK2/STAT3 pathway. Transl. Cancer Res. 2021, 10, 2023–2035. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Joo, S.H.; Seo, J.H.; Kim, J.; Yoon, G.; Jeon, Y.J.; Lee, M.H.; Chae, J.I.; Kim, W.K.; Shim, J.H. Licochalcone H Induces Cell Cycle Arrest and Apoptosis in Human Skin Cancer Cells by Modulating JAK2/STAT3 Signaling. Biomol. Ther. 2022, 30, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, A. Computational modeling and in vitro evaluation identified natural product-Z218 as a novel Janus kinase 2 (JAK2) inhibitor to combat β-thalassemia. Biotechnol. Appl. Biochem. 2023, 70, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Upreti, S.; Muduli, K.; Pradhan, J.; Elangovan, S.; Samant, M. Identification of novel inhibitors from Urtica spp. against TNBC targeting JAK2 receptor for breast cancer therapy. Med. Oncol. 2023, 40, 326. [Google Scholar] [CrossRef] [PubMed]
- Vaziri-Amjad, S.; Rahgosha, R.; Taherkhani, A. Potential JAK2 Inhibitors from Selected Natural Compounds: A Promising Approach for Complementary Therapy in Cancer Patients. Evid. Based Complement. Alternat. Med. 2024, 2024, 1114928. [Google Scholar] [CrossRef]
- Yu, R.N.; Chen, C.J.; Shu, L.; Yin, Y.; Wang, Z.J.; Zhang, T.T.; Zhang, D.Y. Structure-based design and synthesis of pyrimidine-4,6-diamine derivatives as Janus kinase 3 inhibitors. Bioorg. Med. Chem. 2019, 27, 1646–1657. