Abstract
Advanced biotherapeutic systems such as gene therapy, mRNA lipid nanoparticles, antibody–drug conjugates, fusion proteins, and cell therapy have proven to be promising platforms for delivering targeted biologic therapeutics. Preserving the intrinsic stability of these advanced therapeutics is essential to maintain their innate structure, functionality, and shelf life. Nevertheless, various challenges and obstacles arise during formulation development and throughout the storage period due to their complex nature and sensitivity to various stress factors. Key stability concerns include physical degradation and chemical instability due to various factors such as fluctuations in pH and temperature, which results in conformational and colloidal instabilities of the biologics, adversely affecting their quality and therapeutic efficacy. This review emphasizes key stability issues associated with these advanced biotherapeutic systems and approaches to identify and overcome them. In gene therapy, the brittleness of viral vectors and gene encapsulation limits their stability, requiring the use of stabilizers, excipients, and lyophilization. Keeping cells viable throughout the whole cell therapy process, from culture to final formulation, is still a major difficulty. In mRNA therapeutics, stabilization strategies such as the optimization of mRNA nucleotides and lipid compositions are used to address the instability of both the mRNA and lipid nanoparticles. Monoclonal antibodies are colloidally and conformationally unstable. Hence, buffers and stabilizers are useful to maintain stability. Although fusion proteins and monoclonal antibodies share structural similarities, they show a similar pattern of instability. Antibody–drug conjugates possess issues with conjugation and linker stability. This review outlines the stability issues associated with advanced biotherapeutics and provides insights into the approaches to address these challenges.
Keywords:
biologics; stability concerns; stabilization strategies; mRNA lipid nanoparticles; cell therapy; monoclonal antibodies; fusion proteins; conformational instability; colloidal instability; gene therapy; antibody–drug conjugates (ADCs); stabilization mechanisms; non-viral vectors; viral vectors 1. Introduction
Advanced biologic therapeutics have significantly transformed the healthcare landscape by providing effective treatments for various diseases, including cancer, genetic disorders, and autoimmune conditions [1]. Advanced biologic therapeutics include gene therapy, cell therapy, mRNA therapy, and others. In contrast to conventional small molecules, biologic therapies contain complex macromolecules, such as genes, proteins, cells, nucleic acids, etc., that require complex characterization techniques [2,3]. Additionally, the delivery of biologics also poses substantial stability challenges due to their inherent susceptibility towards physical and chemical degradation [4,5].
Among the various biologic therapeutics, gene therapy has emerged as a transformative approach aimed at correcting genetic defects by delivering functional genes into a patient’s cells. It commonly employs viral vectors, such as adeno-associated viruses (AAVs) or lentiviruses, to introduce genetic material into the target cells. Non-viral methods such as lipid nanoparticles (LNPs) are also being explored for gene therapy. However, the stability of gene therapy products is of critical concern. Viral vectors are prone to degradation during storage, which can impact their therapeutic efficacy [6]. Cell therapy involves the transfer of autologous or allogenic cells into the patient for therapeutic purposes [7]. The challenge with cell therapy is maintaining the viability and stability of cells from production to administration. Additionally, cell therapies require cryopreservation and the use of cryoprotectants to ensure that the cells remain active and functional. Maintaining stability is found to be crucial, as cell viability is directly linked to therapeutic success [8]. On the other hand, messenger RNA (mRNA) therapies involve the use of mRNA, which, upon administration, produces specific proteins. LNPs are frequently used as delivery systems for this type of therapy. Both LNPs and mRNA are prone to degradation, thus requiring excipients to maintain their stability [9,10].
Monoclonal antibodies (mAbs) are proteins that target specific antigens and are often administered intravenously or subcutaneously. mAbs are prone to stability issues such as conformational (due to altered protein structure) or colloidal instability, which frequently lead to aggregation [11]. Antibody–drug conjugates (ADCs) are composed of drugs that are chemically attached to mAbs and which specifically target cancer cells [12]. However, the stability of ADCs is critically dependent on the integrity of the chemical linker between the mAb and the drug (payload). This is because the conjugation changes the properties of the attached mAb [13]. Instability in the linker can result in the premature release of the cytotoxic agent, thus leading to reduced therapeutic efficacy and increased toxicity [14,15].
On the other hand, fusion proteins are prepared by combining two or more genes that can encode different proteins. They contain two or more different protein domains in a single molecule [16]. These proteins will have unique stability issues due to the complexity of their structures [17,18]. Given this background, it is evident that advanced biologic therapies are susceptible to stability issues. This review aims to present a detailed examination of the stability challenges associated with these advanced biologic therapeutics, including gene therapy, cell therapy, mRNA therapies, mAbs, ADCs, and fusion proteins (Figure 1). Additionally, we emphasize the various strategies and approaches discussed and employed in the literature, such as the use of novel excipients, lyophilization techniques, optimized linkers, and conjugation methods to address these challenges and ensure the stability and efficacy of these therapeutics throughout development and clinical use.
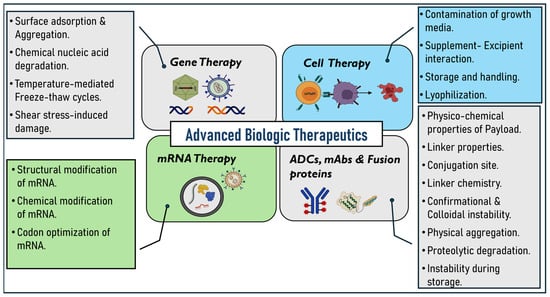
Figure 1.
Instability challenges related to advanced biologic therapeutics.
2. Stability Considerations for Advanced Biologic Therapeutics
2.1. Gene Therapy
Gene therapy is an advanced medical technique that uses deoxy ribonucleic acid (DNA) recombination and gene cloning to treat or prevent genetic problems. This technique has the potential to directly repair or substitute genes that cause genetic conditions such as cancers, cystic fibrosis, acquired immunodeficiency syndrome, cardiovascular ailments, and sickle cell anemia. Gene therapy offers a direct approach to targeting the underlying genetic condition by addressing the root cause, offering a potentially curative solution compared to the traditional treatments that focus on alleviating symptoms. For example, Leber congenital amaurosis, a rare eye disorder, is caused by a mutated RPE65 gene and results in inherited retinal dystrophy. This condition can be treated with gene therapy using a product called Luxturna to deliver a healthy version of the gene to the retinal cells. Similarly, sickle cell disease and beta-thalassemia are caused by heritable, single-gene mutations, leading to complications such as anemia and reduced hemoglobin production, respectively. CRISPR-Cas9-based gene therapies such as CTX001 can be used as a treatment to boost the production of healthy fetal hemoglobin, thereby addressing the root cause and reducing the need for blood transfusions [19,20,21,22]. Gene therapies usually involve the delivery of a functional gene into the cells of the patient, which can either correct a mutation, replace the faulty gene, or help fight a disease (Figure 2). This is accomplished by using vectors, with viral vectors being most frequently used due to their capability to proficiently enter cells. Viral vectors in gene therapy come with limitations such as immunogenicity, insertional mutagenesis, and potential toxicity to host cells [23]. Non-viral methods like nanoparticles offer advantages such as reducing the risk of triggering an immune response and avoiding the integration of genetic material into the host genome, thus enhancing safety. For example, CRISPR-Cas9 can be delivered via non-viral methods like LNPs or electroporation, allowing for targeted gene editing without the risks associated with viral vectors [24].
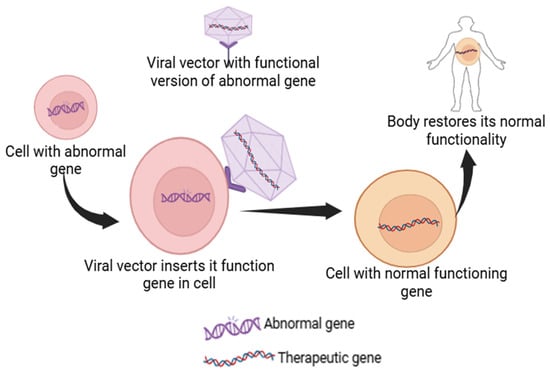
Figure 2.
Schematic representation of a viral vector inserting its functional gene into the abnormal gene-containing cell to restore its native body function.
Gene therapy has already experienced success in clinical trials and has been introduced into the pharmaceutical market with hope for patients with conditions such as metachromatic leukodystrophy, Leber congenital amaurosis, spinal muscular atrophy, sickle cell disease, β-thalassemia, and severe combined immunodeficiency [24]. However, some challenges are associated with gene therapy, which include its off-target effects, cost, and immune response. Moreover, a risk–benefit analysis should be performed to ensure the safety of the treatment [25,26].
2.1.1. Viral Vectors
Gene therapy requires vectors to transfer genetic material effectively and safely into host cells. These are precisely designed to target and enter specific cells, penetrate their membranes, and become an integral part of the genetic material, ensuring the unremitting production of the therapeutic genes. Optimal vector design is indeed essential for achieving favorable gene therapy, with the stability aspects of DNA being a critical consideration. Naked nucleic acids are prone to degradation by nucleases, possess challenges in cellular uptake, and may trigger immune responses, limiting their therapeutic efficacy. Therefore, the optimization of thoughtful vector engineering can improve translation efficiency, refine tissue specificity through promoter and regulatory region optimization, reduce immune response activation, and increase manufacturing yield and quality [27,28]. The selection criteria of viral vectors are key attributes and depend on various factors, such as efficiency, ease of production, safety, toxicity, and stability [29]. The primary categories of viral vectors comprise retroviruses, adenoviruses, adeno-associated viruses, lentiviruses, and herpes simplex viruses, each with distinct characteristics [30].
Adenovirus Vectors (Ad-Based Vectors)
Adenoviral vectors are non-enveloped, double-stranded DNA viruses, widely used in gene therapy due to their high transduction efficiency and ease of scalability. However, a significant challenge with these vectors is their potential to trigger immunological reactions, stemming from pre-existing immunity and adaptive immune responses [31,32]. First-generation vectors can accommodate transgenes up to 6.5 kb but still tend to elicit immunological responses. Second-generation vectors were designed to address the shortcomings of their predecessors by minimizing the expression of viral genes, thereby reducing immunogenicity and further enhancing transgene capacity. Third-generation vectors, capable of holding up to 36 kb of genetic material, offer reduced chances of immune responses and longer sustainability, although they require a helper virus to function properly [33,34,35,36]. A total of 50% of the human gene therapy research worldwide utilizes Ad-based vectors for the development of new treatments [28,37]. However, these vectors enhance inflammatory reactions and do not integrate into the host’s chromosome. Hence, they are better suited for applications that require short-term gene expression, such as the delivery of suicide genes in cancer research [38].
