

Population Pharmacokinetic Modeling of Total and Unbound Pamiparib in Glioblastoma Patients: Insights into Drug Disposition and Dosing Optimization

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Study and PK Data
2.2. Population PK Modeling Analysis
2.2.1. Structure Model Development
2.2.2. Covariate Model Development
2.3. Model Evaluation
2.4. Model Simulations
3. Results
3.1. Population PK Model
3.2. Model Evaluation
3.3. Model Simulation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xiong, Y.; Guo, Y.; Liu, Y.; Wang, H.; Gong, W.; Liu, Y.; Wang, X.; Gao, Y.; Yu, F.; Su, D.; et al. Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia 2020, 22, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ren, B.; Liu, Y.; Jiang, B.; Guo, Y.; Wei, M.; Luo, L.; Kuang, X.; Qiu, M.; Lv, L.; et al. Discovery of Pamiparib (BGB-290), a Potent and Selective Poly (ADP-ribose) Polymerase (PARP) Inhibitor in Clinical Development. J. Med. Chem. 2020, 63, 15541–15563. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhu, J.; Wang, J.; Lin, Z.; Yin, R.; Sun, W.; Zhou, Q.; Zhang, S.; Wang, D.; Shi, H.; et al. Pamiparib Monotherapy for Patients with Germline BRCA1/2-Mutated Ovarian Cancer Previously Treated with at Least Two Lines of Chemotherapy: A Multicenter, Open-Label, Phase II Study. Clin. Cancer Res. 2022, 28, 653–661. [Google Scholar] [CrossRef]
- Lickliter, J.D.; Voskoboynik, M.; Mileshkin, L.; Gan, H.K.; Kichenadasse, G.; Zhang, K.; Zhang, M.; Tang, Z.; Millward, M. Phase 1A/1B dose-escalation and -expansion study to evaluate the safety, pharmacokinetics, food effects and antitumor activity of pamiparib in advanced solid tumours. Br. J. Cancer 2022, 126, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Sun, T.; Shi, Y.; Cui, J.; Yin, Y.; Ouyang, Q.; Liu, Q.; Zhang, Q.; Chen, Y.; Wang, S.; et al. Pamiparib in patients with locally advanced or metastatic HER2-negative breast cancer with germline BRCA mutations: A phase II study. Breast Cancer Res. Treat. 2023, 197, 489–501. [Google Scholar] [CrossRef]