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.G.; Wang, J.A.; Meng, L.; Pei, X.; Zhang, L.; An, L.; Li, C.L.; Miao, Y.L. Design, synthesis, biological activity evaluation of 3-(4-phenyl-1H-imidazol-2-yl)-1H-pyrazole derivatives as potent JAK 2/3 and aurora A/B kinases multi-targeted inhibitors. Eur. J. Med. Chem. 2021, 209, 112934. [Google Scholar] [CrossRef]
- Medvedeva, S.M.; Shikhaliev, K.S. Synthesis of 4,5-Dihydro-1H-[1,2]dithiolo[3,4-c]quinoline-1-thione Derivatives and Their Application as Protein Kinase Inhibitors. Molecules 2022, 27, 4033. [Google Scholar] [CrossRef]
- Wei, J.; Pan, Y.; Shen, Z.; Shen, L.; Xu, L.; Yu, W.; Huang, W. A hybrid energy-based and AI-based screening approach for the discovery of novel inhibitors of JAK3. Front. Med. 2023, 10, 1182227. [Google Scholar] [CrossRef]
- Faris, A.; Cacciatore, I.; Ibrahim, I.M.; Al-Mughram, M.H.; Hadni, H.; Tabti, K.; Elhallaoui, M. In silico computational drug discovery: A Monte Carlo approach for developing a novel JAK3 inhibitors. J. Biomol. Struct. Dyn. 2023, 20, 1–23. [Google Scholar] [CrossRef]
- Faris, A.; Ibrahim, I.M.; Alnajjar, R.; Hadni, H.; Bhat, M.A.; Yaseen, M.; Chakraborty, S.; Alsakhen, N.; Shamkh, I.M.; Mabood, F.M.; et al. QSAR-driven screening uncovers and designs novel pyrimidine-4,6-diamine derivatives as potent JAK3 inhibitors. J. Biomol. Struct. Dyn. 2023, 7, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Gao, Y.Q.; Deng, Y.J.; Zhang, H.H.; Wu, Y.R.; Hu, Y.; Mei, Q.X. Identification of Chinese Herbal Compounds with Potential as JAK3 Inhibitors. Evid. Based Complement. Alternat. Med. 2019, 2019, 4982062. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.M.; Xu, T.; Tu, Z.C.; Zhu, H.J.; Cheng, Y.X. Sulfur and nitrogen-containing compounds from the whole bodies of Blaps japanensis. Bioorg. Chem. 2020, 102, 104086. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Yi, E.H.; Jee, J.G.; Jeong, A.J.; Sandoval, C.; Park, I.C.; Baeg, G.H.; Ye, S.K. Tubulosine selectively inhibits JAK3 signalling by binding to the ATP-binding site of the kinase of JAK3. J. Cell Mol. Med. 2020, 24, 7427–7438. [Google Scholar] [CrossRef]
- Sanachai, K.; Mahalapbutr, P.; Tabtimmai, L.; Seetaha, S.; Kittikool, T.; Yotphan, S.; Choowongkomon, K.; Rungrotmongkol, T. Discovery of JAK2/3 Inhibitors from Quinoxalinone-Containing Compounds. ACS Omega 2022, 7, 33587–33598. [Google Scholar] [CrossRef] [PubMed]
- Sanachai, K.; Mahalapbutr, P.; Tabtimmai, L.; Seetaha, S.; Kaekratoke, N.; Chamni, S.; Azam, S.S.; Choowongkomon, K.; Rungrotmongkol, T. In Silico and In Vitro Study of Janus Kinases Inhibitors from Naphthoquinones. Molecules 2023, 28, 597. [Google Scholar] [CrossRef]















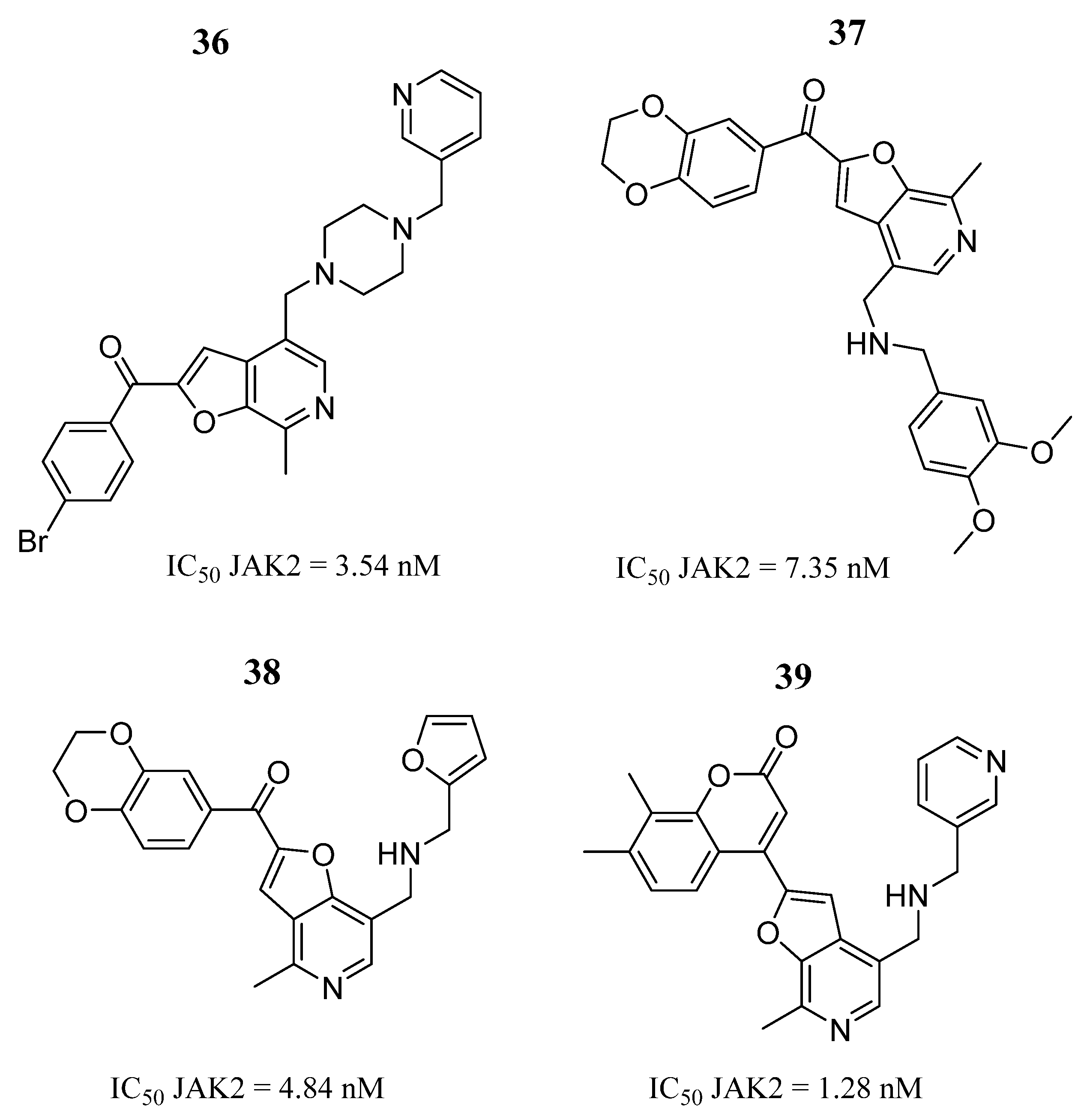
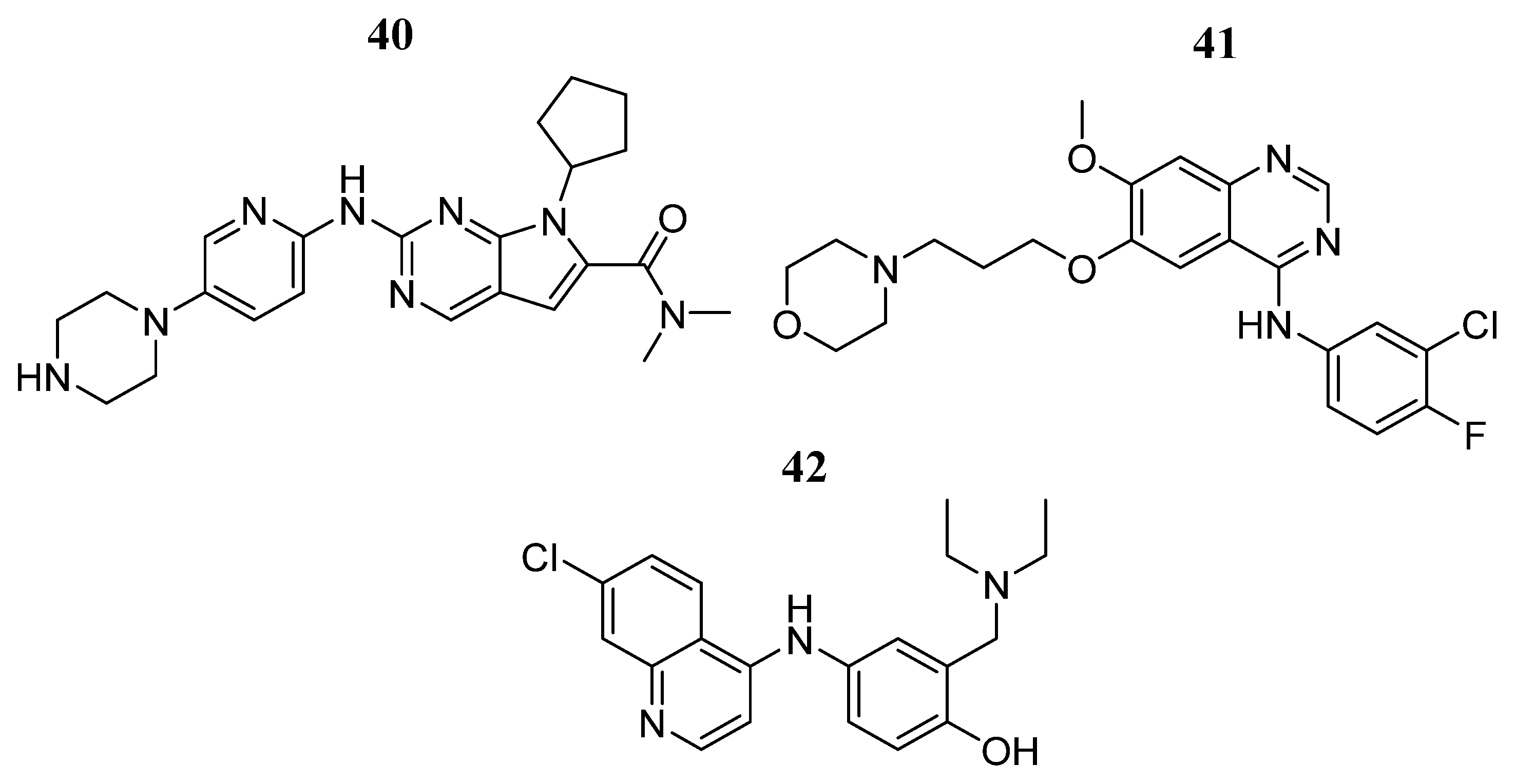
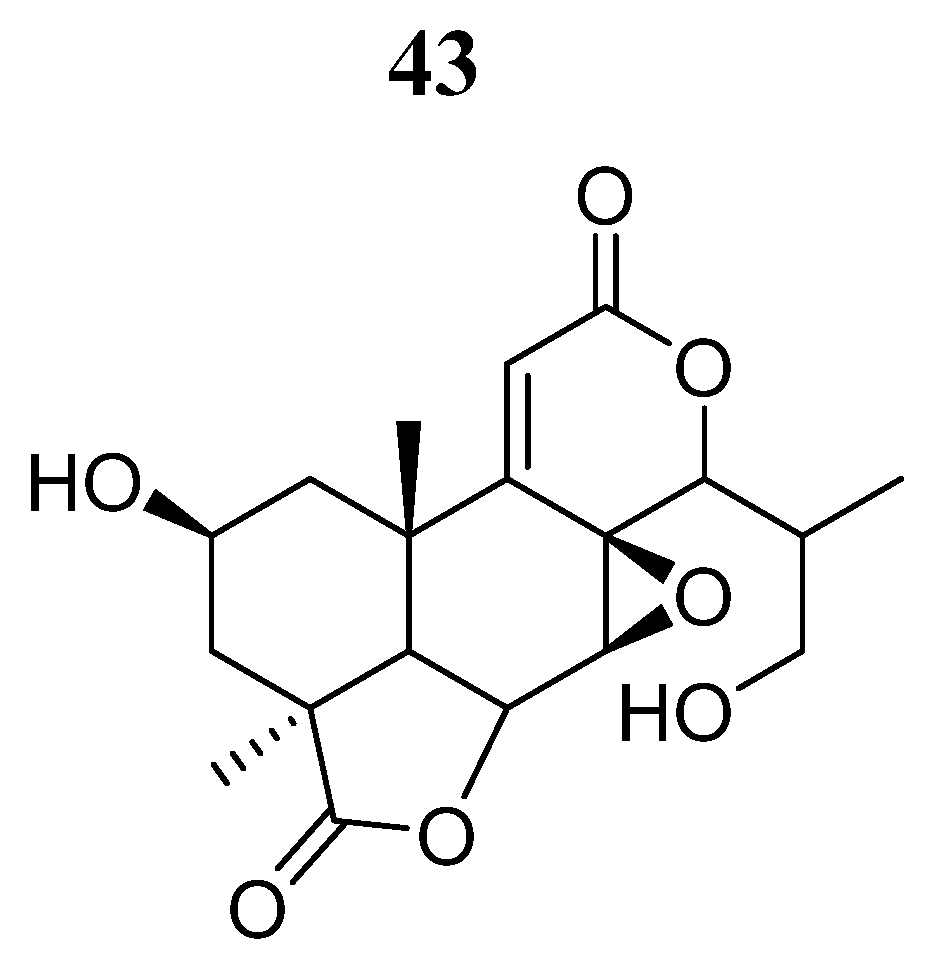