Retrovirus Vectors
Retroviruses encapsulate two copies of positive-strand RNA within their structure. They have gag, pol, and env genes, surrounded by long terminal repeats (LTRs) that act as enhancers and promoters [39]. They are employed in gene therapy due to their ability to integrate transgenes into the genome of the host cells, resulting in sustained expression for an extended period [40]. However, the major limitation of the retrovirus vector includes induced insertional oncogenesis due to random genomic integration and a failure to transduce non-dividing cells [41]. They are used for transferring genes into rapidly dividing cells over an extended period. However, the integration process in the host cell poses certain challenges, like insertional mutagenesis, oncogene activation, immune response, and the dysregulation of host oncogenes [42,43].
Adeno-Associated Virus Vector (AAV)
AAV vectors are used as delivery vehicles due to their safety and effectiveness. They are harmless, non-replicating viruses that can effectively transport therapeutic genes to specific cells and tissues, resulting in sustained gene expression [29]. They have several advantages, such as a wide range of tissue targeting, a minimal immune response, and non-integration into the host genome, minimizing genetic damage. However, they have certain challenges, including pre-existing immunity, restricted cargo capacity, and manufacturing cost [44]. There are three approved AAV-based gene therapies for retinal dystrophy, spinal muscular atrophy, and hemophilia B [45].
Herpes Simplex Virus Vector (HSV)
HSV vectors are large, enveloped viruses classified within the Alphaherpes virinae subfamily of the Herpesviridae family. HSV-1-based vectors can infect proliferating and non-dividing cells, without integrating into the genome of the host cells [46]. They are considered a highly promising technique for delivering genes to the central nervous system (CNS). Additionally, HSV vectors have been used for the treatment of recurrent breast cancer, head and neck cancer, unresectable pancreatic cancer, refractory superficial cancer, melanoma, and various neurological and pain problems. Various studies have demonstrated HSV as a possible remedy for delivering genes to the CNS and other regions where conventional gene therapy vectors may be inadequate [47,48].
2.1.2. Stability Issues Associated with Gene Therapy
Developing a gene-based medication poses significant stability challenges that can impact its safety and efficacy [49]. The different physical and chemical factors that affect the stability of gene-based therapeutics are enumerated below.
Aggregation
Aggregation hinders the effectiveness and safety of viral vectors. AAVs and lentiviruses can undergo aggregation during manufacturing and storage, forming larger complexes that cannot effectively enter into host cell, thereby reducing the efficacy of therapy [50,51]. Aggregation is caused due to factors such as temperature, stress, and pH, often caused by the destabilization of the capsid, leading to the poor therapeutic efficacy of the vector. Moreover, aggregation of the viral vectors can cause poor quality control and may elicit a stronger immune response, causing a severe inflammatory response in patients [42,52].
Adsorption
The adsorption (unwanted binding) of viral vectors on the surface can lead to a significantly decreased efficiency and therapeutic outcome of the gene delivery. Various viral vectors tend to adsorb on various surfaces such as cellular membranes, container material, and purification media during the manufacturing and storage process [53,54]. This can lead to decreased transduction efficiency. Adsorption can cause conformational changes in the viral vector that may compromise the integrity and rate of infectivity [55]. This further complicates the purification process, leading to the uneven distribution of full versus empty capsids, affecting the overall quality of a final product [56].
Deamidation, Oxidation, and Hydrolysis
Deamidation, oxidation, and hydrolysis are amongst the other critical factors that significantly affect the stability and efficacy of a gene therapy product. These can cause altered immunogenicity and an increased risk of adverse events in patients. Deamidation is the most common post-translational modification that can affect the structural integrity of the vector, leading to reduced effectiveness, and can cause altered immunogenicity, as the deamination of proteins may present different epitopes to the immune system, potentially causing unintended immune responses [57,58]. Oxidation can be detrimental to the viral vectors as it can enhance the production of reactive oxygen species, damaging the viral protein and affecting the stability and functionality. Hydrolysis can also impact capsid stability and cause the premature release of genetic material before it reaches target cells, thereby reducing therapeutic efficiency. Overall, the chemical process imposes manufacturing complexity and affects the quality of the product [59,60].
Freeze–Thaw (F/T)
The freeze–thaw process also poses a significant challenge to the stability of AAVs used in gene therapy. AAVs are sensitive to environmental conditions, and uncontrolled handling can lead to degradation, loss of infectivity, and reduced therapeutic efficacy. The mechanisms of instability during freeze–thaw include capsid rupture, DNA release, and aggregation. Freeze–thaw cycles can lead to the rupture of the capsid, which compromises its structural integrity, which will increase the risk of non-encapsulated DNA release [61,62]. A study found that adding ≥0.0005% w/v of poloxamer 188 (P188) eliminated substantial recovery losses (up to ~60% without P188) and minimized rupture to ≤1% per freeze–thaw cycle. Utilizing controlled-rate freezing and thawing cycles can help to ensure the reliability and success of applications with viral vectors [62].
Shear Stress
Shear stress, a force exerted by a fluid on a cell, is observed with gene therapy products that impose a major challenge on the stability [63]. The major production steps that can cause shear stress are caused by agitation, aeration, continuous gassing, and the few microfluid platforms that are used to study cellular responses [64]. It can lead to the aggregation or denaturation of viral capsids, compromising capsid stability and transduction efficiency. The aggregation caused due to shear stress can cause functional losses of vectors, causing altered pharmacokinetics and biodistribution upon administration [23].
Temperature
Temperature plays a critical role in the stability of gene therapy products, with both high and low temperatures posing significant challenges. Variations in the temperature can significantly affect the behavior of these vectors [65,66]. Exposing frozen gene therapy products to ambient temperatures during the transfer from manufacturing to shipping can cause severe thermal shock. This can lead to the denaturation and destabilization of the viral capsid, resulting in a loss of infectivity. Temperature also influences the replication cycle of the viruses, affecting the effectiveness of gene delivery [67]. Thermal sensitivity during the manufacturing process can reduce the yield and potency of the final product. Maintaining the appropriate temperature is critical throughout the storage, transport, and handling of gene therapy products to ensure their stability and efficacy [68,69,70].
2.1.3. Strategies to Address Instabilities in Gene Therapy
Various strategies are employed to address the stability issues of viral vectors to enhance their integrity, efficacy, and shelf life. Some of the approaches that can be employed to mitigate the instability issues for gene therapy are provided below.
Excipients
The utilization of excipients in various formulations of viral vectors is essential for addressing stability issues. They ensure the enhanced stability, efficacy, and overall performance of these biopharmaceutical products [71]. Excipients help viral particles from degrading during storage, processing, and the manufacturing process, thereby ensuring their efficacy and safety [72]. There are several types of excipients used in the preparation of gene delivery products, such as sugars, polyols, gelatine, amino acids, glycerol, and surfactants (Table 1). For example, during the development of a candidate vaccine against HIV-1 using Recombinant Human Cytomegalovirus Vector (rHCMV-1), stability was one of the major challenges. This challenge was resolved with the addition of sucrose, sorbitol, and trehalose in the viral vector formulation for stability enhancement [23]. Similarly, during the development of a formulation for the adeno-associated virus, a major challenge of stability was encountered during manufacturing and extended storage. The addition of poloxamer 188 with a low concentration of sucrose and dextrose significantly enhanced the stability of AAV vectors [62,73].
Lyophilization
Lyophilization is the process of removing water content from the biological product and stabilizing it without altering its functionality, thereby providing long-term storage stability. The lyophilized product [36] remains potent for long durations, ensuring its accessibility and effectiveness. For example, a modified formulation for the vaccinia recombinant vaccine (rTTV-OVA) and the adenovirus vaccine (Ad5-ENV) containing polyethylene glycol, dextran, bovine serum albumin (BSA), and L-glutamic acid (L-Glu) improved thermal stability [74].
Biomaterials
Biomaterials also play a significant role in addressing the stability issues in viral-based gene delivery products. They protect the viral vectors from eliciting immune responses, hence improving the delivery efficiency. For example, alginate-based hydrogels for the sustained release of AAVs show promising results in enhancing the local delivery of AAVs for gene therapy applications [75], improving stability and transduction efficiency. Similarly, chitosan nanoparticles [76] demonstrate improved stability for delivering lentivirus vector-based formulations. Poly (lactic-co-glycolic acid) (PLGA) microspheres encapsulate the adenoviral vectors [77], protecting them from degradation and resulting in the overall stability and efficacy of gene delivery [78].

Table 1.
Summary of key stability issues and excipients used to stabilize AAVs in gene therapy.
Table 1.
Summary of key stability issues and excipients used to stabilize AAVs in gene therapy.
| Instability Parameter | Excipient | Stabilization Mechanism | Outcome | References |
|---|---|---|---|---|
| Low mass concentration of AAV | Sucrose, citrate | Increasing the glass transition temperature of the lyophilized cake | Excipients were crucial for improving the stability of AAV particles in dry formulations, hence lowering aggregation and preserving infectivity during storage. | [36,79] |
| Aggregation due to low ionic strength | Citrate | Providing a minimum ionic strength to inhibit aggregation | Excipients lowered particle aggregation, therefore improving formulation uniformity and transduction efficiency both during storage and usage. | [36,79] |
| Degradation and potency loss due to mannitol crystallization during freezing | Sucrose instead of mannitol | Avoiding crystallization-induced damage | The improved retention of viral vector integrity and potency after freeze–thaw cycles. | [36,79] |
| Capsid damage and genome DNA release due to over-drying | Glycerol | Preventing over-drying by increasing residual moisture content | Use of excipients resulted in reduced capsid damage, ensuring higher transduction efficiency and genome stability. | [36,79] |
| Short-term stability of liquid formulations | Lyophilization | Improving long-term stability at refrigerated storage conditions | Use of excipients resulted in improved long-term stability under cold storage, reducing potency loss over time. | [36,79] |
| Aggregation and degradation of viral vectors | Sucrose, trehalose, glycerol | Increasing glass transition temperature, maintaining structural integrity | Use of excipients resulted in enhanced stability of viral vectors, reducing aggregation and degradation while ensuring consistent therapeutic efficacy. | [80,81] |
| Oxidative stress and loss of potency | Antioxidants (e.g., ascorbic acid, glutathione) | Protecting against oxidative damage | Excipients aided in preserving vector potency, ensuring reliable gene delivery performance. | [80,81] |
| Adsorption to container surfaces | Surfactants (e.g., polysorbate 80) | Reducing surface adsorption | The addition of excipients improved the recovery of viral vectors from containers, enhancing dosing accuracy and therapeutic efficacy. | [80,81] |
| Thermal instability during storage | Lyophilization, spray-drying | Improving long-term stability at refrigerated or ambient temperatures | Both preservation techniques provided improved long-term stability, minimizing potency loss over extended periods. | [80,81] |
| Immune responses against viral vectors | Immunosuppressants (e.g., rapamycin) | Reducing vector-mediated immune reactions | Use of excipients lowered immune responses, enhancing the safety profile of AAV therapies while maintaining efficacy. | [82] |
2.2. Cell Therapy
Cell therapies introduce a whole new paradigm in drug development. The mid-20th century saw red blood cell infusions enhance trauma, medicinal, and surgical outcomes, demonstrating cells’ transformative potential [83]. Subsequently, several cell therapies using T cells, hematopoietic stem cells, progenitor cells, and fibroblasts have received the United States Food and Drug Administration (USFDA) approval [84]. These are also currently in pre-clinical and clinical research on cell therapy for treating various diseases, including infectious diseases, cancer, genetic disorders, neurodegenerative diseases, and autoimmune disorders [85,86,87,88,89].
