
Characterizing Extracellular Vesicles Generated from the Integra CELLine Culture System and Their Endocytic Pathways for Intracellular Drug Delivery

 , , , and
, , , and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and EVs Isolation
2.2.1. Cell Culture Using the Integra CELLine Culture System
2.2.2. Cell Culture Using Conventional Flasks
2.2.3. Methods for EVs Isolation
2.3. Characterization of EVs
2.3.1. Evaluation of the EVs Yield
2.3.2. Nanoparticle Tracking Analysis (NTA)
2.3.3. Dynamic Light Scattering (DLS)
2.3.4. Cryogenic Transmission Electron Microscopy (Cryo-TEM)
2.3.5. Total Protein Content by Bicinchoninic Acid (BCA) Assay
2.4. Evaluation of the Cellular Uptake of EVs
2.4.1. The Kinetics of Cellular Uptake of EVs
2.4.2. Quantification of Cellular Uptake of EVs
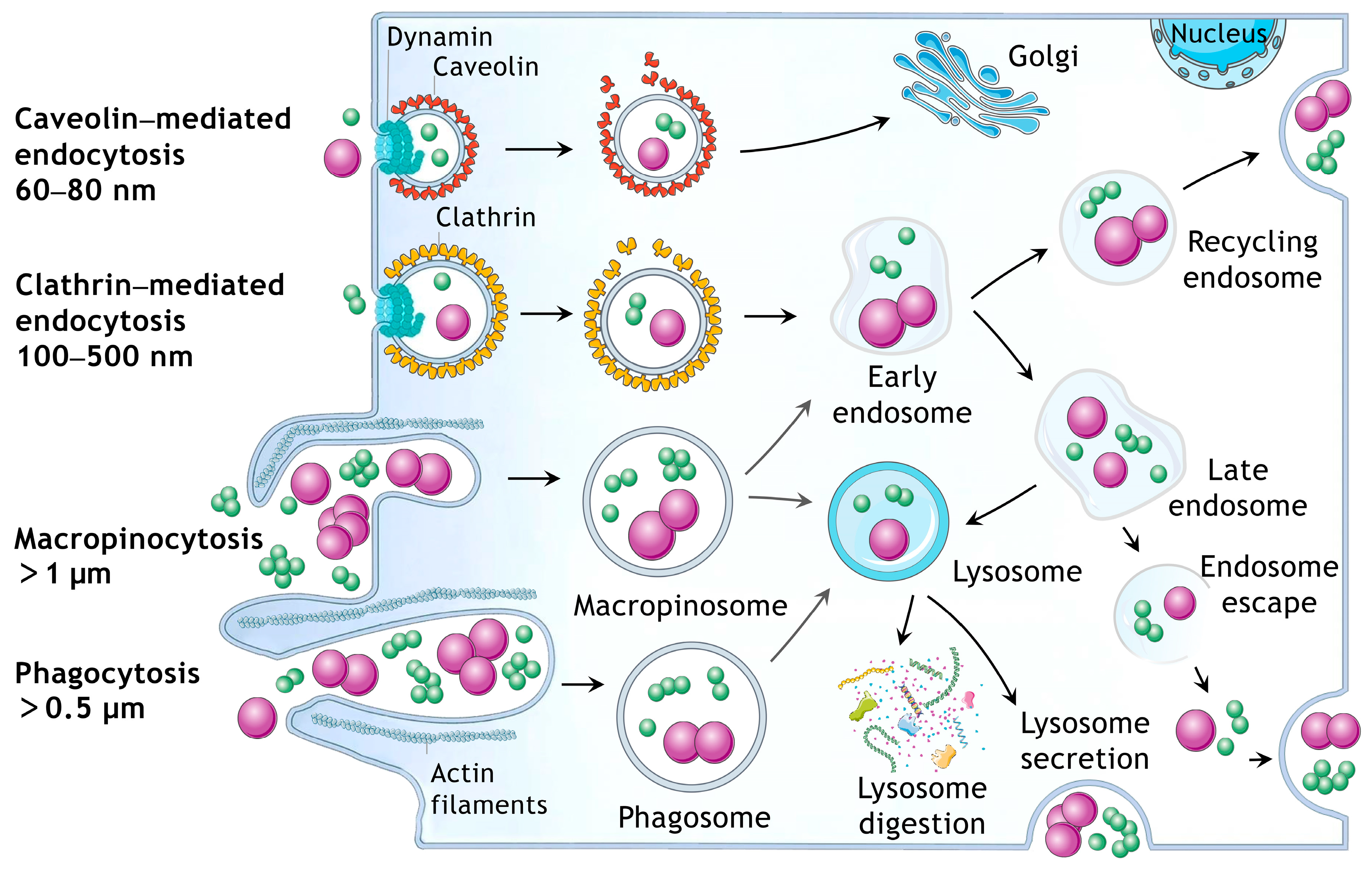
2.5. Investigation of EVs Endocytic Pathways
2.5.1. Optimization of Inhibitor Pre-Treatment Concentration
2.5.2. Determination of Endocytic Pathways of sEVs and lEVs
2.6. Data Analysis
3. Results and Discussion
3.1. Evaluation of EVs Yield
3.1.1. EVs Yield by Measuring EVs Number
3.1.2. EVs Yield by Measuring EVs Protein Content
3.2. Characterization of EVs from the Integra CELLine Culture System
3.2.1. Size Distribution of EVs by NTA
3.2.2. Size Distribution and Zeta Potential of EVs by DLS
3.2.3. Morphology of EVs by Cryo-TEM
3.3. Evaluation of EVs Cellular Uptake
3.3.1. The Kinetics of Cellular Uptake of sEVs and lEVs
3.3.2. Quantification of Cellular Uptake of sEVs and lEVs
3.4. Comparison of Endocytic Pathways of sEVs and lEVs
3.4.1. Optimization of Endocytic Inhibitors Concentration
3.4.2. Different Endocytic Pathways of sEVs and lEVs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cabeza, L.; Perazzoli, G.; Peña, M.; Cepero, A.; Luque, C.; Melguizo, C.; Prados, J. Cancer therapy based on extracellular vesicles as drug delivery vehicles. J. Control. Release 2020, 327, 296–315. [Google Scholar] [CrossRef] [PubMed]
- Rayamajhi, S.; Nguyen, T.D.T.; Marasini, R.; Aryal, S. Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019, 94, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Zhang, H.-X.; He, C.-P.; Fan, S.; Zhu, Y.-L.; Qi, C.; Huang, N.-P.; Xiao, Z.-D.; Lu, Z.-H.; Tannous, B.A.; et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials 2018, 150, 137–149. [Google Scholar] [CrossRef]
- Lee, H.; Park, H.; Noh, G.J.; Lee, E.S. pH-responsive hyaluronate-anchored extracellular vesicles to promote tumor-targeted drug delivery. Carbohydr. Polym. 2018, 202, 323–333. [Google Scholar] [CrossRef]
- Liu, J.; Ye, Z.; Xiang, M.; Chang, B.; Cui, J.; Ji, T.; Zhao, L.; Li, Q.; Deng, Y.; Xu, L.; et al. Functional extracellular vesicles engineered with lipid-grafted hyaluronic acid effectively reverse cancer drug resistance. Biomaterials 2019, 223, 119475. [Google Scholar] [CrossRef]