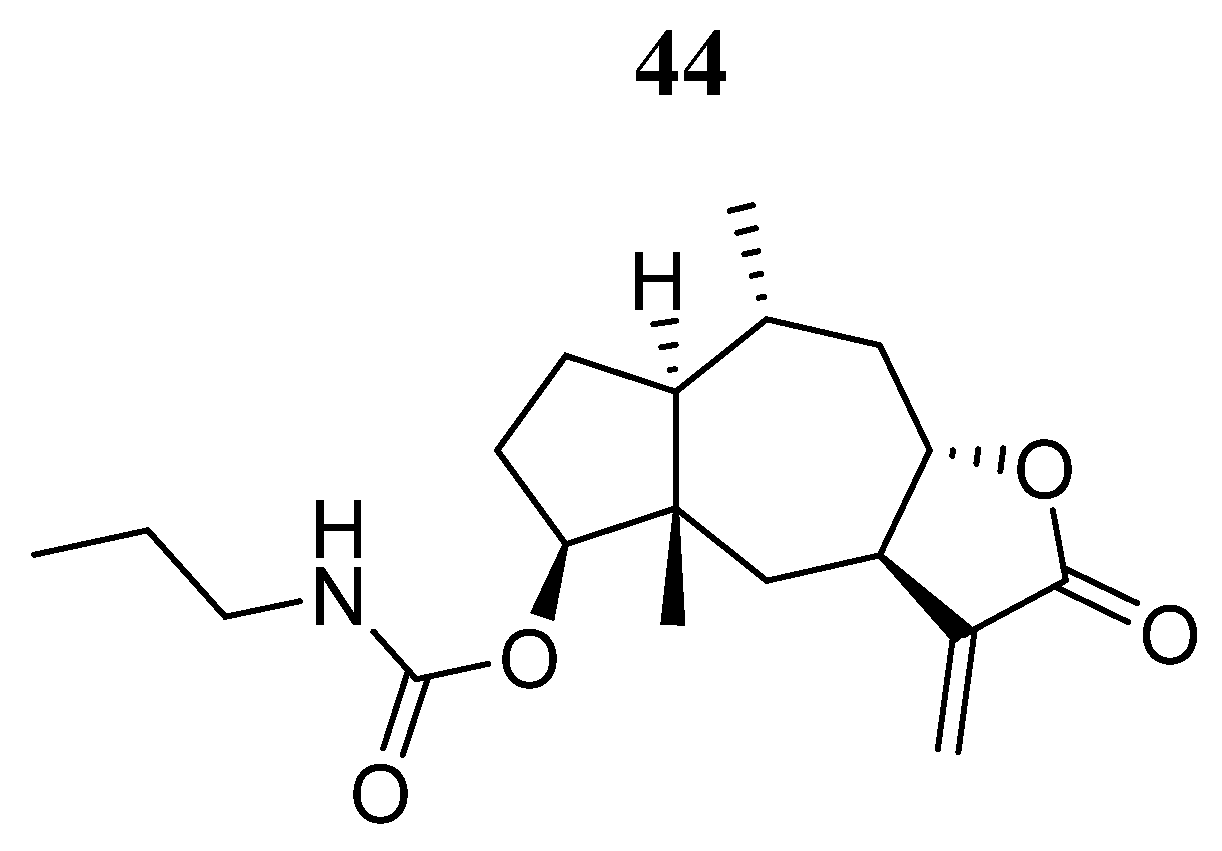

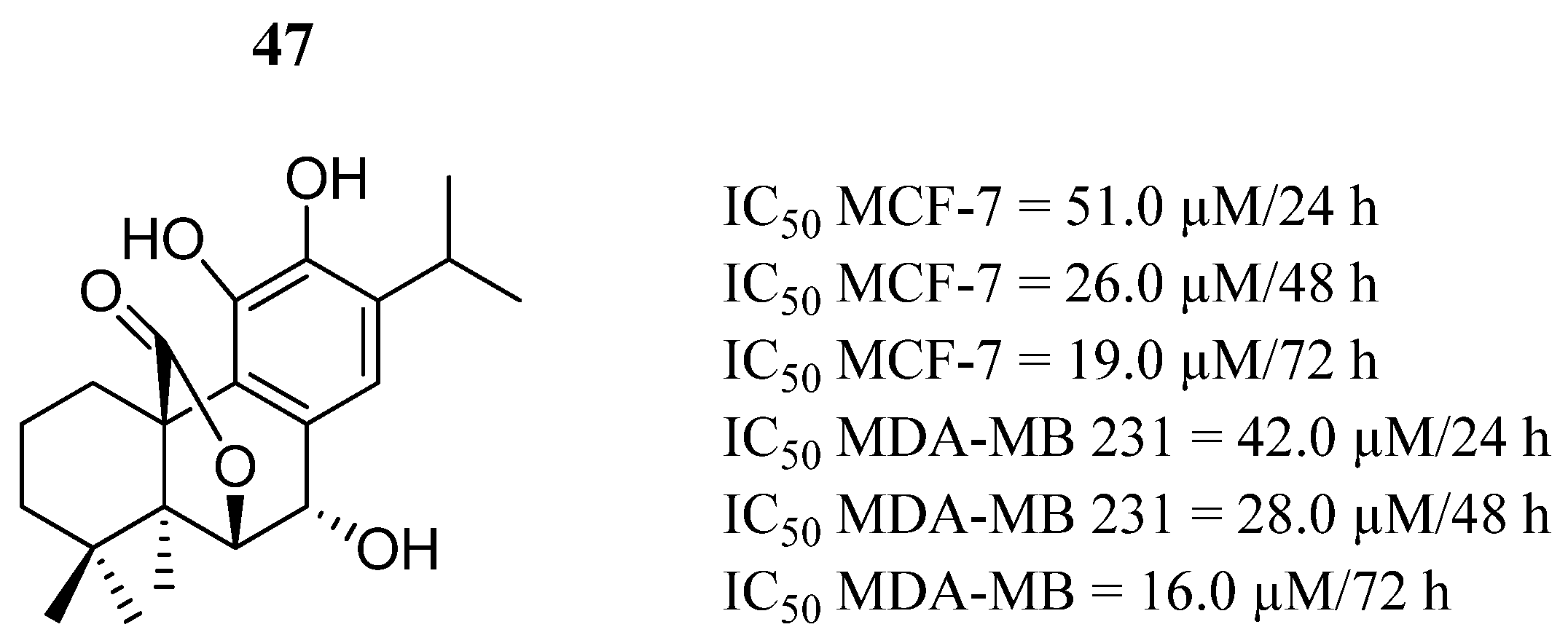












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Activity | Disease | Toxic Effects |
|---|---|---|---|
 Ruxolitinib | IC50 = 2.8 nM | Polycythemia, Myelofibrosis, Various cancers | Diarrhea, abdominal pain, anemia, thrombocytopenia |
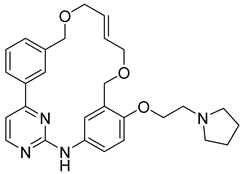 Pacritinib | IC50 = 23 nM | Myeloid leukemias, Myelofibrosis | Cardiovascular and hemorrhagic events |
 AZD1480 | Ki = 0.26 nM | Myeloproliferative diseases, Solid tumors | Dizziness, anxiety, memory loss, ataxia, hallucinations, behavior changes |
| Compound | Activity | Disease | Toxic Effects |
|---|---|---|---|
 Ritlecitinib | IC50 = 33.1 nM | Alopecia areata, Vitiligo, Ulcerative colitis, Rheumatoid arthritis, Crohn’s disease | Hepatotoxicity, Pruritus, Influenza |
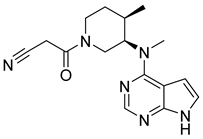 Tofacitinib | IC50 = 1 nM | Transplant patients, Autoimmune disease, Rheumatoid arthritis | Infection, Cytopenias |
| Compound | Anticancer Activity | Enzyme Activity | In Silico Analysis | References |
|---|---|---|---|---|
| JAK2 | ||||
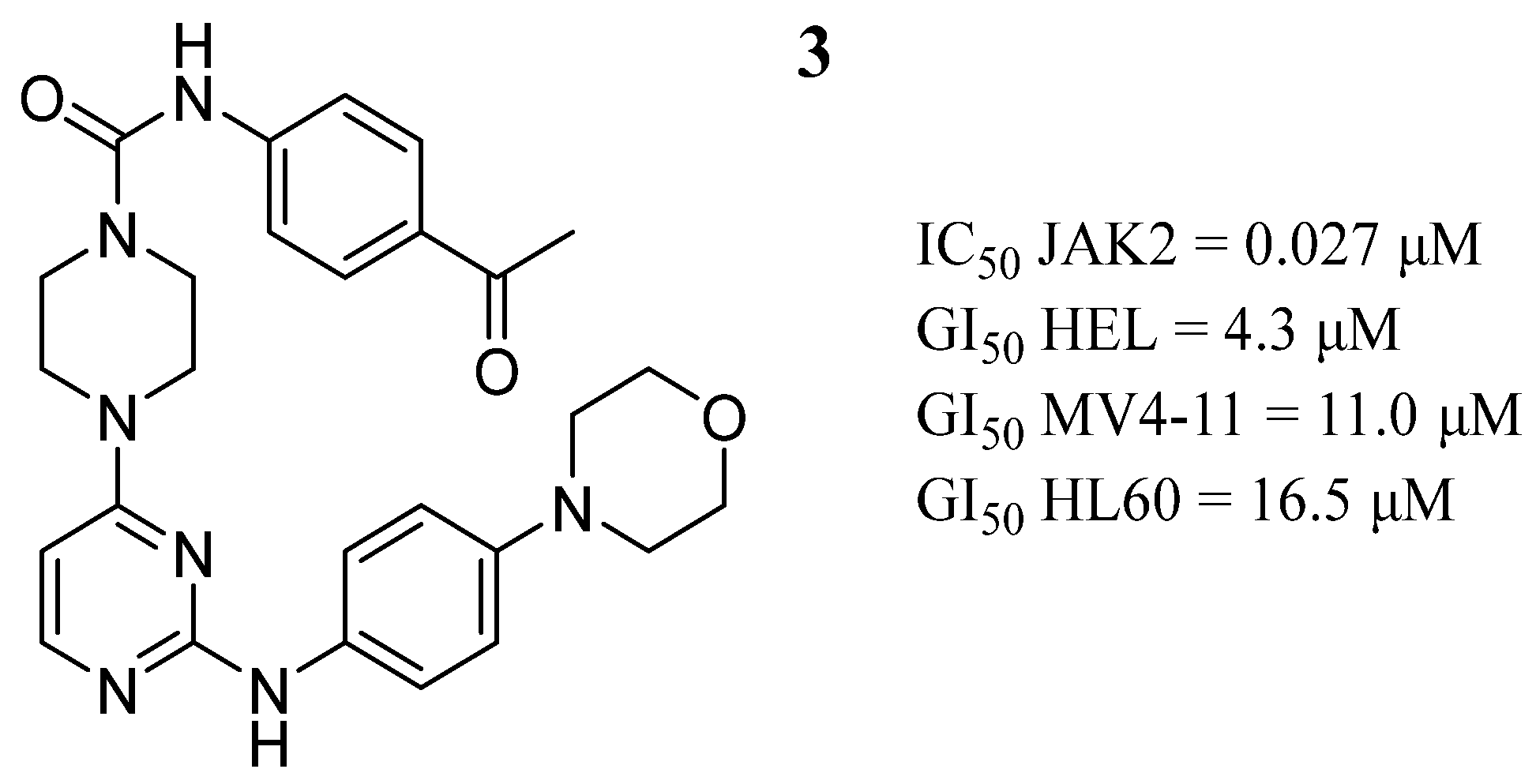
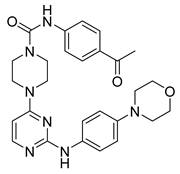 3 | GI50 HEL = 4.3 μM GI50 MV4-11 = 11.0 μM GI50 HL60 = 16.5 μM | IC50 = 0.027 μM | Binds to the ATP-binding site, two hydrogen bonds with Leu932, a network-like alkyl-π interaction with the adjacent Ala880, Leu855, a hydrogen bond with Lys943 and another with Leu855. | [53] |