Often, the dosage forms of cell therapies are formulated as injectable suspensions either in liquid or cryopreserved form, as implantable scaffolds, and as sheets. Briefly, dosage form development involves collecting the cells from the donor and programming them, such as by treating them with artificial antigen-presenting cells or beads in a suitable media [90]. Cells collected from the patients are known as autologous cells, and developing these cells as dosage forms for the same patient is a highly controlled process. The obtained cell suspension, depending on the need, is either reinfused into the patient (for instance, CAR-T cell therapy for a cancer patient) or cryopreserved using various excipients such as buffers (tris, histidine, sodium acetate), salts (sodium chloride, potassium chloride, magnesium chloride), polymers, proteins (albumin), and preservatives and is administered through a suitable administration route [91,92]. Maintaining sterile and aseptic conditions is important during dosage form development and characterization.
Instability Issues of Cell Therapy and Approaches to Overcome These
Stability issues are a major challenge, specifically that of maintaining cell viability during the development and shelf life of cell-based therapies. Cell density, excipient concentrations, and potential degradation products can all impact stability. Cell therapy products are cryopreserved and stored at very low temperatures. During this process, the integrity of the cells may be damaged. Encapsulating different cells (including bacteria, fungi, plant meristems, liver and pancreatic cells, and various human cells) strengthens mammalian cell membranes, minimizes ice damage, and maintains cryopreservation, thereby increasing storage stability. The addition of cryoprotecting agents also preserves cell integrity through the vitrification mechanism. Table 2 summarizes the various stability issues with cell therapies and the approaches employed to address them.
Table 2.
Summary of key stability issues with cell therapies and approaches to overcome the instabilities associated with cell therapy [93,94,95,96,97].
Formulation development involves programming the cells, which can lead to alterations in the genomic stability of the cultured or processed cells [98]. The cause of this genomic heterogeneity is due to variability in cell sources. Cell therapies often use patient-derived cells or donor cells, which can have different interaction potentials with the excipients. Cell-based formulations contain different excipients such as buffers, salts, polymers, proteins, and preservatives to maintain stability or to provide physiological osmolarity. Also, during the manufacturing process, the formulation is exposed to various supplements such as process aids, which are not desired to be included in the final product. Residual levels of these supplements may interact with other excipients or cells and form unintentional products during the product’s shelf life. This will lead to batch-to-batch or supplier-to-supplier variability. To address these issues, the controlled use of excipients, evaluation of novel excipients, and use of cryopreservation media components in the manufacturing of finished medicinal products help in minimizing these issues [90,92].
Instabilities may also occur during the shipment and handling of developed cell therapy products. The transfer of frozen or growth-inhibited cells from central storage to local storage, followed by recovery, formulation, and injection to the patients, must be considered as a whole process at an early stage in the development of the product to generate an effective and efficient delivery chain to enable critical use. Use of non-conventional raw materials; condensed manufacturing timelines, for example, the vein-to-vein in 21 days approach; and contamination due to unintentional products that lead to batch-to-batch variability are some of the common issues that can raise the stability concerns of these cell therapy products [99,100,101,102]. These can be addressed by using consistent cryopreserved media or fit-for-purpose media components. Overall, the stability of cells and excipients must be evaluated in the final formulation. The stability issues can be reduced by following the appropriate measures. For instance, to avoid contamination due to unintentional products, cell culture should be standardized with fit-for-purpose media to maintain the quality. To enhance consistency and reduce batch-to-batch variation, rigorous testing protocols and advanced technologies like single-cell transcriptomics are helpful [102,103]. In addition, optimizing supply chains improves overall product consistency and reduces stability issues [92]. Also, instead of ex vivo cell engineering, developing in situ cell therapy using various biomaterials, such as lipids, polymers, inorganic materials, and others, including polyethylene glycol and peptides (e.g., CD47), as excipients holds great promise in improving stability [84]. For example, for chimeric antigen receptor T (CAR-T) cell therapy, pseudo-typing the viral particles enables the transduction of delivery systems into the specific type of immune cells. This not only avoids the expensive and complex laboratory protocols but also reduces the toxicity.
2.3. mRNA-Based Therapies
LNPs are essential delivery systems in mRNA-based therapeutics, performing critical functions such as encapsulating mRNA to shield it from enzymatic degradation and facilitating cellular uptake. A key aspect of LNP functionality is promoting endosomal escape, which is crucial for successful mRNA delivery. Following administration, LNPs are internalized into cells via endocytosis, entering endosomes [104,105]. To prevent mRNA degradation through fusion with lysosomes, mRNA must escape these vesicles. LNPs are engineered with ionizable lipids that are neutral at physiological pH but become positively charged within the acidic endosomal environment. This charge alteration destabilizes the endosomal membrane, aiding in mRNA release into the cytoplasm for translation [106,107]. This pH-dependent ionization not only protects the mRNA cargo but also enhances transfection efficiency. Additional components such as cholesterol and PEGylated lipids contribute to membrane fusion and improve colloidal stability; both of these components are vital for efficient delivery and intracellular trafficking [108,109]. The delivery process involves several stages: Initially, LNPs enclose mRNA within a lipid structure, typically using ionizable cationic lipids, which form stable complexes with mRNA’s negatively charged phosphate backbone. This encapsulation protects mRNA from extracellular RNases, enhancing its stability during circulation and storage [104,108]. Following administration, LNPs in the systemic circulation are protected by PEGylated lipids that prevent rapid clearance by the mononuclear phagocyte system, reduce aggregation, and enhance colloidal stability [110]. Subsequently, LNPs enter cells largely via clathrin-mediated endocytosis, resulting in LNP internalization within an early endosome (Figure 3). The ionizable lipids within LNPs remain relatively neutral at physiological pH to minimize toxicity during circulation. After endocytosis, the LNP-mRNA complex is contained within an endosomal compartment. As the endosome acidifies, the ionizable lipids in LNPs undergo protonation and acquire a positive charge. These positively charged lipids interact with the negatively charged endosomal membrane, disrupting its integrity through mechanisms such as the proton sponge effect and membrane destabilization, facilitating mRNA release into the cytoplasm. This interaction disrupts the endosomal membrane by promoting the formation of non-bilayer structures, leading to the release of the encapsulated mRNA into the cytoplasm for translation [109,110]. This escape is critical; failure results in mRNA degradation by lysosomes. Once in the cytoplasm, mRNA is accessible to the host cell’s ribosomal machinery and is translated into the desired protein, inducing a therapeutic or immune response. Research continues to optimize LNP composition, focusing on ionizable lipids, cholesterol derivatives, and helper lipids, to improve mRNA stability, reduce cytotoxicity, and enhance tissue-specific delivery [111,112].
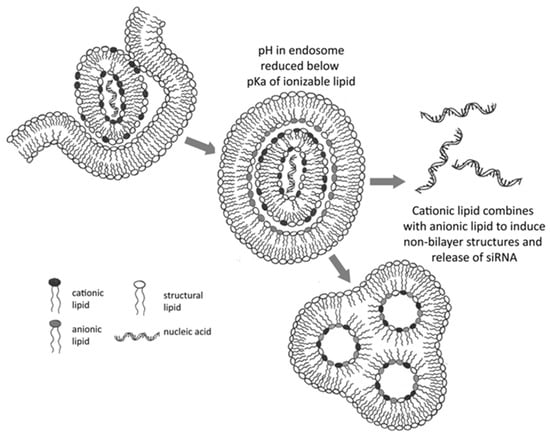
Figure 3.
Mechanism of RNA or DNA delivery to the cytoplasm by LNPs containing ionizable cationic lipids. The acidic endosomal environment encourages lipid protonation, leading to non-bilayer phase formation and subsequent endosomal disruption, thereby facilitating mRNA escape. Adapted with permission from [110]. 2025, Anusha Thumma.
2.3.1. Stability Issues with mRNA-Based Therapies and Approaches to Address These Issues
The efficient delivery of mRNA remains a significant challenge in mRNA-based therapies. Due to its large size and negative charge, mRNA molecules require specialized carriers to facilitate their entry into cells. LNPs are widely employed carriers that help in protecting mRNA from degradation, thereby favoring cellular uptake [113]. Despite their benefits, LNPs have heat resistance and target specificity issues. LNPs are inefficient in fields without cold storage because of these restrictions. This requires heat-resistant LNPs or other delivery mechanisms. Research is now targeted on the optimization of LNP composition to improve durability without compromising mRNA integrity [114,115]. Advances in nanobiotechnology offer new paths to improve the overall stability and efficiency of the mRNA delivery system. Some molecular and nanotechnology methods, such as using polymeric micelles as well as particles of hybrid nano-lipoproteins, offer further protection against mRNA degradation via the use of an enzyme, i.e., RNase, that favors the overall delivery. For instance, polymeric micelles having pH-responsive cross-linked cores not only limit mRNA degradation but also allow its specific release in target cells [116]. It has been observed that cationic nano-lipoprotein particles (exhibiting high density) play a vital role in the protection of large mRNA constructs (Figure 4), as well as favoring effective in vivo expression [117].