- Saari, H.; Lázaro-Ibáñez, E.; Viitala, T.; Vuorimaa-Laukkanen, E.; Siljander, P.; Yliperttula, M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J. Control. Release 2015, 220, 727–737. [Google Scholar] [CrossRef]
- Tang, K.; Zhang, Y.; Zhang, H.; Xu, P.; Liu, J.; Ma, J.; Lv, M.; Li, D.; Katirai, F.; Shen, G.-X.; et al. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat. Commun. 2012, 3, 1282. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Soleymani, S.; Yari, F.; Bolhassani, A.; Bakhshandeh, H. Platelet microparticles: An effective delivery system for anti-viral drugs. J. Drug Deliv. Sci. Technol. 2019, 51, 290–296. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.-S.; Roh, T.-Y.; Park, J.; Nilsson, J.; Lötvall, J.; Kim, Y.-K.; Gho, Y.S. Bioinspired Exosome-Mimetic Nanovesicles for Targeted Delivery of Chemotherapeutics to Malignant Tumors. ACS Nano. 2013, 7, 7698–7710. [Google Scholar] [CrossRef] [PubMed]
- Panariti, A.; Miserocchi, G.; Rivolta, I. The effect of nanoparticle uptake on cellular behavior: Disrupting or enabling functions? Nanotechnol. Sci. Appl. 2012, 5, 87–100. [Google Scholar] [PubMed]
- Geng, T.; Tang, M.; Yee Paek, S.; Leung, E.; Chamley, L.W.; Wu, Z. A simple approach to re-engineering small extracellular vesicles to circumvent endosome entrapment. Int. J. Pharm. 2022, 626, 122153. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef]
- Kaźmierczak, Z.; Szostak-Paluch, K.; Przybyło, M.; Langner, M.; Witkiewicz, W.; Jędruchniewicz, N.; Dąbrowska, K. Endocytosis in cellular uptake of drug delivery vectors: Molecular aspects in drug development. Bioorg. Med. Chem. 2020, 28, 115556. [Google Scholar] [CrossRef]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef]
- Mosquera, J.; García, I.; Liz-Marzán, L.M. Cellular Uptake of Nanoparticles versus Small Molecules: A Matter of Size. Acc. Chem. Res. 2018, 51, 2305–2313. [Google Scholar] [CrossRef]
- Sousa de Almeida, M.; Susnik, E.; Drasler, B.; Taladriz-Blanco, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Understanding nanoparticle endocytosis to improve targeting strategies in nanomedicine. Chem. Soc. Rev. 2021, 50, 5397–5434. [Google Scholar] [CrossRef]
- Donahue, N.D.; Acar, H.; Wilhelm, S. Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Adv. Drug Deliv. Rev. 2019, 143, 68–96. [Google Scholar] [CrossRef]
- Firdessa, R.; Oelschlaeger, T.A.; Moll, H. Identification of multiple cellular uptake pathways of polystyrene nanoparticles and factors affecting the uptake: Relevance for drug delivery systems. Eur. J. Cell Biol. 2014, 93, 323–337. [Google Scholar] [CrossRef]