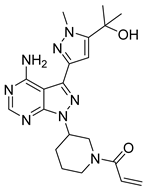 5 | IC50 HEL = 6.46 μM | IC50 = 0.0065 μM | Two hydrogen bonds with Leu932 and two π-π interactions with Tyr931, a hydrogen bond with Lys857, two hydrogen bonds with residue Tyr931, and hydrogen bonding with Ser936 and Asp939 via a water molecule, near the ATP-binding site. | [39] |
 15 | IC50 MCF-7 = 6.39 µM IC50 A549 = 6.9 µM | - | Hydrogen bond with the residue Leu932, lipophilic interactions with the nonpolar amino acid residues Leu83, Leu855, Val863, Pro933, Met929, Ala880 and Leu932 within the receptor pocket. | [70] |
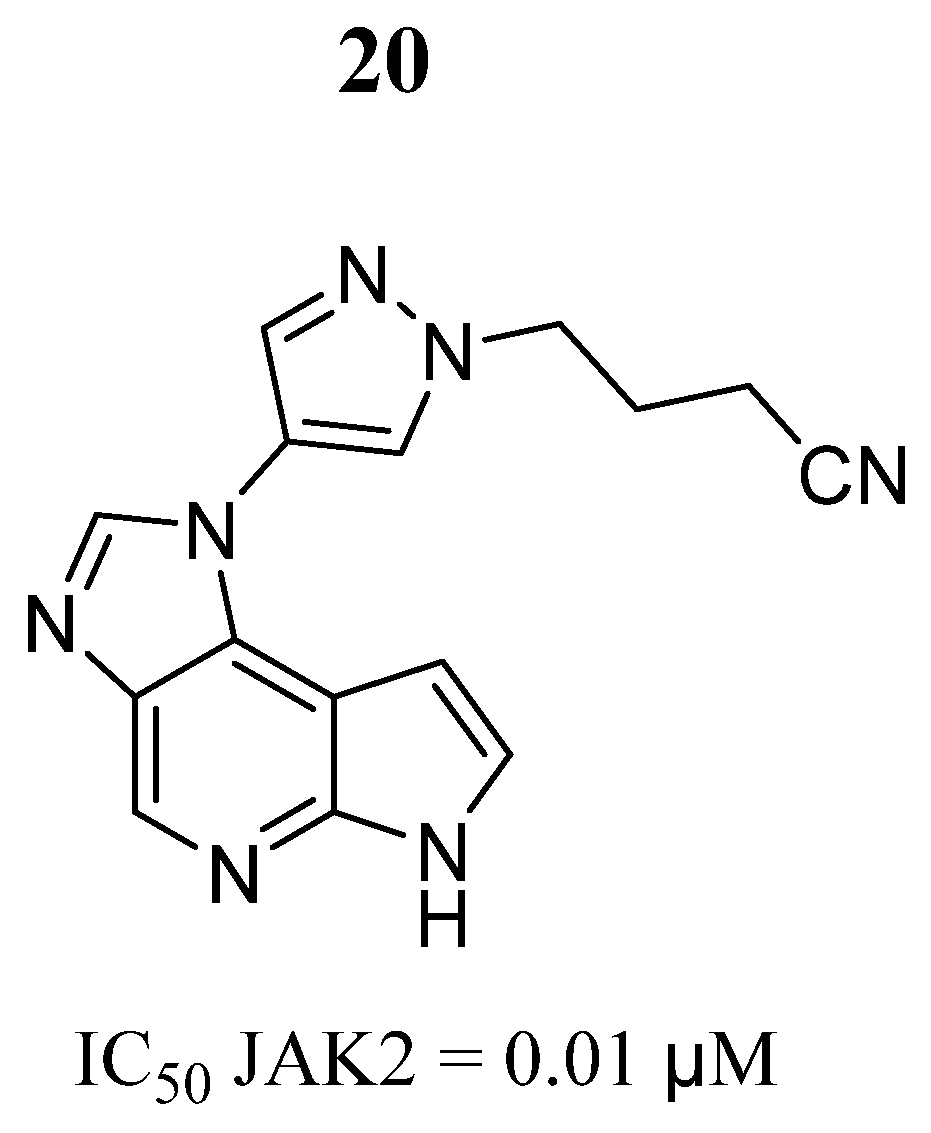
 20 | IC50 = 0.01 μM | Hydrogen bonds with Glu930 and Leu932, hydrogen bonds with Lys882. | [74] | |
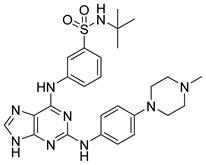 30 | - | IC50 = 0.022 μM | Hydrogen bonds with Pro375, Lys378, Asp381, Tyr390, and Asn433 | [80] |
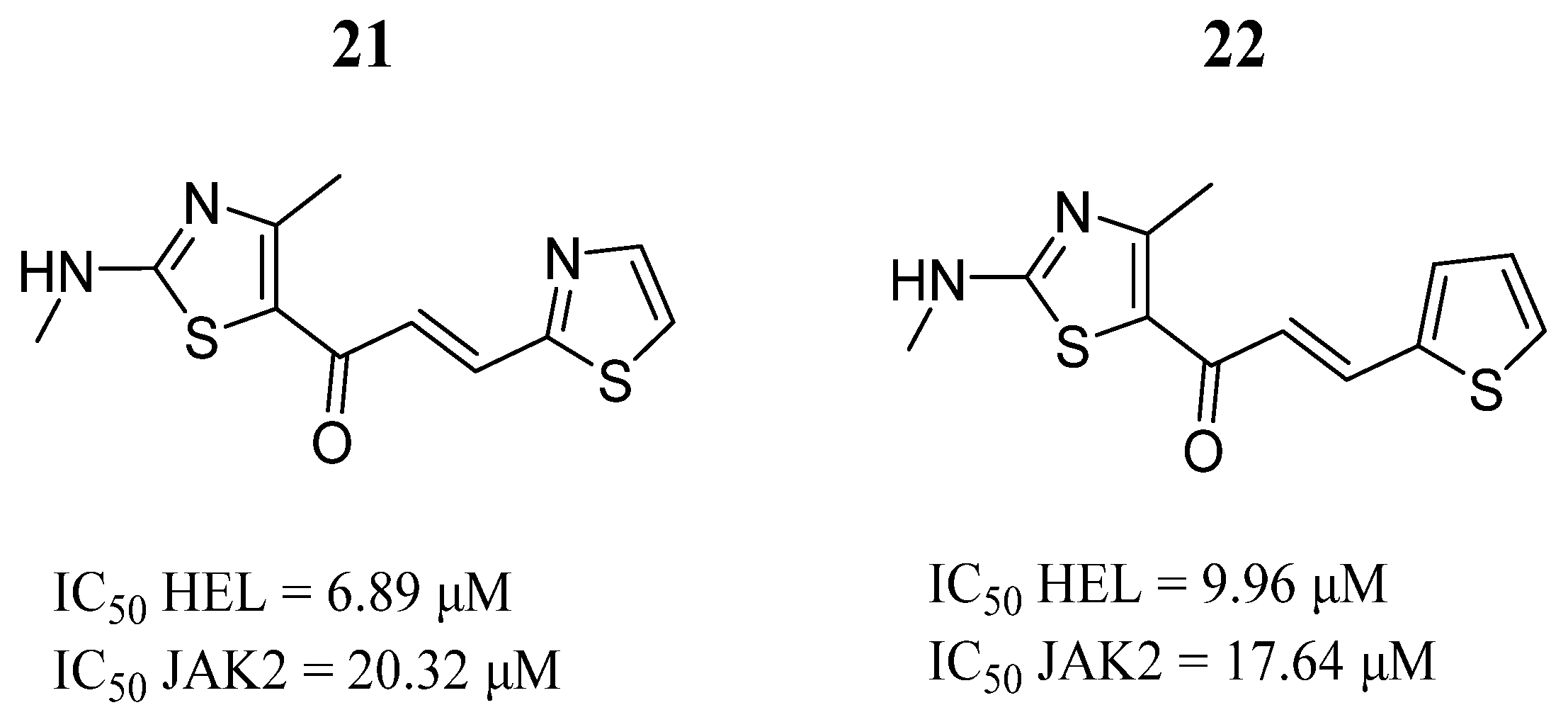
 34 | IC50 HEL = 5.6 µM IC50 SET-2 = 5.8 µM | IC50 = 0.04 µM | Two hydrogen bonds with Val629 and Glu627, hydrogen bond with the Lys640 side chain of the αD helix. | [28] |
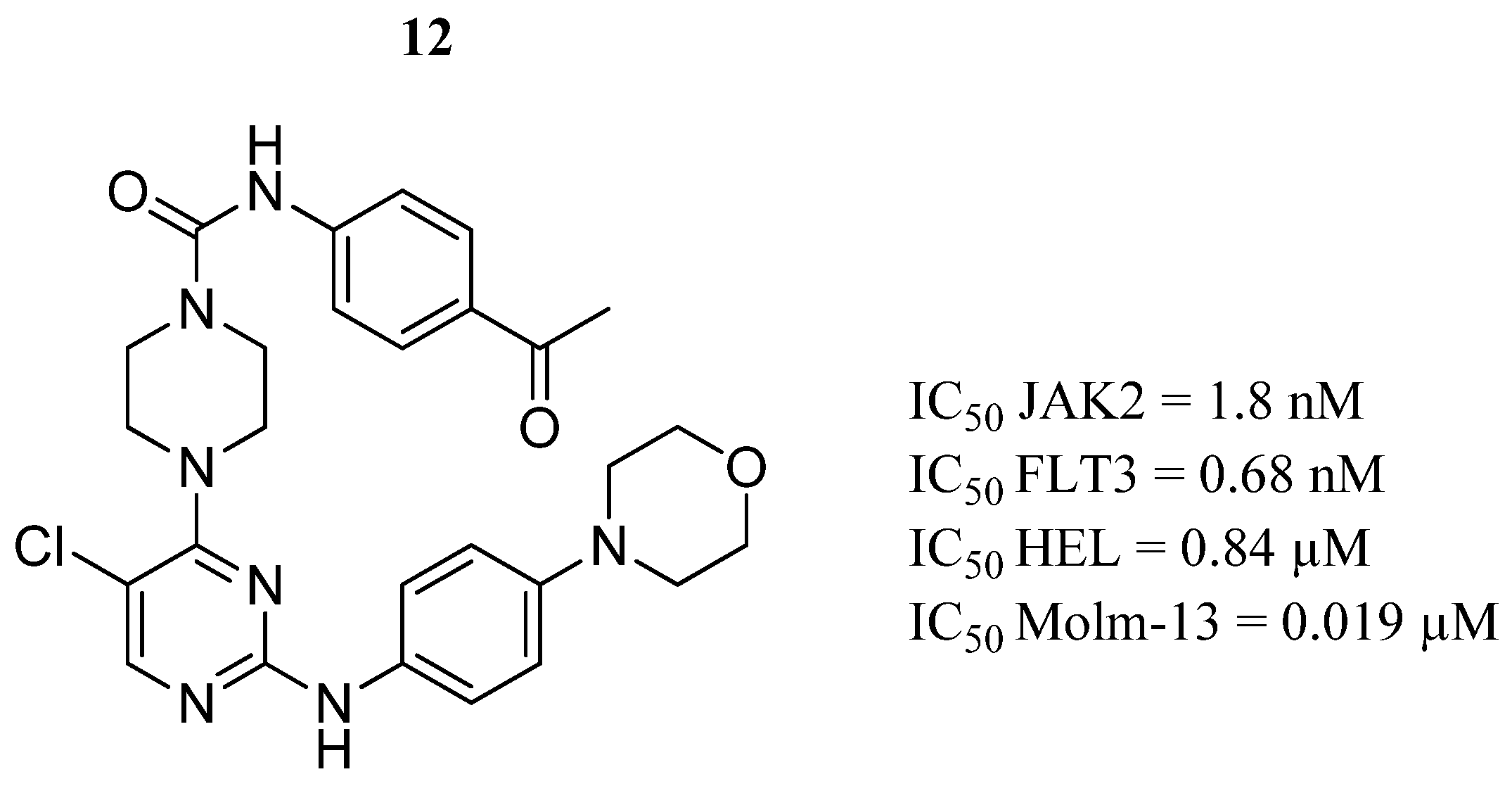
 46 | - | - | Hydrogen bonding interactions with Lys882, Arg980, Ser936, Asp939, and Lys943, carbon hydrogen bonding interactions with Leu932 and Leu855, and a sulfur bonding interaction with Lue855 | [71] |