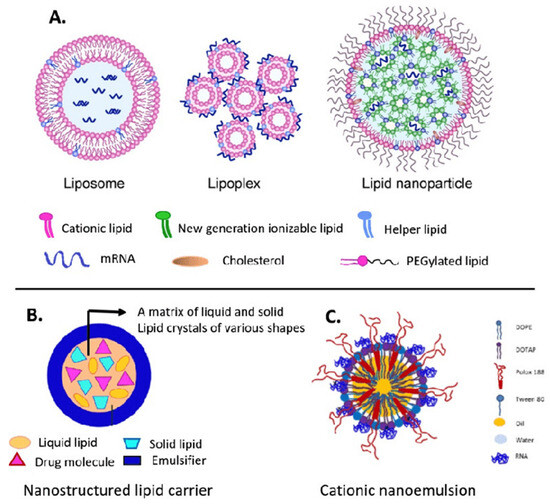
Figure 4.
Main lipid nanocarriers of mRNA: (A) liposome, lipoplex, lipid nanoparticle; (B) nanostructured lipid carrier; and (C) cationic nanoemulsion. Adapted with permission from [118,119]. 2025, Shashank Reddy Pasika.
In general, the development of more stable as well as specific targeted delivery systems plays an important role in increasing the uses of mRNA therapeutics for all over clinical applications worldwide. The stability along with the translation efficiency of mRNA used in therapeutics are highly affected by several key structural modifications including the 5′ cap, poly(A) tail, internal nucleotide substitutions, as well as secondary structure optimization that play a vital role in the protection of mRNA from degradation, enhancing protein translation, and eventually optimizing mRNA’s therapeutic potential.
Structural Modifications of mRNA
mRNA structural components such as the 5′ cap, ARCA, ORF, and poly(A) tail can be modified (Table 3). Anti-reverse cap analogs (ARCAs) like dimethylated guanosine dinucleotide (2mGpppG) can prevent reverse incorporation and stabilize mRNA, protecting it from degradation and promoting the translation process. In SARS-CoV-2 mRNA vaccine development, this change is crucial [120,121,122,123,124]. The literature suggests that an ideal poly(A) tail length of 100 nucleotides maximizes translation rates, while shorter tails (~75 nucleotides) promote closed-loop structures for better translation initiation and termination efficiency [125,126,127,128,129]. In therapeutic fields, adding non-adenine residues to poly(A) tails improves mRNA translation [129,130]. 2′-O-methylation, mainly by fibrillarin, improves mRNA stability by maintaining its integrity over time [131,132]. These changes reduce the degradation and stabilize mRNA in different cellular contexts [128,133].
Table 3.
mRNA vaccine structural modifications that help in improving the stability and translation efficiency [124].
Secondary structure modifications, particularly in the form of “superfolder” mRNAs, aid in stabilizing the mRNA by making structures less susceptible to hydrolysis as well as degradation. This modification process helps in improving the half-life of mRNA for use in a variety of therapeutic modalities [134,135,136]. The encapsulation of mRNA in nanoparticles, along with combining stabilizing chemical groups as thiophosphates, suggests thermal stability as well as the efficient delivery of mRNA [128,132,137,138]. Furthermore, the optimization of codon usage, modified UTRs, and poly(A) tail modifications have also been carried out for improving mRNA stability, translation efficiency, and a achieving a faster antibody response from vaccines (Figure 5A,B) [124].
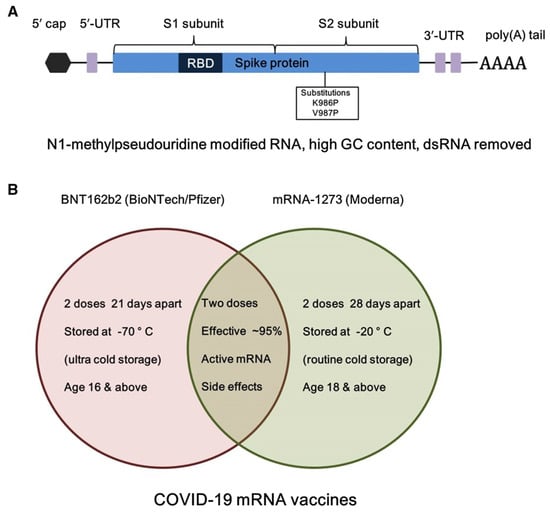
Figure 5.
(A) Structural element of BNT162b2 (BioNTech/Pfizer) mRNA vaccine, and (B) comparison of storage and dose information of the two used BNT162b2 and mRNA-1273 COVID-19 mRNA vaccines. Adapted with permission from [124]. 2025, Shashank Reddy Pasika.
Chemical Modification of mRNA
Modifying these mRNA elements aids in improving the overall stability of mRNA by providing strength to its structure and enhancing its effectiveness in translation. Pseudouridine (Ψ) improves the mRNA stability via increasing base stacking interactions and maintaining RNA duplex integrity, making it less vulnerable to degradation. In this type of modification, a universal base paired with adenine, guanine, uracil, or cytosine strengthens the structure of RNA and its overall performance in different cellular environments [139,140,141]. However, this process of mRNA pseudouridylation is highly affected by nutrient deficiency, temperature stress, cellular metabolism, as well as disease states, thus affecting protein translation in various ways.
In addition to Pseudouridine (Ψ) modification, N1-methylpseudouridine (m1Ψ) also aids in increasing mRNA stability by improving its molecular polarizability and base stacking interactions. This property makes it effective for incorporation in COVID-19 vaccines [141,142,143,144]. Computational studies have revealed that m1Ψ-modified RNA duplexes possess more stable nearest-neighbor interactions, which are crucial for secondary structure formation and for designing their therapeutic use [140,145] without causing any adverse immune reactions. The delivery carriers for this system are generally LNPs incorporated with a modified nucleobase, m1Ψ, which improves mRNA stability and decreases immune activation. Figure 6 shows the process of mRNA uptake by cells, protein synthesis, and immune response, while Figure 6 also reveals the chemical structure of modified nucleobase m1Ψ, which exchanges the natural nucleobase uracil for improving mRNA stability [146].
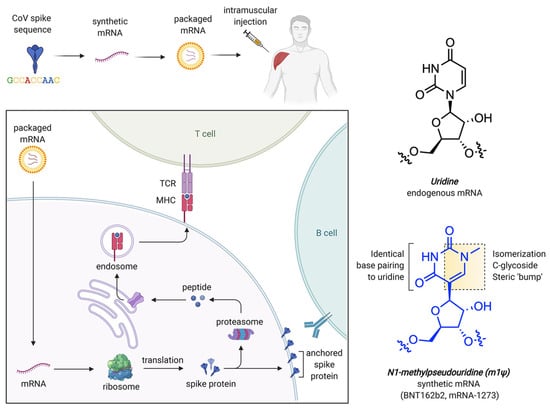
Figure 6.
mRNA-based COVID-19 vaccine approach and structural properties of uridine and m1Ψ. Here, TCR denotes T cell receptor while MHC refers to major histocompatibility complex. Adapted with permission from [146]. 2025, Shashank Reddy Pasika.
5-Methylcytidine (5mCyd) plays a key role in improving mRNA stability and protecting it from photodamage [146,147]. This type of RNA modification method has been widely studied across different organisms, such as mammals, plants, bacteria, as well as yeast. However, mRNA methylation is comparatively rare. NSUN2 has been identified as a key methyltransferase that is involved in mRNA methylation (Figure 7) [137,148].
Figure 7.
Distribution patterns and roles of 5-methylcytosine that is distributed among all regions in mRNA, primarily enriched within the 3′UTR and around the translation start sites. It helps in improving mRNA stability and its nuclear–cytoplasmic shuttling and translation. Adapted with permission from [148]. 2025, Shashank Reddy Pasika.
Codon Optimization of mRNA
For the design of stable mRNA for therapeutic purposes, codon optimization is important to improve both stability and protein synthesis by the translation of mRNA. Codon optimization increases mRNA stability, reduces the chance of translating mistakes, and reduces destruction. Additionally, it modifies the secondary structures of mRNA to avoid unstable folds that could lead to destruction. Furthermore, it guarantees effective translation rates, encouraging appropriate co-translational protein folding that improves the stability and activity of proteins. In this process of modification, the selection of synonymous codons is optimized for the host organism, thereby improving the effectiveness of mRNA-based therapies [149]. Studies have revealed that advancements in computational algorithms remarkably contributed to codon optimization efforts, suggesting the importance of computational algorithms for the development of mRNA vaccines and therapeutic proteins (Figure 8) [150]. The algorithmic method of codon optimization works by optimizing the sequence of mRNA through a balance of codon usage and structural stability. This leads to the transformation of a problem into a formal language theory model. This method showed remarkable advances in mRNA half-life and protein expression, especially in large sequences such as the SARS-CoV-2 Spike protein, which improved in vivo antibody titers in comparison to conventional benchmarks [149,151]. A model, i.e., the nearest-neighbor (NN) model, makes use of statistical physics principles for the optimization of codon usage by considering neighboring codon interactions. For effective protein expression in vivo, this model is useful, suggesting its potential for broader biotechnological usage with mRNA-based therapies [152].
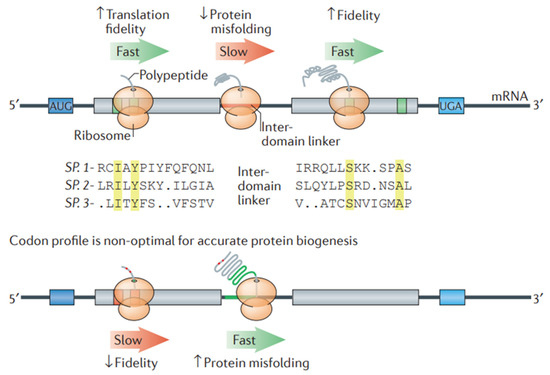
Figure 8.
Codon optimization in a transcript helps in optimizing protein folding. A stretch of non-optimal codons (red line) can slow down the process of translation elongation. At a higher level of codon optimization (green line), protein misfolding can take place. On the contrary, ribosomes can effectively aid in translating optimal codons (green boxes) that generally encode highly conserved residues (yellow colored). Optimal codons are less susceptible to reading errors as they relate to tRNA species having a higher tRNA ratio, resulting in the high translation fidelity of the most functionally crucial residues. When amino acids are encoded via non-optimal codons (highlighted in red boxes), missense errors can take place during translation. Adapted with permission from [150]. 2025. Shashank Reddy Pasika.