- Stradella, A.; Johnson, M.; Goel, S.; Park, H.; Lakhani, N.; Arkenau, H.T.; Galsky, M.D.; Calvo, E.; Baz, V.; Moreno, V.; et al. Phase 1b study to assess the safety, tolerability, and clinical activity of pamiparib in combination with temozolomide in patients with locally advanced or metastatic solid tumors. Cancer Med. 2024, 13, e7385. [Google Scholar] [CrossRef]
- Friedlander, M.; Mileshkin, L.; Lombard, J.; Frentzas, S.; Gao, B.; Wilson, M.; Meniawy, T.; Baron-Hay, S.; Briscoe, K.; McCarthy, N.; et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: Results from the dose-expansion stage of a multicentre, open-label, phase I trial. Br. J. Cancer 2023, 129, 797–810. [Google Scholar] [CrossRef]
- Ciardiello, F.; Bang, Y.J.; Cervantes, A.; Dvorkin, M.; Lopez, C.D.; Metges, J.P.; Sanchez Ruiz, A.; Calvo, M.; Strickland, A.H.; Kannourakis, G.; et al. Efficacy and safety of maintenance therapy with pamiparib versus placebo for advanced gastric cancer responding to first-line platinum-based chemotherapy: Phase 2 study results. Cancer Med. 2023, 12, 13145–13154. [Google Scholar] [CrossRef]
- Li, J.; Wickramasinghe, C.; Jiang, J.; Wu, A.; Jiang, Y.; Tovmasyan, A.; Kim, S.; Sanai, N. Mechanistic Modeling of Spatial Heterogeneity of Drug Penetration and Exposure in the Human Central Nervous System and Brain Tumors. Clin. Pharmacol. Ther. 2025, 117, 690–703. [Google Scholar] [CrossRef]
- Kennedy, W.R.; Chang, Y.W.; Jiang, J.; Molloy, J.; Pennington-Krygier, C.; Harmon, J.; Hong, A.; Wanebo, J.; Braun, K.; Garcia, M.A.; et al. A Combined Phase 0/2 “Trigger” Trial Evaluating Pamiparib or Olaparib with Concurrent Radiotherapy in Patients with Newly-Diagnosed or Recurrent Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2023, 117, e115. [Google Scholar] [CrossRef]