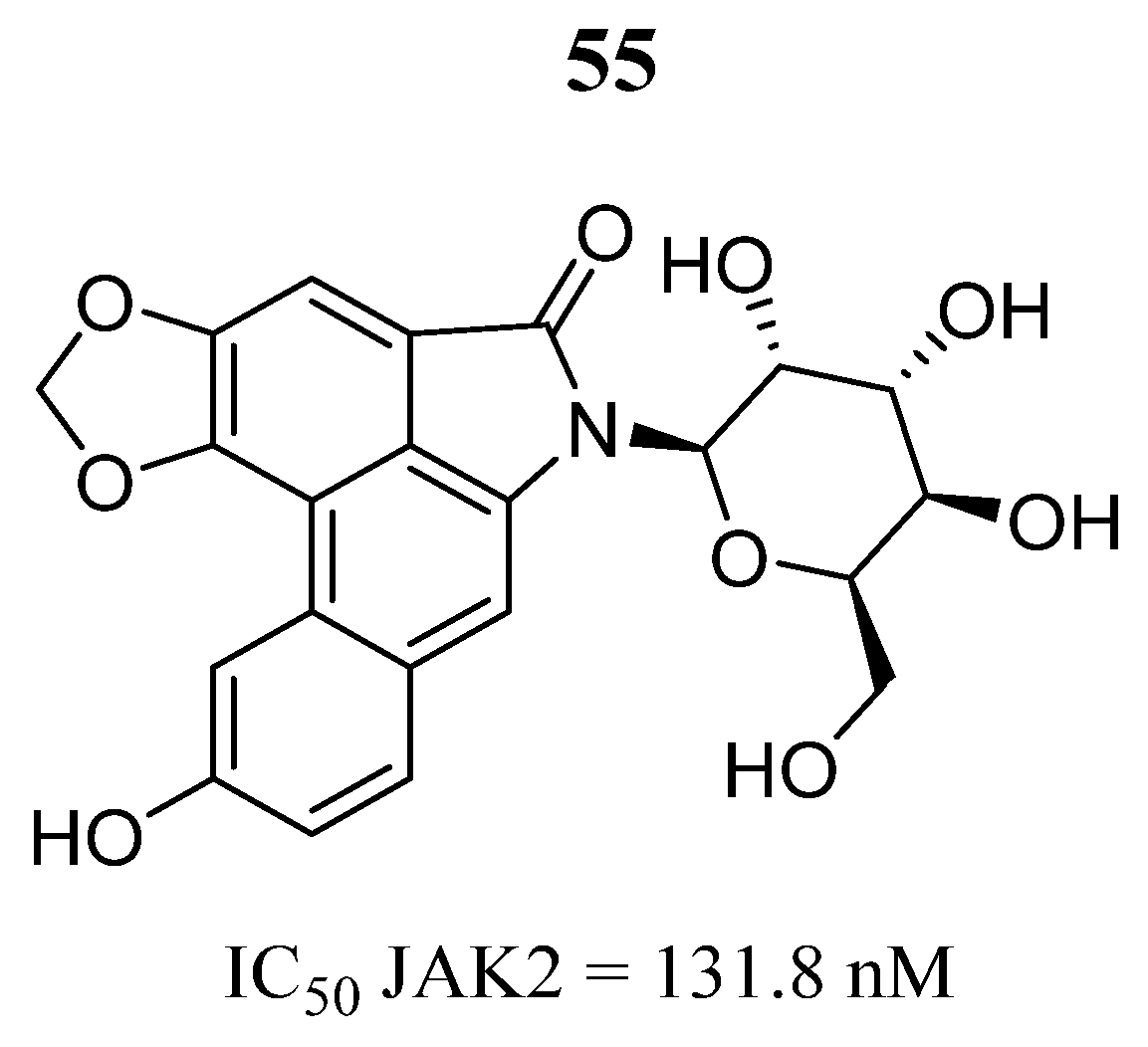
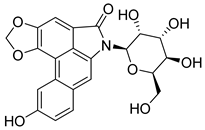 55 | - | IC50 = 131.8 nM | Interacts with the amino acid residues Ala880, Val863, Tyr931, Leu855 Asp994, Leu983, Asn981 and Arg980 | [93] |
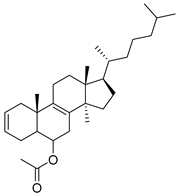 56–67 | IC50 TNBC = 90.09 to 172.16 μg/mL IC50 HEK293T = 732.52 to 1367.25 μg/mL | - | Interaction with residues of catalytic sites such as Leu983, Leu855, Val863, Arg980, Val863, Ala880, Leu855 and Leu932, in the binding pocket | [94] |
| JAK3 | ||||
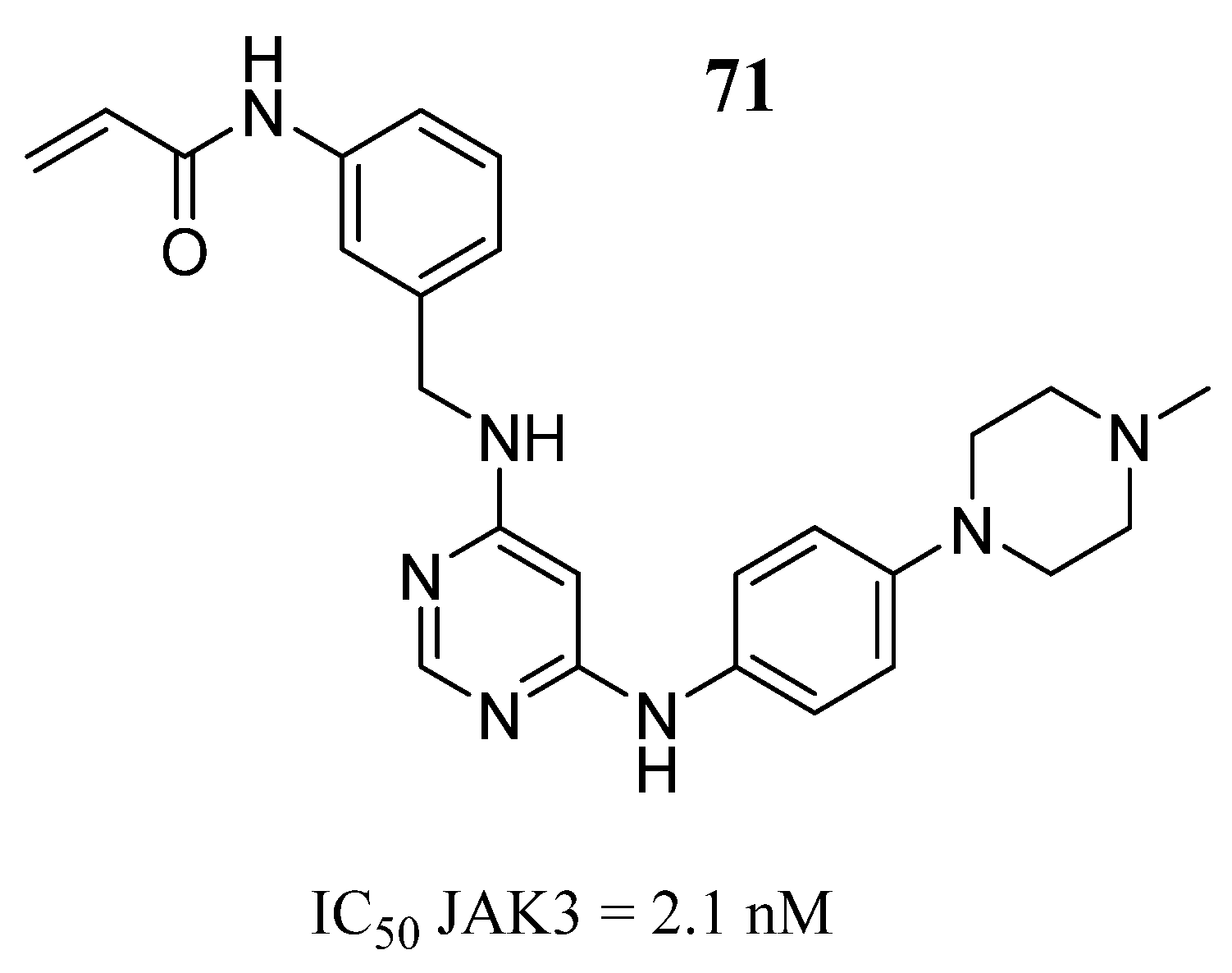
 71 | - | IC50 = 2.1 nM | Bidentate hydrogen bonds with Leu905, and the Cys909, van der Waals contact with Leu956 and Leu828 in the ATP-binding pocket, a hydrogen bond with Arg953 | [96] |
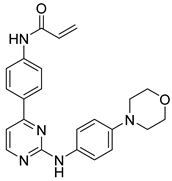 72 | - | IC50 = 1.7 nM | Bidentate hinge hydrogen bonds with Leu905, covalent bonds with Cys909, two σ-π interactions and one σ-π interaction with amino acid residues Leu828 and Gly908, a hydrogen bond with Asp912 | [20] |
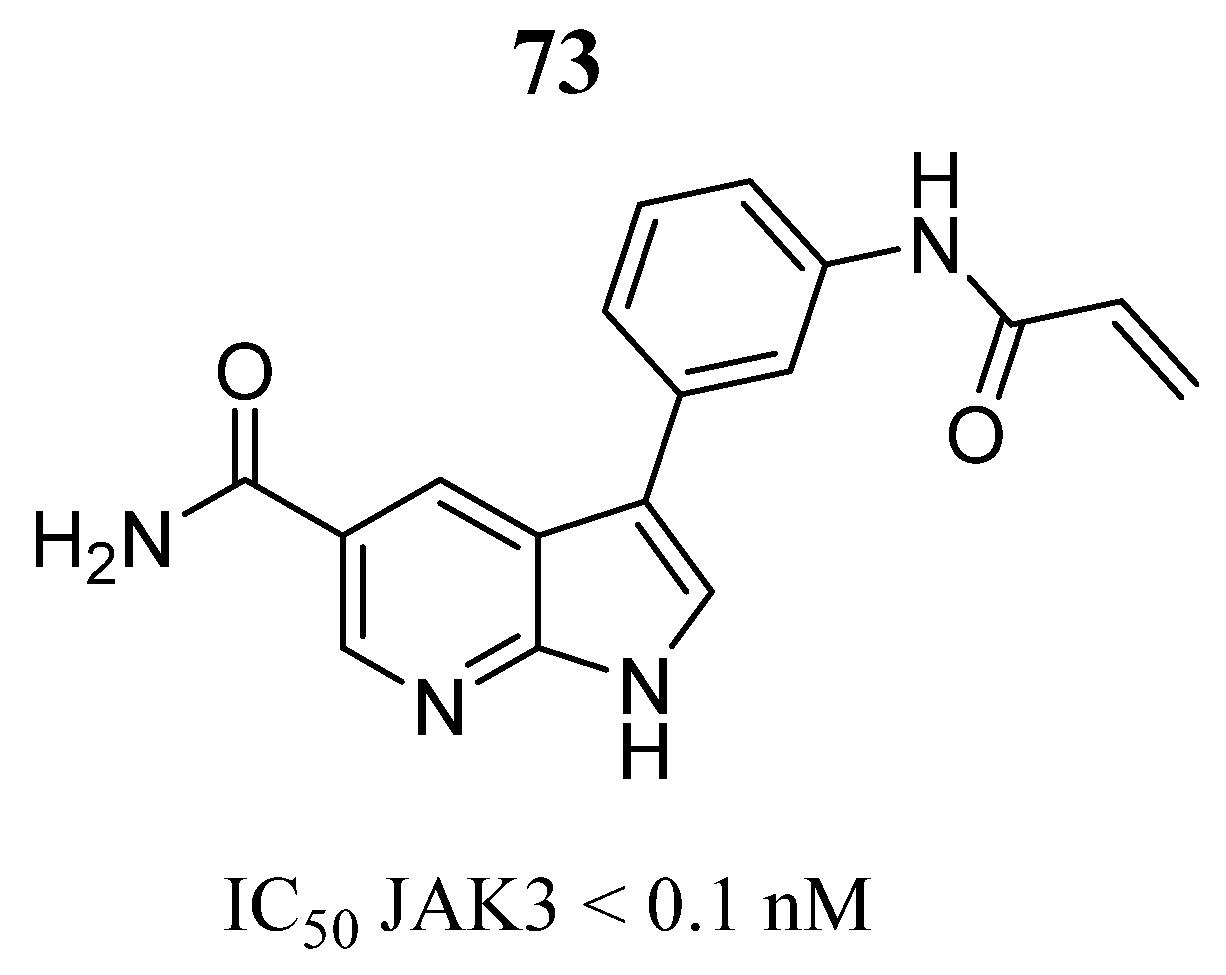
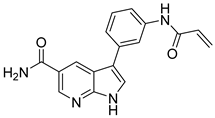 73 | - | IC50 < 0.1 nM | Hinge interaction pattern and the covalent binding of Cys909, two hydrogen bonds with Lys905, a hydrogen bond with Glu903. | [17] |
 74 | IC50 K562 = 6.72 µM | IC50 = 0.057 µM | Interaction at the binding site with Glu930, Tyr931, Leu932, Ser936 and Gly993. | [97] |