It has been reported that the modification of the 3′ untranslated regions (3′ UTRs) of mRNA is a promising method for improving the stability of mRNA. The main elements of mRNA, including AU-rich regions (AREs), affect its stability via binding to RNA-binding proteins (ARE-BPs). This helps in modulating the degradation of mRNA and its translation. For example, AREs in 3′ UTRs of Ebola virus mRNAs show interactions with particular proteins, such as tristetraprolin (TTP), which help in stabilizing or destabilizing mRNA [153]. When particular dinucleotide sequences, such as TA-rich regions, are present in mRNA, RNA destabilization promotes mRNA degradation. On the other hand, when sequences are GC-rich and show interactions with protective proteins, it improves the stability of mRNA. For the design of the desired mRNA, researchers nowadays are working on manipulating these sequences [154] via the incorporation of structured viral RNA elements into mRNA constructs. This aids in improving the mRNA transcript stability as well as its expression by inhibiting the efficiency of exoribonuclease (as seen in recombinant AAV vectors) [155,156].
2.3.2. Challenges and Future Directions
Codon optimization and mRNA stability engineering have improved the stability aspects of mRNA-based therapeutics; however, several limitations still remain. LNPs and polymeric nanoparticles are essential for enhancing mRNA stability, transport, and efficacy. These delivery technologies preserve mRNA against degradation, enhancing cellular absorption and tailored distribution [157,158].
LNPs protect mRNA against hydrolysis and enzymatic degradation. Nanoparticles and lipids, notably ionizable lipids, transport mRNA to target cells safely and efficiently [157,158]. These lipid particles are crucial both for effective encapsulation and for improving the endosomal release of mRNA. Cholesterol stabilizes the LNP bilayer and fuses membranes. PEG–lipids improve colloidal stability but must be tuned to prevent transfection difficulties [159]. Histidinamide-conjugated cholesterol improves endosomal escape, mRNA transport, and stability [158]. Microfluidic mixing has been employed to formulate LNPs with a consistent particle size and composition, leading to enhanced stability and delivery efficiency [160]. Regulating LNP surface charge helps in upgrading colloidal stability, particularly for inhaled delivery [161,162].
The use of the spray drying technique to powder LNPs enhances their storage stability. This SD technique has the potential to preserve mRNA functioning and eliminate cold chain storage [163]. Even tiny lipid particle contaminants like ALC-0315 affect the stability of LNPs. To maintain mRNA stability and reduce degradation, lipid particle quality must be considered [164]. Further, to improve the thermal stability of LNPs, the use of cryoprotectants, mineral encapsulation, and novel formulation approaches could be employed [165]. In the absence of cryoprotectants, LNP aggregation could occur, which may hinder protein translation [166]. Some other ways of improving the thermal stability of LNPs may include the incorporation of biomimetic minerals [167], silk fibroin [168], alcohol dilution–lyophilization techniques [169], solvent-free liquid polyplexes [170], and lyophilization. [171,172,173,174,175,176].
Polymeric nanoparticles are responsible for protecting mRNA via shielding it from degradation. Poly(amidoamine) (PAA)-based polymers in combination with chloroquinoline moieties enable endosomal escape for effective cytoplasmic release, thereby improving their therapeutic efficiency [177,178,179]. In general, the development of LNPs and polymeric nanoparticles plays a vital role in the field of mRNA delivery, stability, as well as effective immune modulation [53,180,181,182,183,184,185]. RNA-binding proteins (RBPs) and microRNAs (miRNAs) are the main regulators of mRNA stability. RBPs bind to specific RNA sequences that may protect or degrade mRNA. miRNAs, on the other hand, may target mRNA for degradation or limit translational performance. The interplay between RBPs and miRNAs is complex and essential for fine-tuning gene expression. Understanding these regulatory mechanisms is critical for developing therapeutic strategies that may target the stability of mRNA [186].
RBPs play a crucial role in cancer development and drug resistance via the regulation of stability and the translation of cancer-related transcripts [187]. RNA-binding proteins (RBPs) influence the expression of genes implicated in cancer processes like metastasis, apoptosis, and proliferation by regulating the stability of mRNA. RBP dysregulation can either destabilize tumor suppressor mRNAs or stabilize oncogenic mRNAs, which promotes the initiation and spread of cancer. In drug resistance, RBPs improve the stability of mRNAs that encode repair proteins, survival factors, or drug efflux pumps. This allows tumor cells to withstand stress and avoid treatment, which makes them viable candidates for therapeutic intervention. Engineered RBPs offer potential therapeutic uses [188]. miRNAs, via the formation of RISC complexes, prevent the translation of mRNA and also promote its degradation. The effects of miRNAs are specific; some miRNAs hinder translation, while others may promote it [189]. Codon composition can influence mRNA stability, which may be modulated via miRNAs. For example, miR-430 may reverse the stabilizing effect of optimal codons [190]. RBPs can also modulate miRNA function, affecting the stability of mRNA [190]. Modifications like methylation and acetylation may vary the structure of RNA, thereby influencing the overall mRNA decay rates [191,192]. However, more elaborate research on improved delivery systems and mRNA modification methods, and an improved understanding of interactions within biological systems will aid in the development of mRNA-based therapies.
2.4. Monoclonal Antibodies (mAbs)
Monoclonal antibodies have gained interest from the first approved muromonab-CD3 in 1986 to the recent approval of lecanemab in 2023, and several are currently in the clinical trial pipeline [193,194]. Intravenous administration is the most favorable approach for delivering mAbs because of instabilities in the gastrointestinal tract. The highly hydrophilic nature of protein molecules will also contribute to formulating an intravenous dosage form. Although well-versed intravenous dosages were developed, the frequent administration rates of intravenous routes paved the way for newer ones. Innovative approaches are being explored, mainly through subcutaneous routes of administration, to increase patient compliance and reduce invasiveness [195,196,197]. The subcutaneous administration of mAbs can bypass intravenous infusion with shorter administration times and is more convenient and patient centric. Currently, subcutaneous administration is a fast-growing field in mAb therapeutics, and antibodies are being formulated in high concentrations, along with matrix materials, to release the molecule for longer and maintain steady-state concentrations [197].
Commercially available intravenous monoclonal antibody formulations have buffering agents (acetate, histidine, and phosphate), tonicity modifiers (sodium chloride, sucrose), stabilizers (polysorbates), along with bulking agents/ cryoprotectants for lyophilized products [198,199,200]. The subcutaneous route is considered advantageous compared with the intravenous route, as the injection volume is limited to 2 mL for subcutaneous administration. These small volumes require highly concentrated mAbs (50–100 mg/mL), often resulting in the aggregation of proteins in the formulations. This aggregation is due to the decrease in intermolecular distances between adjacent molecules and possible electrostatic interactions. mAbs instability is particularly concerning as it may not only limit the therapeutic efficiency but may additionally possess immunogenic reactions [201]. Moreover, highly concentrated formulations exhibit increased viscosity, causing manufacturing complications. Factors including temperature, pressure, ionic strength, pH, and excipients have a positive and negative effect on the aggregation propensity in mAbs. Consequently, the formulation composition is crucial for supporting the physicochemical stability of mAbs during their manufacture, storage, and delivery [202].
The development of mAbs, stability, and structural integrity is a key aspect that needs to be investigated throughout the formulation journey. Intrinsic protein–protein cohesive interactions, the addition of external agents like excipients [203], and the processing conditions will affect the stability of the antibody in a formulation [204,205]. Mechanical stress occurs at various stages during the production and handling of mAbs (mAbs), potentially affecting their stability and efficacy [206]. Agitation stress during stirring in bioreactors and the exposure of mAbs to lower pH conditions during chromatographic purification and filtration will affect the overall stability of mAbs [207]. A study conducted by Utkarsh Tathe et al. explored instabilities related to agitation in bioreactors during production [208] and the instability that occurred by the formation of dimers and tetramers, with the effects of temperature on storage [209]. Similarly, formulations are also prone to shaking stress and aggregation during transportation, resulting in the formation of subvisible particles, leading to instabilities [210]. A comparative study conducted by Himanshu Malani et al. on the degradation of bevacizumab and trastuzumab upon different stresses, i.e., thermal stress, low pH, and shaking stress, identified aggregation mainly in thermal and chemical stress, and that could be nullified using suitable excipients [211]. The additional incorporation of surfactants in monoclonal antibody formulations mitigates the development of interfacial tension at air–liquid and solid–liquid interfaces [212], while buffering agents maintain ionic equilibrium within the solutions [213]. The aggregation is due to the conformational and colloidal instability of the proteins in the formulation. These instabilities are evaluated by the amino acid sequencing of antibodies, higher order structure analysis of proteins, purity profiles, aggregates, intact and reduced mass, potency, and binding affinity.
2.4.1. Instability Issues Associated with Monoclonal Antibodies
Conformational Instability
The tendency of mAbs to completely unfold or partially unfold from the non-native structure in formulations leads to conformational instabilities, causing aggregation with a loss of biological activity [214]. Aggregation is an assembly of originally native and folded proteins into high molecular weight species (multimers), irrespective of their size or the nature of their peptide–peptide linkages. Hence, conformational stability is the primary contributor to protein structure; disrupting secondary and tertiary structures leads to the aggregation of proteins. Conformational instability triggers aggregation, which may arise exclusively through weak nonspecific bonds (van der Waals interactions, hydrogen bonding, and hydrophobic interactions) without altering the primary structure, a phenomenon also called self-association [215]. The formulation concerns of conformational instability include temperature stress, exposure to hydrophobic environments, and alterations in pH. The elevated protein concentrations in intravenous or subcutaneous formulation, including the surface tension between the interfaces, will affect the conformational stability.
Monoclonal antibodies are more susceptible to conformational changes due to chemical degradation by hydrolysis and deamidation. The chemical degradation is triggered by factors like exposure to light, oxidation, enzymes, and the presence of other excipients in the formulation, which are susceptible to react covalently with the primary structures of proteins [216]. Particularly, the deamination (asparagine, glutamine) and oxidation of amino acids (methionine, tryptophan) and the cleavage of disulfide bonds [217] possess instabilities. Many studies have provided significant evidence of the impact of methionine and tryptophan oxidation, along with asparagine deamidation, on the stability of mAbs [218]. Hence, there is a need for the careful screening of newer excipients apart from conventional materials and oxidation profiling [219]. Studies have reported that the addition of common peroxides in mAb formulations prevents conformational instability by inhibiting disulfide bond reduction [220,221]. Alongside the most used stabilizers like polysorbate 20, these stabilizers are also susceptible to hydrolytic degradation. The host cell proteins convert PS20 into a free fatty acid (FFA) form that can form subvisible particles over long-term storage [222]. Carle et al. studied the root cause analysis to elucidate the degradation of PS20 and PS80 and developed a mitigation strategy. This approach effectively differentiated between hydrolytic and oxidative degradation, due to increase in FFAs and non-esterified FFAs. Hence, FFA increase patterns can be used as a predictive tool for the analysis of the long-term chemical-based confirmational instability of mAbs [223].