- Jiang, J.; Bao, X.; Yue, Y.; Schiff, D.; Bindra, R.; Li, J. Development and validation of a liquid chromatography coupled with tandem mass spectrometry method for determining total and unbound pamiparib in human plasma and brain tumors. Biomed. Chromatogr. 2022, 36, e5478. [Google Scholar] [CrossRef] [PubMed]
- Yafune, A.; Ishiguro, M. Bootstrap approach for constructing confidence intervals for population pharmacokinetic parameters. I: A use of bootstrap standard error. Stat. Med. 1999, 18, 581–599. [Google Scholar] [CrossRef]
- Efron, B. The bootstrap and Markov-chain Monte Carlo. J. Biopharm. Stat. 2011, 21, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Efron, B. Bayesian inference and the parametric bootstrap. Ann. Appl. Stat. 2012, 6, 1971–1997. [Google Scholar] [CrossRef]
- Bergstrand, M.; Hooker, A.C.; Wallin, J.E.; Karlsson, M.O. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011, 13, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Beal, S.L.; Sheiner, L.B. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J. Pharmacokinet. Pharmacodyn. 2001, 28, 171–192. [Google Scholar] [CrossRef]
- Comets, E.; Brendel, K.; Mentre, F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: The npde add-on package for R. Comput. Methods Programs Biomed. 2008, 90, 154–166. [Google Scholar] [CrossRef]
- Mu, S.; Palmer, D.; Fitzgerald, R.; Andreu-Vieyra, C.; Zhang, H.; Tang, Z.; Su, D.; Sahasranaman, S. Human Mass Balance and Metabolite Profiling of [(14) C]-Pamiparib, a Poly (ADP-Ribose) Polymerase Inhibitor, in Patients With Advanced Cancer. Clin. Pharmacol. Drug Dev. 2021, 10, 1108–1120. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2019, 17, 409–419. [Google Scholar] [CrossRef]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Cheng, F.; Sun, L.; Shao, F.; Wang, J.; Yang, L.; Jiang, G. 778P Efficacy and safety of pamiparib monotherapy in recurrent ovarian cancer (rOC) after prior PARPi exposure: A prospective, open label, single-arm, phase II study. Ann. Oncol. 2024, 35, S585. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Krivak, T.C.; Kabil, N.; Munley, J.; Moore, K.N. PARP Inhibitors in Ovarian Cancer: A Review. Target. Oncol. 2023, 18, 471–503. [Google Scholar] [CrossRef] [PubMed]