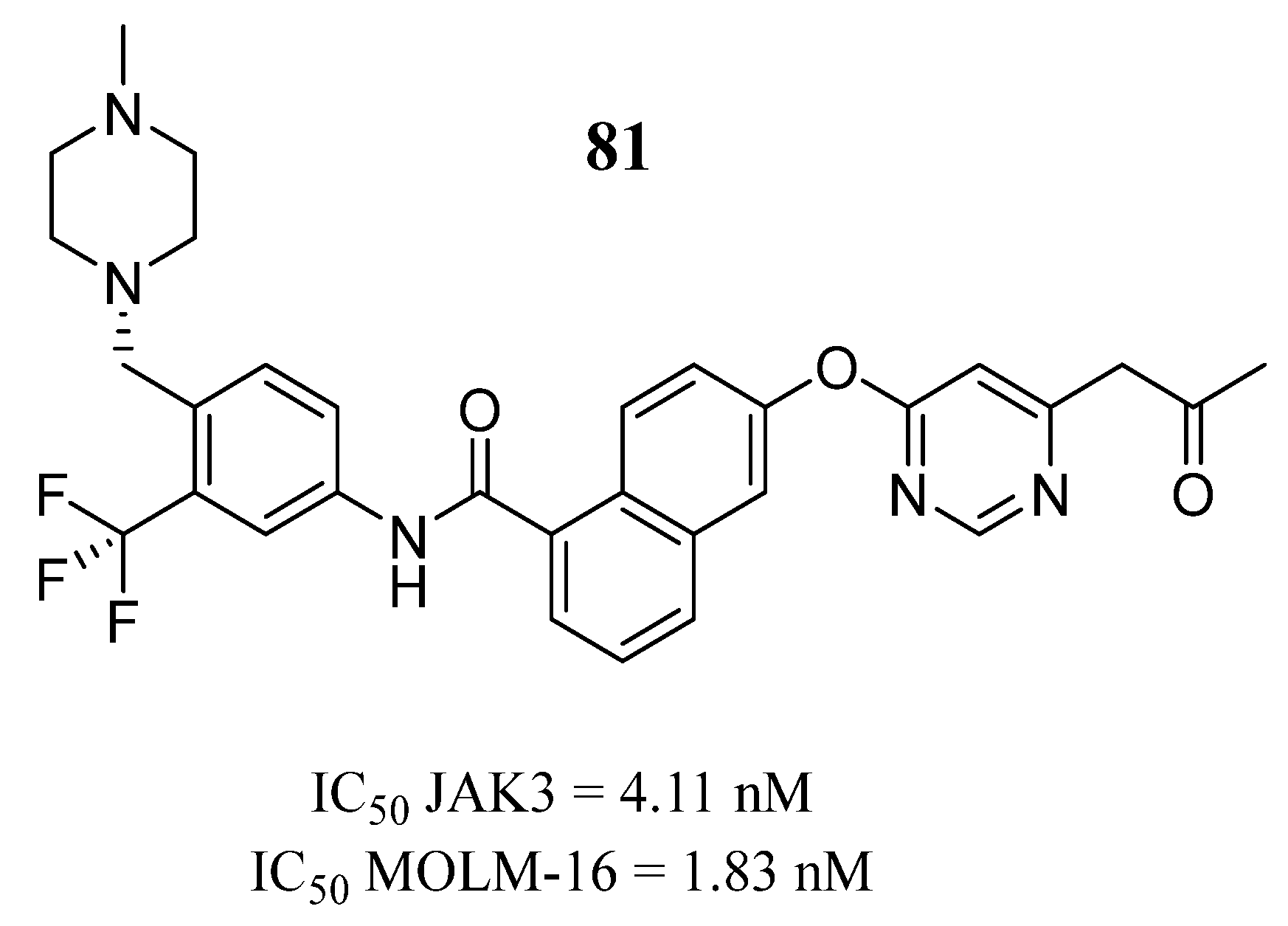
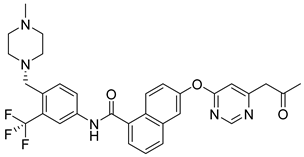 81 | IC50 MOLM-16 = 1.83 nM | IC50 = 4.11 nM | Hydrogen bond interaction with Lys855, hydrogen bond interaction with Leu905, hydrophobic interactions with Leu905, Gly906, and Arg953 | [99] |
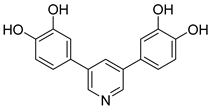 92 | IC50 A549 = 1.68 µM IC50 Huh-7 = 4.88 µM IC50 K562 = 2.13 µM | IC50 = 1.72 µM | - | [103] |
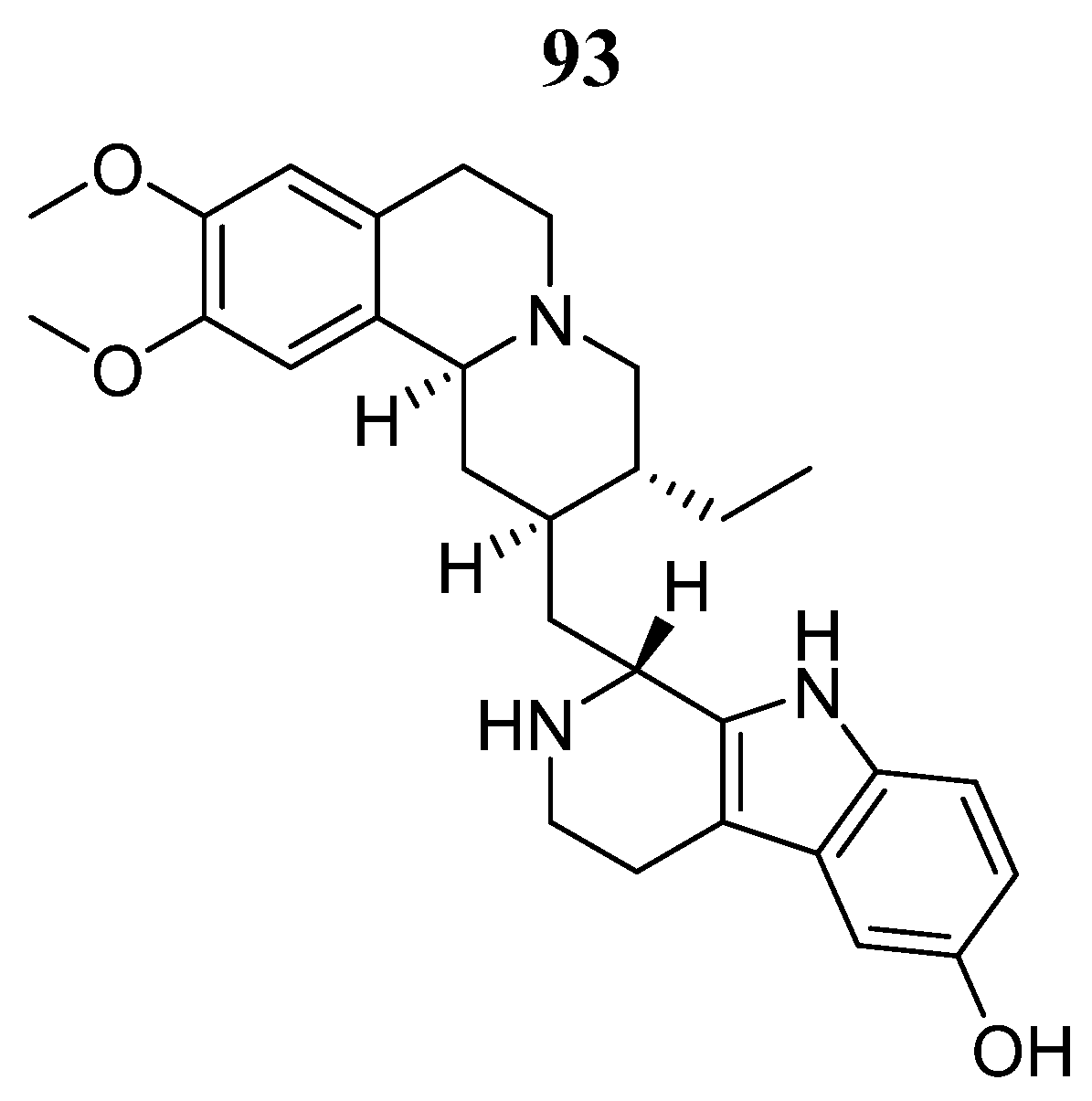
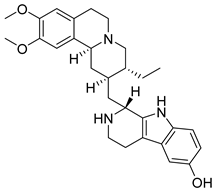 93 | - | EC50 = 1.4 μmol/L | Interaction with Val812, Ala829, Glu847, Met878, Leu881, Leu932 y Asp943. | [104] |
| JAK2/3 | ||||
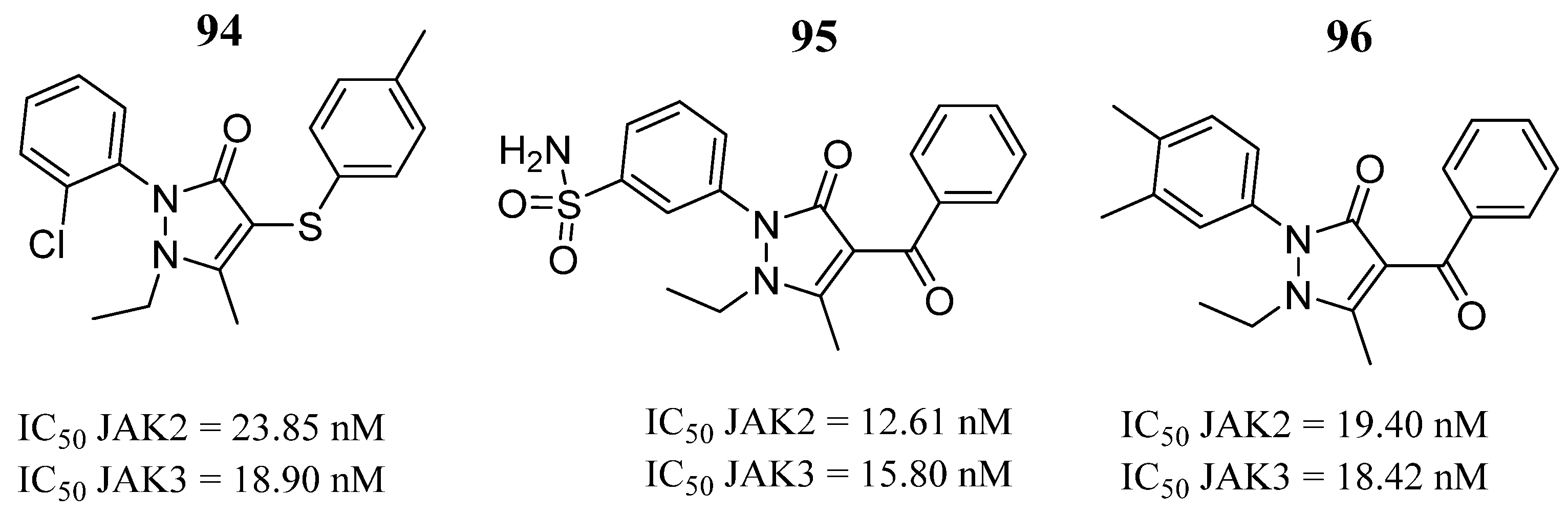
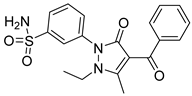 95 | - | IC50 JAK2 = 12.61 nM IC50 JAK3 = 15.80 nM | Hydrogen bonds in the hinge region with residues Glu930 and Lys932 of JAK2 and Glu903 and Lys905. | [49] |
 97 | IC50 TF1 = 15.53 μM IC50 HEL = 17.90 μM | IC50 JAK2 = 13.00 nM IC50 JAK3 = 14.86 nM | Hydrogen bonds in the hinge region with residues Ser936 and Arg938. | [105] |
 99 | IC50 TF1 = 18.10 μM IC50 HEL = 6.65 μM | IC50 JAK2 = 11.11 nM IC50 JAK3 = 10.24 nM | Hydrogen bonds with residues Y931 and L932 and hydrophobic contact with the hinge region, the G loop and the catalytic loop. | [106] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Jiménez, L.K.; Rivera, G.; Juárez-Saldivar, A.; Ortega-Balleza, J.L.; Ortiz-Pérez, E.; Jaime-Sánchez, E.; Paz-González, A.; Lara-Ramírez, E.E. Biological