Colloidal Instability
Colloidal instability occurs due to protein–protein interactions that lead to aggregation and phase separation [224]. Attractive forces like hydrophobic or van der Waals will lead to colloidal instability. The balance between these electrostatic repulsions and attractive forces in a system is crucial for maintaining the colloidal stability of mAbs in the formulation [225]. The highly concentrated mAb formulation imparts colloidal instability due to the proximity effect, i.e., individual antibody molecules are forced into closed spaces, decreasing intermolecular distances. This phenomenon adversely affects the subcutaneous delivery of mAbs, which need very high concentrations in small volumes [226]. The solution conditions, i.e., the presence of pH modifiers and ionic strength modifiers, will propagate to colloidal instability. Specifically, lower pH conditions will affect the surface charge on mAbs, resulting in a reduction in electrostatic repulsions, promoting colloidal instability. This was shown in studies performed with IgG4-N1, where colloidal instability was caused due to the unfolding of mAbs below pH 3.3 [227]. Salts affect the ionic strength in formulations; salt effects often differ based on the mAbs’ surface charge. The total impact of salt on physical stability is a balance of the salt’s interactions with water and mAbs. Salts can affect physical stability by modifying the characteristics of the mAbs–solvent system (Hofmeister effects) and by limiting electrostatic interactions (Debye–Hückel effects) [228]. A comparative study of nine mAbs demonstrated that the ionic strength affected the colloidal interactions, altering the surface charge properties. Colloidal instabilities were assessed utilizing the effective surface charge through zeta potential, diffusion interaction parameter (kD), second virial coefficient, and B22 [202,229]. Also, highly hydrated ions (kosmotropes) adversely influence the hydration shell of mAbs, promote hydrophobic protein–protein interactions, and detrimentally affect the protein colloidal stability (e.g., Na2SO4). This colloidal instability occurs due to the kosmotropic effect strengthening mAbs-mAbs interactions, leading to an increase in the viscosity of the formulation [230]. A comparative study was performed to assess the stability of highly concentrated mAbs; the hydroxyl group of polyethylene glycol was shown to interact with positively charged amino acid residues in mAbs. This particular binding alters the net charge of the protein and may adversely affect its colloidal stability below the isoelectric point [230,231]. This instability is due to the liquid–liquid phase separation phenomenon in which mAbs in dense fractions are separated and concentrated [231]. The chaotropic salts, including potassium iodide, diminish hydrophobic interactions by disrupting the aqueous framework surrounding solubilized molecules. They exert a colloidal impact on mAbs. However, these salts may enhance colloidal stability at low to moderate quantities [232]. The colloidal and conformational instabilities occur in a cascade fashion, and they are mostly interrelated, resulting in the aggregation and destabilization of mAbs. Figure 9 gives an overview of the mechanisms of colloidal and conformational instabilities.
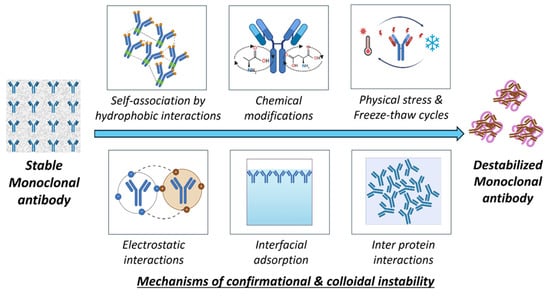
Figure 9.
Mechanisms of conformational and colloidal instability in mAbs.
Miscellaneous Instabilities
Photo-oxidation is also a significant concern in the growing field of mAb therapeutics. Upon light exposure, pharmaceutical proteins may change their conformation. A study by Shah et al. reported that the exposure of mAbs to light as per the ICH guidelines leads to photo-oxidation. This is due to the destabilization of two methionine groups on the CH2 domain, which is responsible for forming an aggregate [233]. Similarly, in a study conducted by Du et al., the presence of visible light impacted the stability of mAb therapeutics by the production of reactive oxygen species, leading to the oxidative degradation of tryptophan and methionine [234]. The presence of histidine-based buffers in the formulation promoted even more destabilization. Another investigation provided strong evidence that the formulation with histidine buffers increased hydrogen peroxide production due to photo-oxidation, resulting in instability [235]. The formation of oxidants in the formulation will affect conformational stability by disrupting hydrogen bonds with oxidative deamidation [236]. This promotes the exposure of hydrophobic regions, posing conformational instability. The study provides significant pieces of evidence comparing three commonly used oxidants, namely, hydrogen peroxide, tert-butyl hydroperoxide, and 2,2′-Azobis(2-amidinopropane) dihydrochloride (AAPH). The results showed that the presence of these oxidants significantly affected the stability of mAbs and promoted destabilization [220]. To mitigate oxidation-mediated instability issues, high-throughput oxidation profiling can be employed to evaluate the oxidation susceptibility of mAbs [219]. The formulation’s composition must be chosen carefully to avoid conformational and colloidal instabilities, as well as instabilities related to photochemical and mechanical stress.
2.4.2. Approaches for Overcoming These Instability Challenges
Buffering Agents
Despite several studies on understanding mAbs’ instabilities, buffer-specific effects are consistently disregarded. Buffer composition is a critical factor in maintaining the pH and ionic equilibrium in mAb formulations. Buffer components, upon hydration, adsorb on the surface of the mAb, promoting electrostatic stabilization [213]. Tweaking these electrostatic interactions will enhance the colloidal stability of proteins. Alongside this, buffer components function as a pseudo-substrate by facilitating component binding and proton transfer [237]. Nevertheless, conventional buffers such as phosphate and succinate buffers may not always be the right choice for all mAb formulations [238]. In a study, various buffer systems were identified as alternative buffers for the stabilization effect under freeze–thaw conditions [239]. The replacement of chloride-containing buffering agents with aspartate, glutamate, acetate, sugars, and glucuronate significantly improved conformational stability [240]. The stabilization effect is due to the light-absorbing nature of buffering agents that prevent hydrolysis by obstructing activation energy. These buffering agents preserve ionic strength and enhance surface stabilization through a charge shielding mechanism, thereby enhancing colloidal stability [238]. The mAbs formulations contain chains of individual amino acids such as aspartate (pKa 3.7), glutamate (pKa 4.3), and histidine (pKa 6.5). They potentially act as buffering agents in the pharmaceutically relevant range (pH 4.0 to 7.0) [241]. The combination of histidine (50mM concentration) and arginine in formulations provides characteristic shreds of evidence for stable mAbs. In this case, histidine provides buffer capacity at a pH below the pI of the antibody, while arginine promotes increased stability and protein integrity due to chaotropic effects [242]. The chaotropic effect reduces interference with the hydrogen bonding network between water and mAb molecules and develops more intra-structural protein interactions [243]. This unique property of amino acids, preventing colloidal instability over a wide range of pH values, paved the way to explore similar excipients like arginine and glutamic acid [244]. For example, histidine with pka 6.0 and buffering capacity synergistically works with carbohydrates (sucrose, trehalose), even in freeze-drying. The native conformation of proteins is stabilized by the preferential hydration nature of carbohydrates [245]. The use of di, tri carboxylic acids (citric acid, tartaric acid) enhances the thermal stability of antibodies during freeze-drying [246]. Excipient interactions also need to be considered, as the imidazole ring on histidine accelerates the catalytic degradation of polysorbates 20 or 80 through ester hydrolysis [247]. The accumulation of fatty acids as a byproduct may induce adverse immune reactions. Therefore, multiple approaches have been provided in the literature to stabilize mAbs. Preferential molecular interactions need to be understood for the stabilization of mAbs in formulations.
Surfactants
The interfacial adsorption of mAbs at liquid–gas and liquid–liquid interfaces may induce structural deformation, leading to undesired colloidal and conformational instabilities. The surfactants inhibit the adsorption of monoclonal antibodies at the air–water interfaces, which often results in elevated protein concentrations at these interfaces [248]. These elevated concentrations are accountable for colloidal instabilities. This surface adsorption reveals hydrophobic areas of proteins and may also result in conformational instability [249]. Surfactants engage with exposed hydrophobic areas, inhibiting protein absorption in solutions and at the air/water interfaces [250]. Non-ionic surfactants such as PS20, PS80, and P188 are found to be effective even at lower concentrations because of their better ability to replace mAbs for adsorption at the interfaces [251]. The interfaces might be between air and water or a solid glass surface and water. The surfactant stabilization mechanism involves the formation of a protective layer on the glass surface that prevents interactions with the vial surface [252]. This stabilization effect may be due to the competitive adsorption of surfactants at the air/water interfaces, forming a monomeric layer surrounding each protein molecule. This stabilization mechanism was proved using X-ray reflectivity measurements and surface tension measurements [253]. In contrast, the gelation of mAbs at the air/water interface will also contribute to the instability. This physical gelation will be further retarded by the addition of polysorbate above the critical micellar concentration [254]. Comprehending the aggregation tendencies of various mAb–surfactant interactions and comparing them with interfacial behavior can significantly enhance the understanding of the instability process and assist in reducing aggregate formation by optimizing surfactant type and concentration in the formulation [227]. Polysorbate 80 and polysorbate 20 are the predominant surfactants utilized for stabilizing proteins in formulations. Nonetheless, challenges related to surfactants, such as contamination, degradation, and the potential induction of undesirable immunological responses, have been observed [255].
Viscosity Modifiers
The high-concentration mAb formulations have the potential to have high intermolecular associations. Electrostatic, hydrophobic interactions with charged molecules will contribute to these instabilities [256]. It is important to establish control over the ratio of viscosity-modifying excipients with mAbs for protein stabilization. The addition of amines such as arginine, histidine, lysine, proline, and pyridoxamine can interact with hydrophobic patches of the mAbs surface via cation–π interactions [257]. This interaction covers the hydrophobic patches on mAbs, preventing protein–protein hydrophobic associations and lowering the viscosity [258]. Another study evaluated the effect of arginine monohydrochloride, proline, and lysine monohydrochloride on the viscosity and shear-thinning ability at different high and ultra-high mAb concentrations. The outcomes stated that formulation viscosity followed the trend of arginine > proline > lysine for lowering the viscosity [259]. In a study conducted by Dear et al. [260], viscosity reduction was achieved by reducing the pH using co-solutes (camphor sulfonic acid, imidazole) and cationic amino acids (arginine, histidine, lysine), each of which contains hydrophobic groups attributed to the weakening of local anisotropic electrical and hydrophobic attractions. These electrostatic and hydrophobic attractions are responsible for network formation with increasing viscosity [260]. Charge-based (electrostatic) interactions significantly influence molecules over greater distances than hydrophobic interactions, resulting in conformational instability [261]. Sodium chloride (150 mM concentration) can reduce the viscosity of a formulation by neutralizing protein charge, hence diminishing electrostatically induced protein–protein interactions [262]. In contrast with traditional salts, newer salts like ethanolamine-DPA and diethanolamine-DPA were identified for their viscosity-reducing properties by altering hydrophobic pockets of proteins. Also, small molecules like caffeine in mAb formulations significantly reduced their viscosity. The reduction in viscosity may be due to the protein interface binding of caffeine inhibiting short-range protein–protein interactions [263]. This concludes that the careful screening of newer excipients can achieve highly stable and highly concentrated mAb formulations. Table 4 represents some of the excipients studied to enhance the stability of mAb formulations.