- Tu, Z.; Achazi, K.; Schulz, A.; Mülhaupt, R.; Thierbach, S.; Rühl, E.; Adeli, M.; Haag, R. Combination of Surface Charge and Size Controls the Cellular Uptake of Functionalized Graphene Sheets. Adv. Funct. Mater. 2017, 27, 1701837. [Google Scholar] [CrossRef]
- Yameen, B.; Choi, W.I.; Vilos, C.; Swami, A.; Shi, J.; Farokhzad, O.C. Insight into nanoparticle cellular uptake and intracellular targeting. J. Control. Release 2014, 190, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Shi, Y.; Qi, T.; Qiu, S.; Huang, Y.; Zhao, X.; Sun, Q.; Lin, G. Precise design strategies of nanomedicine for improving cancer therapeutic efficacy using subcellular targeting. Signal Transduct. Target. Ther. 2020, 5, 262. [Google Scholar] [CrossRef]
- Costa Verdera, H.; Gitz-Francois, J.J.; Schiffelers, R.M.; Vader, P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J. Control. Release 2017, 266, 100–108. [Google Scholar] [CrossRef]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef]
- Silva, A.K.; Luciani, N.; Gazeau, F.; Aubertin, K.; Bonneau, S.; Chauvierre, C.; Letourneur, D.; Wilhelm, C. Combining magnetic nanoparticles with cell derived microvesicles for drug loading and targeting. Nanomedicine 2015, 11, 645–655. [Google Scholar] [CrossRef]
- He, C.; Jaffar Ali, D.; Xu, H.; Kumaravel, S.; Si, K.; Li, Y.; Sun, B.; Ma, J.; Xiao, Z. Epithelial cell -derived microvesicles: A safe delivery platform of CRISPR/Cas9 conferring synergistic anti-tumor effect with sorafenib. Exp. Cell Res. 2020, 392, 112040. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Rathore, S.; Munshi, A.; Ramesh, R. Organically derived exosomes as carriers of anticancer drugs and imaging agents for cancer treatment. Semin. Cancer Biol. 2022, 19, 80–100. [Google Scholar] [CrossRef] [PubMed]
- Geng, T.; Pan, P.; Leung, E.; Chen, Q.; Chamley, L.; Wu, Z. Recent Advancement and Technical Challenges in Developing Small Extracellular Vesicles for Cancer Drug Delivery. Pharm. Res. 2021, 38, 179–197. [Google Scholar] [CrossRef]
- Mitchell, J.P.; Court, J.; Mason, M.D.; Tabi, Z.; Clayton, A. Increased exosome production from tumour cell cultures using the Integra CELLine Culture System. J. Immunol. Methods. 2008, 335, 98–105. [Google Scholar] [CrossRef]
- Tian, T.; Zhu, Y.-L.; Zhou, Y.-Y.; Liang, G.-F.; Wang, Y.-Y.; Hu, F.-H.; Xiao, Z.-D. Exosome Uptake through Clathrin-mediated Endocytosis and Macropinocytosis and Mediating miR-21 Delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef] [PubMed]