- Bhamidipati, D.; Haro-Silerio, J.I.; Yap, T.A.; Ngoi, N. PARP inhibitors: Enhancing efficacy through rational combinations. Br. J. Cancer 2023, 129, 904–916. [Google Scholar] [CrossRef]
- Kim, D.; Nam, H.J. PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them. Int. J. Mol. Sci. 2022, 23, 8412. [Google Scholar] [CrossRef]
- Bruin, M.A.C.; Sonke, G.S.; Beijnen, J.H.; Huitema, A.D.R. Pharmacokinetics and Pharmacodynamics of PARP Inhibitors in Oncology. Clin. Pharmacokinet. 2022, 61, 1649–1675. [Google Scholar] [CrossRef]
- Carney, B.; Kossatz, S.; Lok, B.H.; Schneeberger, V.; Gangangari, K.K.; Pillarsetty, N.V.K.; Weber, W.A.; Rudin, C.M.; Poirier, J.T.; Reiner, T. Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nat. Commun. 2018, 9, 176. [Google Scholar] [CrossRef]
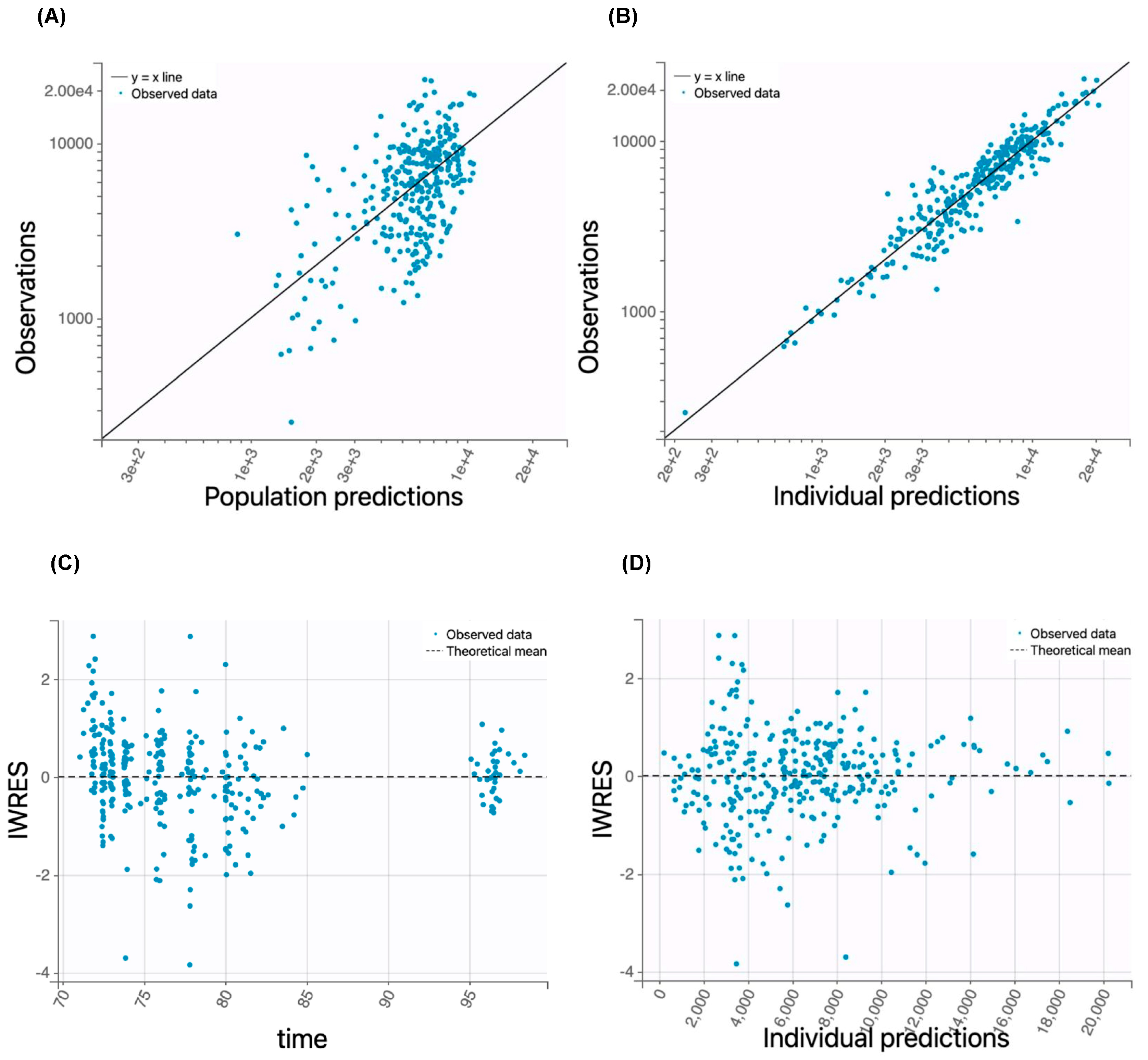


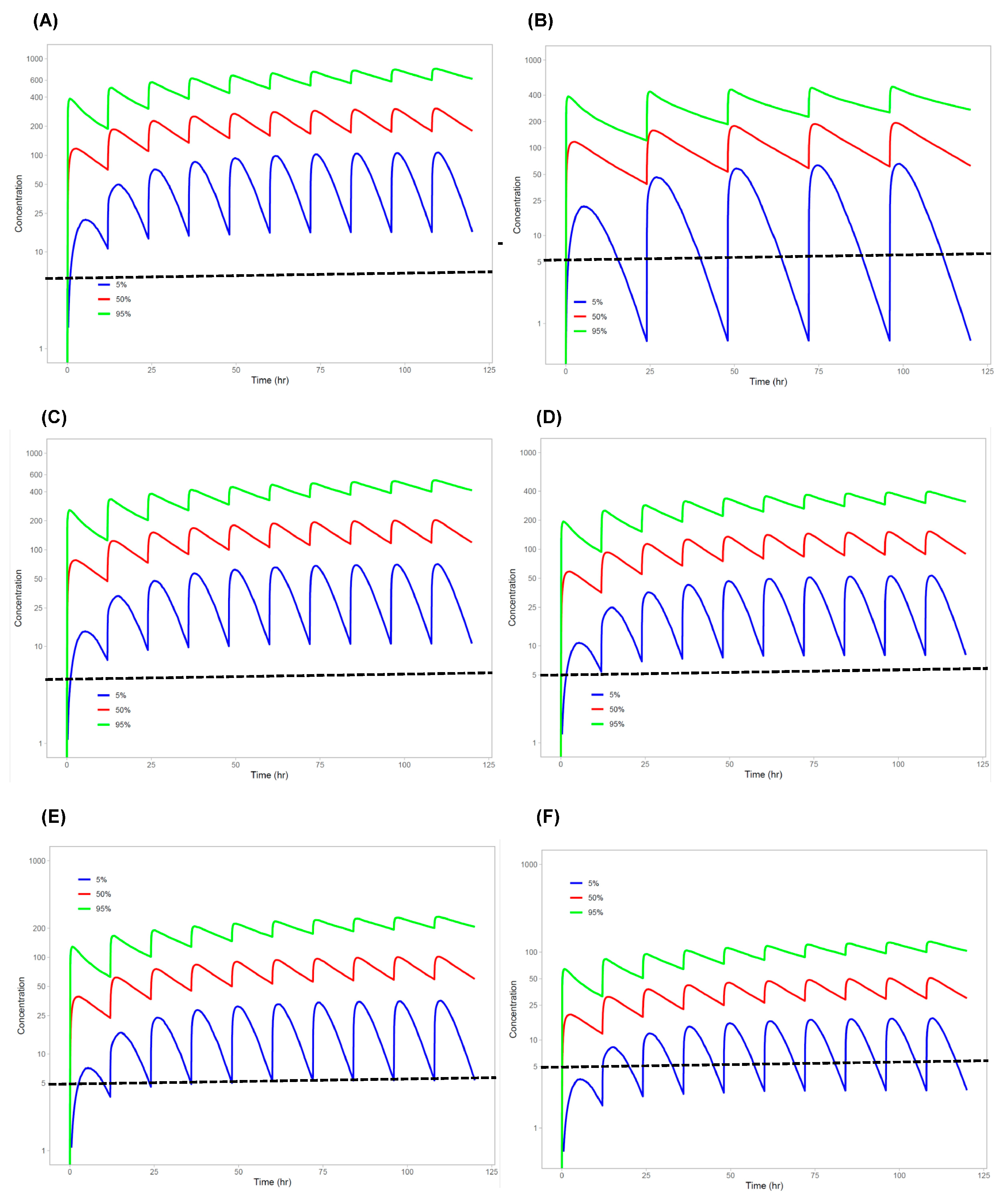

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Median (Range) or Number of Patients | |
|---|---|
| Baseline characteristics | |
| Race (white/non-white) † | 38/3 |
| Sex (male/female) † | 20/21 |
| Age (years) * | 60 (31–80) |
| Weight (kg) * | 80 (45–129) |
| Height (cm) * | 173 (155–193) |
| BSA () * | 1.99 (1.41–2.53) |
| Liver function * | |
| Predose total bilirubin (mg/dL) | 0.5 (0.3–1.8) |
| Post-operation total bilirubin (mg/dL) | 0.5 (0.2–1.5) |
| Predose AST (IU/L) | 19 (9–44) |
| Post-operation AST (IU/L) | 23 (9–106) |
| Predose ALT (IU/L) | 24 (8–135) |
| Post-operation ALT (IU/L) | 45 (14–193) |
| Predose plasma albumin (mg/dL) | 4.1 (3.5–5) |
| Post-operation plasma albumin (mg/dL) | 3.9 (3–4.5) |
| Kidney function * | |
| Predose serum creatine (mg/dL) | 0.83 (0.56–1.34) |
| Post-operation serum creatine (mg/dL) | 0.75 (0.41–1.3) |
| Predose creatine clearance (mL/min) | 98 (39–154) |
| Post-operation creatine clearance (mL/min) | 111 (59–169) |
| Predose GFR (mL/min) | 92 (48–116) |