Table 4.
Common and emerging excipients investigated for the stabilization of mAbs.
2.5. Fusion Proteins
Fusion proteins are engineered proteins created by joining two or more genes that originally coded for separate proteins, merging the functional domains from different proteins, and enhancing their specificity and therapeutic efficiency [273]. One of the classic examples is the Fc-fusion protein, also known as a chimeric protein, which associates the Fc region of an antibody with another protein, such as enzymes or receptors [274]. This addition improves the functional characteristics of the resulting hybrid protein with advanced pharmacokinetics and therapeutic perspectives (Figure 10). Currently, they are utilized across various therapeutic fields, including cancer therapies and autoimmune disorders, due to their integrated structure and assorted mechanisms of action. The flexibility of hybrid proteins has opened new avenues and bridged the gap between diverse biological functions [275,276,277,278,279,280,281].
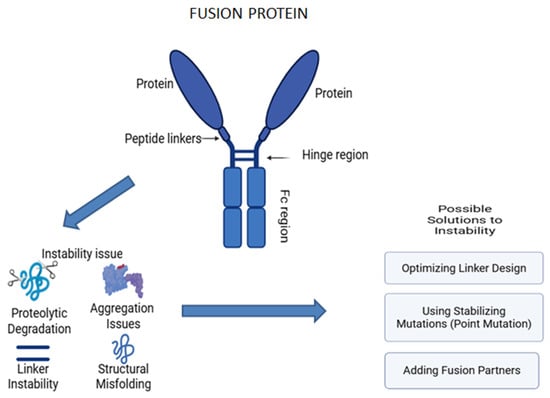
Figure 10.
Structure and instability issues associated with fusion protein.
Recombinant technology introduces monoclonal antibodies (mAbs) alongside fusion proteins. They are both interrelated in terms of their production approaches, structural components, and therapeutic functions. The mAbs are naturally occurring, highly specific, immune molecules with long, stable half-lives, whereas fusion proteins are engineered hybrid proteins with several functional domains, having flexibility and multi-functionality [282,283].
2.5.1. Instability of Fusion Proteins and Approaches to Address Stability Issues
Fusion proteins are hybrid proteins that are engineered by joining two or more proteins into one polypeptide chain. Fusion proteins and monoclonal antibodies (mAbs) have similar therapeutic applications, such as targeting specific molecules or pathways with great specificity and efficacy. However, both types of biologics encounter stability issues during production and storage, such as aggregation and structural instability. Both have comparable stabilizing mechanisms, which rely on common excipients to prevent aggregation, maintain proper folding, and enhance colloidal stability [18,281]. These instability issues arise due to heterogeneity, as fusion proteins are created through the combination of different protein domains, allowing them to target specific molecules or pathways. However, this non-natural combination can result in instability, potentially leading to aggregation. Achieving proper folding for each component in fusion proteins is a significant challenge, as combining domains that do not naturally occur together can lead to instability, such as aggregation [284,285]. The major factors that cause stability issues with fusion proteins are aggregation and protein structure breakdown, both chemically and during the storage process.
Aggregation
Fusion proteins contain multiple domains with variable physicochemical properties. The fusion of different domains can significantly disrupt the folding patterns. The hydrophobic regions of the fusion proteins are also exposed, causing hydrophobic interactions that lead to intermolecular interactions, facilitating aggregations of the proteins. Alterations in environmental factors like pH, stress, temperature, and concentration directly affect the stability of the protein. One of the effective ways to eliminate aggregation is to genetically fuse stabilizing peptides while preparing the fusion protein, as these maintain proper folding and mask the hydrophobic regions, thereby reducing the aggregation [286]. The addition of suitable sugars such as trehalose, mannitol, or amino acids like glycine helps in stabilizing the structural conformity of proteins by forming a protective layer [287,288].
Proteolytic Degradation
Fusion proteins that link the domains via flexible linkers are more sensitive to proteolytic degradation, which affects the therapeutic activity and leads to unwanted protein loss and dysfunction. Fusion proteins, during production in host cells, can be a prime target for endogenous proteases that cleave specific peptide bonds, causing the formation of non-functional proteins. The improper folding in the fusion protein makes it more susceptible to degradation by the ubiquitin–proteasome system. The utilization of genetically modified host cells that lack or have reduced specific protease activity can be a way to minimize proteolytic damage. The addition of protease inhibitors like MG132 during cell cultures will also prevent degradation by inhibiting the host-cell-derived proteases. Mutating the recognition sites of the known proteases in the fusion protein can prevent degradation, offering the structural stability of the fusion protein [281,289,290,291].
Instability During Storage
During the storage process, fusion proteins, especially in liquid formulations, are unstable due to multiple freeze–thaw cycles, temperature fluctuations, and inappropriate buffer conditions. This leads to unfolding, aggregation, or degradation over time, leading to a loss in protein efficacy. Therefore, lyophilization can be an effective way to tackle these instability issues [4]. The removal of water content provides a lower risk of microbial growth and degradation. Utilizing various cryoprotectants like mannitol, sucrose, or trehalose provides a protective glassy matrix to the formulation, preventing further damage [281]. Understanding the mechanism behind the instability issue can aid in choosing the proper targeting strategies to enhance the stability of the fusion protein, thereby increasing the therapeutic efficacy [291,292].
2.6. Antibody–Drug Conjugates (ADCs)
Antibody–drug conjugates are characterized as targeted therapeutics, sustaining a steady increase in the biopharmaceutical sector due to the increasing requirement for combination therapies. These ADCs exhibit stability concerns related to storage and in vivo conditions. The instabilities arise from high-dose formulations and combination conjugates developed by attaching highly potent small molecules to mAbs. The first ADC developed was Mylotarg (gemtuzumab ozogamcin), approved in the year 2000 for the treatment of myeloid leukemia. Moreover, 15 FDA-approved ADCs are currently available on the market up to October 2023, and several hundreds are in clinical trials [293,294]. A typical ADC will have an mAb and a payload molecule linked with a chemical linker. The payloads are 100–1000 times more potent than small molecules, can be termed “warheads“, and are the final effector component of ADCs. These payloads are inhibiting transcription, targeting DNA (duocarmycins, calicheamicins, pyrrolobenzodiazepines, irinotecan), or inhibiting tubulin (auristatin monomethyl auristatin E, monomethyl auristatin F) or maytansinoids [295]. The prerequisites for selecting these payloads include solubility, stability, high conjugation ability, and high potency. The high potency reflects that the payload shows its cytotoxicity in nanomolar concentrations. The payload is designed to show its targeting toxicity inside the cells with a low immunogenicity and a longer half-life [296,297]. Thus, the payload should be stable and intact until it reaches the site and during cell uptake.
The payload molecules interact chemically with the linkers (cleavable and non-cleavable linkers). The cleavable linkers release the payload molecules at the tumor sites, which is mediated by the pH conditions (hydrazone linkers), glutathione concentration (disulfide linkers), and other enzymatic cleavable linkers. The other class of linkers is non-cleavable, which is only susceptible to releasing the payload molecule after enzymatic degradation of the antibody in the vicinity of the cell [298]. Examples of ADCs utilizing the cleavable mechanism include brentuximab vedotin, polatuzumab vedotin, and enfortumab vedotin with a pH-sensitive hydrazone linker. Conversely, non-cleavable linkers resist cleavage and stay intact even when attached to the lysosome. This lysozyme promotes protein degradation and releases payload molecules via cathepsin B or plasmin-mediated mechanisms. T-DM1 and mafodotin belantamab are examples of FDA-approved ADCs utilizing non-cleavable linkers [299,300].
2.6.1. Instability in ADCs
Colloidal instability is one of the attributes caused by the self-aggregation of ADCs with the alteration of net charge, pI, and local charge distribution. Bioconjugation can alter the native charge on the surface of the protein when cross-linked with a linker and a payload [301]. These factors reduce the solubility of the mAbs in aqueous media, increasing the viscosity of the formulation [302]. In such formulations, long-term stability is impacted by a decrease in the repulsive forces between adjacent ADCs after conjugation. Multiple studies have investigated the impact of formulation buffers, pH, and salts influencing ionic strength in stability [303,304]. These studies provide the effects of changes in solution conditions that may influence ADCs’ stability. For instance, the addition of 0.9% NaCl retarded the colloidal stability of trastuzumab with maytansinoids’ DM1 conjugate. The instability was reported due to variations in electrostatic interactions [305].
Apart from physical stability, chemical stability is crucial to maintaining the efficacy of ADCs. The first-generation ADCs like Adcetris and Mylotarg are less stable. The stability of these ADCs is affected by the properties of the conjugation chemistry of the linker-toxin, along with the conjugation site. To mitigate these issues, second-generation ADCs like Kadcyla and Polivy were developed. The focus was on site-specific conjugation chemistry to promote homogeneity [306] and maintain linker–payload hydrophobicity. In ADCs, aggregation and fragmentation occur under environmental stress and high ionic strength environments similar to traditional mAbs. Apart from prior factors, linker stability and the breakdown of thiol bonds via conjugation pose additional instability issues [307]. Bioanalytically, instability is characterized by using various spectroscopic and physical techniques, including circular dichroism, UV-Vis, fluorescence, and dynamic light scattering. Furthermore, Differential Scanning Calorimetry (DSC) has proven to be crucial in comprehending the stability of ADCs. Size Exclusion Chromatography (SEC), hydrophobic interaction chromatography (HIC) with Ultraviolet (UV), or Multi-Angle Light Scattering (MALS) detection are necessary techniques for monitoring the existence of aggregation [308]. The antibody, linker, and payload contribute to the stability of the entire ADCs.