- Hazan-Halevy, I.; Rosenblum, D.; Weinstein, S.; Bairey, O.; Raanani, P.; Peer, D. Cell-specific uptake of mantle cell lymphoma-derived exosomes by malignant and non-malignant B-lymphocytes. Cancer Lett. 2015, 364, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Shan, X.; Long, M.; Ahmed, K.S.; Zhao, L.; Mao, J.; Zhang, H.; Sun, C.; You, C.; Lv, G.; et al. Elucidation of cellular uptake and intracellular trafficking of heparosan polysaccharide-based micelles in various cancer cells. Int. J. Biol. Macromol. 2019, 130, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.M.; Wilson, W.R.; Wu, Z. pH-Sensitive PEGylated liposomes for delivery of an acidic dinitrobenzamide mustard prodrug: Pathways of internalization, cellular trafficking and cytotoxicity to cancer cells. Int. J. Pharm. 2017, 516, 323–333. [Google Scholar] [CrossRef]
- El Leithy, E.S.; Abdel-Bar, H.M.; Ali, R.A.-M. Folate-chitosan nanoparticles triggered insulin cellular uptake and improved in vivo hypoglycemic activity. Int. J. Pharm. 2019, 571, 118708. [Google Scholar] [CrossRef]
- He, B.; Yang, D.; Qin, M.; Zhang, Y.; He, B.; Dai, W.; Wang, X.; Zhang, Q.; Zhang, H.; Yin, C. Increased cellular uptake of peptide-modified PEGylated gold nanoparticles. Biochem. Biophys. Res. Commun. 2017, 494, 339–345. [Google Scholar] [CrossRef]
- Guerreiro, E.M.; Vestad, B.; Steffensen, L.A.; Aass, H.C.D.; Saeed, M.; Øvstebø, R.; Costea, D.E.; Galtung, H.K.; Søland, T.M. Efficient extracellular vesicle isolation by combining cell media modifications, ultrafiltration, and size-exclusion chromatography. PLoS ONE 2018, 13, e0204276. [Google Scholar] [CrossRef]
- Geng, T.; Paek, S.Y.; Leung, E.; Chamley, L.W.; Wu, Z. Comparing extracellular vesicles from four different cell origins for intracellular drug delivery to pancreatic cancer cells: Small or large vesicles? J. Drug Deliv. Sci. Technol. 2024, 93, 105416. [Google Scholar] [CrossRef]
- Nigro, A.; Finardi, A.; Ferraro, M.M.; Manno, D.E.; Quattrini, A.; Furlan, R.; Romano, A. Selective loss of microvesicles is a major issue of the differential centrifugation isolation protocols. Sci. Rep. 2021, 11, 3589. [Google Scholar] [CrossRef]
- Kanchanapally, R.; Deshmukh, S.K.; Chavva, S.R.; Tyagi, N.; Srivastava, S.K.; Patel, G.K.; Singh, A.P.; Singh, S. Drug-loaded exosomal preparations from different cell types exhibit distinctive loading capability, yield, and antitumor efficacies: A comparative analysis. Int. J. Nanomed. 2019, 14, 531–541. [Google Scholar] [CrossRef]
- Webber, J.; Clayton, A. How pure are your vesicles? J. Extracell. Vesicles 2013, 2, 19861. [Google Scholar] [CrossRef] [PubMed]
- Grangier, A.; Branchu, J.; Volatron, J.; Piffoux, M.; Gazeau, F.; Wilhelm, C.; Silva, A.K.A. Technological advances towards extracellular vesicles mass production. Adv. Drug Deliv. Rev. 2021, 176, 113843. [Google Scholar] [CrossRef]