| Post-operation GFR (mL/min) | 101 (48–117) |
| Concomitant drugs | |
| Coadministered drugs during the trial (yes/no) † | 36/5 |
| Dexamethasone (given/not given) † | 34/7 |
| Total dexamethason dose (mg) * | 19 (0–66) |
| Parameter * | Total Pamiparib | Unbound Pamiparib | ||
|---|---|---|---|---|
| Base Model | Final Model | Base Model | Final Model | |
| OFV | 9226 | 9210 | 9226 | 9212 |
| TV_KA (h−1) | 1.64 | 1.58 | 1.55 | 1.7 |
| TV_V/F (L) | 44 | 44 † | 1017 | 1060 † |
| TV_CL/F (L/h) | 2.73 | 2.59 ‡ | 65.0 | 62.5 ‡ |
| TV_Fu | 0.042 | 0.041 | 0.042 | 0.042 |
| () | 1.64 (42) | 1.58 (42) | 1.55 (51) | 1.7 (44) |
| () | 44 (9.7) | 15 (28) | 1017 (9.1) | 402 (28) |
| (CL/F) | 2.73 (8.0) | 6.76 (38) | 65.0 (8.2) | 163 (39) |
| () | 0.042(2.8) | 0.041 (2.8) | 0.042 (2.7) | 0.042 (2.6) |
| (PCC on V/F) | - | 0.0094 (25) | - | 0.0087(26) |
| (Age on CL/F) | - | −0.016 (37) | - | −0.016 (38) |
| IIV of Ka (%) | 397 (18) | 410 (20) | 612 (21) | 380 (20) |
| IIV of V/F (%) | 53 (16) | 41 (17) | 50 (17) | 41 (19) |
| IIV of CL/F (%) | 53 (11) | 50 (11) | 54 (11) | 52(11) |
| IIV of Fu (%) | 12 (22) | 12 (20) | 112 (24) | 11 (28) |
| Total Pamiparib | Unbound Pamiparib | |||||
|---|---|---|---|---|---|---|
| Parameter | Population Mean | Bootstrap Mean | Bootstrap 95% CI | Population Mean | Bootstrap Mean | Bootstrap 95% CI |
| () (h−1) | 1.58 | 1.71 | (0.64, 4.15) | 1.7 | 1.86 | (0.67, 4.07) |
| () (L) | 15 | 17 | (8, 30) | 402 | 441 | (219, 787) |
| (CL/F) (L/h) | 6.76 | 7.76 | (3.6, 15.9) | 163 | 185 | (83, 454) |
| () | 0.041 | 0.041 | (0.039, 0.044) | 0.042 | 0.041 | (0.039, 0.044) |
| (PCC) | 0.0094 | 0.0089 | (0.0035, 0.015) | 0.0087 | 0.0083 | (0.0027, 0.014) |
| (Age) | −0.016 | −0.017 | (−0.03, −0.0054) | −0.016 | −0.016 | (−0.032, −0.0053) |
| Ka_SD | 1.7 | 1.6 | (1.0, 2.3) | 1.66 | 1.66 | (1.1, 2.34) |
| V/F_SD | 0.39 | 0.35 | (0.16, 0.5) | 0.39 | 0.35 | (0.16, 0.5) |
| CL/F_SD | 0.47 | 0.46 | (0.35, 0.56) | 0.49 | 0.46 | (0.37, 0.37) |