Evaluations and Computer-Aided Approaches of Janus Kinases 2 and 3 Inhibitors for Cancer Treatment: A Review. Pharmaceutics 2024, 16, 1165. https://doi.org/10.3390/pharmaceutics16091165
Vázquez-Jiménez LK, Rivera G, Juárez-Saldivar A, Ortega-Balleza JL, Ortiz-Pérez E, Jaime-Sánchez E, Paz-González A, Lara-Ramírez EE. Biological Evaluations and Computer-Aided Approaches of Janus Kinases 2 and 3 Inhibitors for Cancer Treatment: A Review. Pharmaceutics. 2024; 16(9):1165. https://doi.org/10.3390/pharmaceutics16091165
Chicago/Turabian StyleVázquez-Jiménez, Lenci K., Gildardo Rivera, Alfredo Juárez-Saldivar, Jessica L. Ortega-Balleza, Eyra Ortiz-Pérez, Elena Jaime-Sánchez, Alma Paz-González, and Edgar E. Lara-Ramírez. 2024. "Biological Evaluations and Computer-Aided Approaches of Janus Kinases 2 and 3 Inhibitors for Cancer Treatment: A Review" Pharmaceutics 16, no. 9: 1165. https://doi.org/10.3390/pharmaceutics16091165
APA StyleVázquez-Jiménez, L. K., Rivera, G., Juárez-Saldivar, A., Ortega-Balleza, J. L., Ortiz-Pérez, E., Jaime-Sánchez, E., Paz-González, A., & Lara-Ramírez, E. E. (2024). Biological Evaluations and Computer-Aided Approaches of Janus Kinases 2 and 3 Inhibitors for Cancer Treatment: A Review. Pharmaceutics, 16(9), 1165. https://doi.org/10.3390/pharmaceutics16091165