- Vestad, B.; Llorente, A.; Neurauter, A.; Phuyal, S.; Kierulf, B.; Kierulf, P.; Skotland, T.; Sandvig, K.; Haug, K.B.F.; Øvstebø, R. Size and concentration analyses of extracellular vesicles by nanoparticle tracking analysis: A variation study. J. Extracell. Vesicles 2017, 6, 1344087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-F.; Zhou, Y.-Z.; Zhang, B.; Huang, S.-F.; Li, P.-P.; He, X.-M.; Cao, G.-D.; Kang, M.-X.; Dong, X.; Wu, Y.-L. Pancreatic cancer-derived exosomes promoted pancreatic stellate cells recruitment by pancreatic cancer. J. Cancer 2019, 10, 4397–4407. [Google Scholar] [CrossRef]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276. [Google Scholar] [CrossRef]
- Khan, M.A.; Anand, S.; Deshmukh, S.K.; Singh, S.; Singh, A.P. Determining the Size Distribution and Integrity of Extracellular Vesicles by Dynamic Light Scattering. Methods Mol. Biol. 2022, 2413, 165–175. [Google Scholar] [PubMed]
- Matsumura, S.; Minamisawa, T.; Suga, K.; Kishita, H.; Akagi, T.; Ichiki, T.; Ichikawa, Y.; Shiba, K. Subtypes of tumour cell-derived small extracellular vesicles having differently externalized phosphatidylserine. J. Extracell. Vesicles 2019, 8, 1579541. [Google Scholar] [CrossRef]
- Noble, J.M.; Roberts, L.M.; Vidavsky, N.; Chiou, A.E.; Fischbach, C.; Paszek, M.J.; Estroff, L.A.; Kourkoutis, L.F. Direct comparison of optical and electron microscopy methods for structural characterization of extracellular vesicles. J. Struct. Biol. 2020, 210, 107474. [Google Scholar] [CrossRef]
- Chuo, S.T.-Y.; Chien, J.C.-Y.; Lai, C.P.-K. Imaging extracellular vesicles: Current and emerging methods. J. Biomed. Sci. 2018, 25, 91. [Google Scholar] [CrossRef]
- Griese, M.; Reinhardt, D. Smaller sized particles are preferentially taken up by alveolar type II pneumocytes. J. Drug Target. 1998, 5, 471–479. [Google Scholar] [CrossRef]
- Augustine, R.; Hasan, A.; Primavera, R.; Wilson, R.J.; Thakor, A.S.; Kevadiya, B.D. Cellular uptake and retention of nanoparticles: Insights on particle properties and interaction with cellular components. Mater. Today Commun. 2020, 25, 101692. [Google Scholar] [CrossRef]
- Franzen, C.A.; Simms, P.E.; Van Huis, A.F.; Foreman, K.E.; Kuo, P.C.; Gupta, G.N. Characterization of uptake and internalization of exosomes by bladder cancer cells. Biomed. Res. Int. 2014, 2014, 619829. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Wu, J.-Y.; Wang, J.-M.; Hu, X.-B.; Xiang, D.-X. Emerging strategies for labeling and tracking of extracellular vesicles. J. Control. Release 2020, 328, 141–159. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, T.; Seino, R.; Umezaki, K.; Shimoda, A.; Ezoe, T.; Ishiyama, M.; Akiyoshi, K. New Lipophilic Fluorescent Dyes for Labeling Extracellular Vesicles: Characterization and Monitoring of Cellular Uptake. Bioconjug. Chem. 2021, 32, 680–684. [Google Scholar] [CrossRef]