| Fu_SD | 0.12 | 0.11 | (0.06, 0.16) | 0.11 | 0.11 | (0.059, 0.15) |
| Dosing Regimen | Css,max | Css,min | Css,ave | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 5th | 50th | 95th | 5th | 50th | 95th | 5th | 50th | 95th | |
| 60 mg BID | 105 | 302 | 774 | 16 | 175 | 581 | 47 | 233 | 673 |
| 60 mg QD | 63 | 193 | 485 | 0.6 | 61 | 253 | 14 | 115 | 357 |
| 40 mg BID | 68 | 201 | 515 | 11 | 117 | 388 | 31 | 155 | 448 |
| 30 mg BID | 53 | 151 | 387 | 8 | 88 | 291 | 24 | 116 | 336 |
| 20 mg BID | 35 | 101 | 258 | 5 | 58 | 194 | 16 | 78 | 224 |
| 10 mg BID | 18 | 50 | 129 | 3 | 29 | 97 | 8 | 39 | 112 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wickramasinghe, C.; Kim, S.; Jiang, Y.; Bao, X.; Yue, Y.; Jiang, J.; Hong, A.; Sanai, N.; Li, J. Population Pharmacokinetic Modeling of Total and Unbound Pamiparib in Glioblastoma Patients: Insights into Drug Disposition and Dosing Optimization. Pharmaceutics 2025, 17, 524. https://doi.org/10.3390/pharmaceutics17040524
Wickramasinghe C, Kim S, Jiang Y, Bao X, Yue Y, Jiang J, Hong A, Sanai N, Li J. Population Pharmacokinetic Modeling of Total and Unbound Pamiparib in Glioblastoma Patients: Insights into Drug Disposition and Dosing Optimization. Pharmaceutics. 2025; 17(4):524. https://doi.org/10.3390/pharmaceutics17040524
Chicago/Turabian StyleWickramasinghe, Charuka, Seongho Kim, Yuanyuan Jiang, Xun Bao, Yang Yue, Jun Jiang, Amy Hong, Nader Sanai, and Jing Li. 2025. "Population Pharmacokinetic Modeling of Total and Unbound Pamiparib in Glioblastoma Patients: Insights into Drug Disposition and Dosing Optimization" Pharmaceutics 17, no. 4: 524. https://doi.org/10.3390/pharmaceutics17040524
APA StyleWickramasinghe, C., Kim, S., Jiang, Y., Bao, X., Yue, Y., Jiang, J., Hong, A., Sanai, N., & Li, J. (2025). Population Pharmacokinetic Modeling of Total and Unbound Pamiparib in Glioblastoma Patients: Insights into Drug Disposition and Dosing Optimization. Pharmaceutics, 17(4), 524. https://doi.org/10.3390/pharmaceutics17040524