- Caponnetto, F.; Manini, I.; Skrap, M.; Palmai-Pallag, T.; Di Loreto, C.; Beltrami, A.P.; Cesselli, D.; Ferrari, E. Size-dependent cellular uptake of exosomes. Nanomedicine 2017, 13, 1011–1020. [Google Scholar] [CrossRef]
- Garcin, P.O.; Panté, N. The minute virus of mice exploits different endocytic pathways for cellular uptake. Virology 2015, 482, 157–166. [Google Scholar] [CrossRef]
- Hsu, C.Y.M.; Uludağ, H. Cellular uptake pathways of lipid-modified cationic polymers in gene delivery to primary cells. Biomaterials 2012, 33, 7834–7848. [Google Scholar] [CrossRef]
- Yan, G.-H.; Song, Z.-M.; Liu, Y.-Y.; Su, Q.; Liang, W.; Cao, A.; Sun, Y.-P.; Wang, H. Effects of carbon dots surface functionalities on cellular behaviors—Mechanistic exploration for opportunities in manipulating uptake and translocation. Colloids Surf. B Biointerfaces 2019, 181, 48–57. [Google Scholar] [CrossRef]
- Barrias, E.; Reignault, L.; de Carvalho, T.M.U.; de Souza, W. Clathrin coated pit dependent pathway for Trypanosoma cruzi internalization into host cells. Acta Trop. 2019, 199, 105057. [Google Scholar] [CrossRef]
- Eom, H.-J.; Choi, J. Clathrin-mediated endocytosis is involved in uptake and toxicity of silica nanoparticles in Caenohabditis elegans. Chem. Biol. Interact. 2019, 311, 108774. [Google Scholar] [CrossRef]
- Agarwal, S.; Rastogi, R.; Gupta, D.; Patel, N.; Raje, M.; Mukhopadhyay, A. Clathrin-mediated hemoglobin endocytosis is essential for survival of Leishmania. Biochim. Biophys. Acta 2013, 1833, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, T.K.; Khorenko, M.; Moussavi, A.; Engelke, M.; Boretius, S.; Feldmann, C.; Reichardt, H.M. Highly selective organ distribution and cellular uptake of inorganic-organic hybrid nanoparticles customized for the targeted delivery of glucocorticoids. J. Control. Release 2020, 319, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Cronqvist, T.; Erlandsson, L.; Tannetta, D.; Hansson, S.R. Placental syncytiotrophoblast extracellular vesicles enter primary endothelial cells through clathrin-mediated endocytosis. Placenta 2020, 100, 133–141. [Google Scholar] [CrossRef]
- Müller, K.H.; Motskin, M.; Philpott, A.J.; Routh, A.F.; Shanahan, C.M.; Duer, M.J.; Skepper, J.N. The effect of particle agglomeration on the formation of a surface-connected compartment induced by hydroxyapatite nanoparticles in human monocyte-derived macrophages. Biomaterials 2014, 35, 1074–1088. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef]
- Banushi, B.; Joseph, S.R.; Lum, B.; Lee, J.J.; Simpson, F. Endocytosis in cancer and cancer therapy. Nat. Rev. Cancer 2023, 23, 450–473. [Google Scholar] [CrossRef]
- Bonsergent, E.; Grisard, E.; Buchrieser, J.; Schwartz, O.; Thery, C.; Lavieu, G. Quantitative characterization of extracellular vesicle uptake and content delivery within mammalian cells. Nat. Commun. 2021, 12, 1864. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Guo, H.; Edman, M.; Hamm-Alvarez, S.F. Application of advances in endocytosis and membrane trafficking to drug delivery. Adv. Drug Deliv. Rev. 2020, 157, 118–141. [Google Scholar] [CrossRef]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.R.; Such, G.K. The Endosomal Escape of Nanoparticles: Toward More Efficient Cellular Delivery. Bioconjug. Chem. 2019, 30, 263–272. [Google Scholar] [CrossRef]
- Dutta, D.; Donaldson, J.G. Search for inhibitors of endocytosis: Intended specificity and unintended consequences. Cell. Logist. 2012, 2, 203–208. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor/Structure | Pathway | Mechanism of Activity | Concentrations |
|---|---|---|---|
Genistein | Caveolin-mediated endocytosis | A tyrosine kinase inhibitor, blocks the phosphorylation of caveolin-1 | 50 μg/mL [56] 75 nM [57] |
Chlorpromazine (CPZ)  | Clathrin-mediated endocytosis | A cationic amphiphilic drug, inhibits clathrin-coated pit formation by relocating clathrin and its adapter proteins from the plasma membrane to the endosomes | 10 μg/mL [58,59] 1 μg/mL [60] 1 μM [61] |
Cytochalasin D (CytoD) | Actin-dependent endocytosis, i.e., phagocytosis and macropinocytosis | Block the actin polymerisation by occupying a faster-growing “barbed” end of actin filaments | 10 μg/mL [36] 1 μg/mL [62] 20 μM [63] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geng, T.; Tian, L.; Paek, S.Y.; Leung, E.; Chamley, L.W.; Wu, Z. Characterizing Extracellular Vesicles Generated from the Integra CELLine Culture System and Their Endocytic Pathways for Intracellular Drug Delivery. Pharmaceutics 2024, 16, 1206. https://doi.org/10.3390/pharmaceutics16091206
Geng T, Tian L, Paek SY, Leung E, Chamley LW, Wu Z. Characterizing Extracellular Vesicles Generated from the Integra CELLine Culture System and Their Endocytic Pathways for Intracellular Drug Delivery. Pharmaceutics. 2024; 16(9):1206. https://doi.org/10.3390/pharmaceutics16091206
Chicago/Turabian StyleGeng, Tianjiao, Lei Tian, Song Yee Paek, Euphemia Leung, Lawrence W. Chamley, and Zimei Wu. 2024. "Characterizing Extracellular Vesicles Generated from the Integra CELLine Culture System and Their Endocytic Pathways for Intracellular Drug Delivery" Pharmaceutics 16, no. 9: 1206. https://doi.org/10.3390/pharmaceutics16091206
APA StyleGeng, T., Tian, L., Paek, S. Y., Leung, E., Chamley, L. W., & Wu, Z. (2024). Characterizing Extracellular Vesicles Generated from the Integra CELLine Culture System and Their Endocytic Pathways for Intracellular Drug Delivery. Pharmaceutics, 16(9), 1206. https://doi.org/10.3390/pharmaceutics16091206