Stability Aspects with Payloads of ADCs
The hydrophobicity and charge of the payload will affect the stability of ADCs during synthesis and storage, restraining the shelf life of the formulated ADCs. They are impacted by the number of payload molecules attached to the antibody, which is called the drug-to-antibody (DRA) ratio. Guo et al. studied how a higher DRA with six payload molecules increased the hydrophobicity of the ADCs compared to DRA2-DRA4 and unconjugated antibodies in cysteine-based conjugation [308]. Another study conducted by Buecheler et al. [309] determined that the increase in DRA simultaneously retarded the stability of ADCs. The same study correlated the effect of the log p value of the payload molecule on aggregation by in silico modeling [309,310]. Excess conjugation with more payloads leads to the removal of disulfide bonds and a further decrease in conformational stability [311,312]. To mitigate this stability, altering the chemical structure of the payload molecule can also give positive outcomes, as the study by Johann et al. demonstrated that the negatively charged payload could exacerbate instabilities in final ADCs compared to neutral payload molecules [310]. The conjugation of relatively hydrophobic drugs to the native lysine/cysteine residues of antibodies enhances their aggregation rates. This aggregation occurs by a reduction in electrostatic repulsions and by promoting hydrophobic protein–protein interactions, leading to conformational instabilities. These interactions can be modulated by altering the ionic strength, temperature, or the presence of surfactants in the formulations [305]. In some cases, the payload imposes stability; in a case study, a developed maytansinoid-based payload showed less hydrophobicity than MMAE-based payloads. However, regardless of the drug linker and DARs, the stability was decreased with the type of payload employed [313,314].
Stability Aspects with Linkers in ADCs
The linker is an often-disregarded component of ADCs, which has the same importance as the payload and mAbs. The linker binds the cytotoxic payload to the antibody and ensures the stability of the ADCs, facilitating the targeted release of the payload within the tumor cell. As hydrophobic drugs (payloads) pose instabilities, there are also similar concerns in regard to linkers and antibodies. The hydrophobicity of linkers leads to the faster renal clearance of ADCs in vivo. Also, by including a hydrophobic component as a linker, the polar surface area can be decreased, hence increasing its susceptibility to colloidal association [15,315]. For example, when highly hydrophobic molecules like PBD (pyrrole benzodiazepine) are conjugated to design ADCs, they can prevent the polar areas of the antibody from interacting in a solution state and cause a loss of colloidal stability. This depends on the nature of the hydrophobicity of the linker as well as the conjugate. Moreover, the process of attaching hydrophobic components might also result in alterations in solubility [308]. PEG derivatives are among the most used linkers in targeted therapy, offering a variety of features like reducing hydrophobicity and pH regulation. Such improvements in solubility are found with the conjugation of the toxin SN38, which enhanced the solubility of ADCs by up to 23-fold due to the presence of four PEG arms [316]. Glycols provide a range of PEG linkers to assist in developing ADCs, such as PEGylated ADCs. All PEG linkers possess a purity level of over 95% and serve as the fundamental components for developing a successful ADC [317]. Two recently approved ADCs, Trodelvy and Zynlonta, were designed with a PEG moiety incorporated into their linker technology to enhance their solubility and stability [318]. In the context of PEG-based linkers, a study was conducted by AstraZeneca to stabilize ADCs with the pegylation of PBD (pyrrole benzodiazepine) using MV- MV-modified molecules. This enabled the hydrophilicity to PBD (binary complex) at the C2 position and was compared against a conventional ADC (T-C239i−SG3249), which was previously reported as being highly potent and stable. The dimerized form of macrocycle CB-8 with PEG showed more stability compared to the ADC (T-C239i−SG3249). This suggests the necessity of PEG linkers in maintaining hydrophilicity by hindering the hydrophobic pocket of the ADCs [319,320]. Another study explored the use of PEG linkers for the preparation of highly hydrophilic lysine conjugates. The comparison was made between the wide range of PEG linkers, i.e., PEG12 (MAP12PS or MAP12NP) and the PEG24 linear linker (MAL24PS). The PEG-modified linker with 12 units provided the optimum hydrophilic load for conjugation, which retarded the aggregation tendency compared to marketed Kadcyla® (24 PEG units) [321]. The addition of other hydrophilic linkers like glutamic acid, disulfide linkers, and the val-cit-PAB [322] linker, and the placement and arrangement of PEG units should be carefully tuned to achieve ADCs with improved stability and pharmacokinetics.
Stability Aspects Concerning Conjugation Sites in ADCs
The structure and stability can be correlated with the site of conjugation for maintaining the intrinsic strength of ADCs. Eliminating a solitary interchain disulfide link results in substantial reductions in conformational stability. The site-specific cysteine conjugation in the light chain using the pyrrolobenzodiazepine linker and hydrogen-exchange mass spectrometry (HX-MS) show increased backbone stability near the conjugation site [323]. Conjugation strategies at glycated sites of antibodies may potentially improve the in vivo efficacy and strength of ADCs in animal models [324]. Also, thermal stability is influenced by site-specific conjugation, as explained by a study conducted by Kaempf et al. [325], which revealed that conjugation on the site of a highly stable CH3 domain did not lead to any conformational instability. In contrast, conjugation at the C-terminus close to the CH2 domain at L328C and T-S239C seemed to have a strong impact on the thermal stability of secukinumab [325]. Another study revealed that the lysine conjugation of trastuzumab at the CH2 domain had significantly retarded thermal stability. The stability was impacted by the increased activation energy at the CH2 domain with thermal stress [326]. It can be explained that the disulfide bonds present in the CH2 domain are responsible for partial unfolding, leading to a high aggregation propensity of ADCs. Also, the increased hydrophobic contacts and charge neutralization at these sites favor the colloidal instability of ADCs. The study was conducted using different types of mAbs targeting the effects of the conjugation site and linker on aggregation. The results suggested that the colloidal stability was influenced by the hydrophobic linker, and that the conformational stability was influenced by the conjugation site. The breakdown of a single interchain disulfide link resulted in substantial conformational instabilities of the ADC itself. The production of conjugates with a higher drug-to-antibody ratio (DAR) will further lead to the elimination of more interchain disulfide bonds and a further decrease in conformational stability, as discussed earlier [311]. To mitigate these issues, the hydrophobicity of the linker and payload conjugation can be optimized inside the buried regions of the 3D protein structure. This could be called a “Trojan horse” in the Fab region of mAbs [327]. Apart from physical instability, in vivo stability is also required to prevent the breakdown of payload and antibodies through site-specific conjugation. Transglutaminase-catalyzed isopeptide linkage showed enhanced stability in in vivo mouse models; this report specifically provided the stability of auristatin-based payloads attached to the specific site [328].
2.6.2. Approaches to Overcome Instabilities
Altering the molecular nature of the antibody can increase the stability of ADCs by reducing hotspots for the chemical degradation of proteins. As mentioned earlier, site-specific conjugation has remained a pioneering approach to overcoming instability. Enhancing the glycation can potentially increase the thermal stabilization of antibodies when the payloads are attached to the C’E loop at the Fc CH2 domain [326]. Due to glycation, the masking effect observed by carbohydrate aromatic interactions with the payload at the CH2 domain enhances thermal stability [329]. Further, the data reveal a correlation between the site of conjugation, the Fc glycosylation site, and the position of the glycan with the stability of the ADC [330]. The site-directed mutagenesis and molecular dynamics simulations demonstrate that the change in Lys 207 aids the stability of the ADCs. For example, Lys 207 in trastuzumab (thiomab Tras-LCV205C). This is crucial for stabilizing linkers with acid-sensitive groups like acetals, which promote the oxidation of succinimide groups in linkers. This modification is time-consuming but very effective, and modifying the specific composition of proteins near the conjugation site can enhance the stability of ADCs [331].
The instability of linker conjugates is a major concern, as it retards the in vivo efficacy of ADCs with off-target toxicity. This off-target toxicity is due to the linker breakdown in physiological conditions, i.e., pH, isotonicity, etc. The stability is attained by modifying the linker chemistry and developing novel linkers, such as in a study which reported DNA linker-based ADCs developed with trastuzumab. The antibody was conjugated with a 37-mer oligonucleotide (ON) modified at the 5-end with MMAE (cON-MMAE) to obtain trastuzumab–DNA–MMAE [332]. The developed conjugate showed complete solubility in aqueous media and decreased the overall hydrophobicity of the toxic MMAE payload. The findings suggested an increase in cytotoxicity of trastuzumab–DNA–MMAE on HER2-negative cells, even in picogram concentrations. The conjugate remained intact for 5 days when incubated in simulated human plasma at 37 °C [332].
A study by Song et al. performed an N-terminal selective conjugation and showed that the conjugate was more stable than the thiol and lysine conjugation of the trastuzumab ADC. The N-terminal antibody conjugate was more stable in vitro and in vivo with comparatively high efficacy and low systemic toxicity [333]. Similarly, a thiol-linked antibody–drug conjugate using maleamic methyl ester was developed using cysteine-based conjugation. This method improved the in vivo stability of the ADC compared with the traditional maleimide containing unstable closed-ring thiosuccimide. Despite marketed formulations such as Adcetris, Polivy, and Padcev being developed with the traditional maleimide linking method, the former approach could be a necessary alternative for next-generation ADCs [334]. The marked in vivo stability of the linker was due to the open ring, the other group of MMAE. Furthermore, albumin solution stability was determined for the maleamic methyl ester linker (ADC mil40-12c) and the traditional marketed linker (ADC mil40-12c’) in a physiological environment. About 3.8% of the ADC mil40-12c payload was shredded compared to the traditional ADC mil40-12c, which was as high as 13.3% in 14 days. These studies provide necessary insights for developing novel linkers and linker-based chemistry to mitigate stability issues. It is important to note that among the 13 approved ADCs, 11 use cleavable linkers and 7 use peptide-based valine-citrulline-p-amino benzyloxycarbonyl (VCit-PABC) for payload delivery. Similarly, glutamic acid–valine–citrulline linkers have been explored to prevent premature cleavage in circulation as compared with valine–citrulline; the modulation in the linker gave this positive outcome [310]. This shows the importance of linkers in the stability of ADCs in vivo, and a further few linker-based stabilizations are summarized in Table 5.
Table 5.
Various types of linkers used in the literature with their stability mechanisms in vivo.
The wide range of protein structural diversities creates the need for a distinct formulation approach. When it comes to ADCs, finding the best formulation depends mainly on linker chemistry, payload properties, and conjugation at specific sites (Figure 11). As the delivery route of ADCs is mostly via IV infusions, buffer agents and the process of lyophilization are the key aspects. The excipients, such as NaCl, dextrose, Ringer’s lactate, sodium phosphate, succinic acid, citrate, TRIS, sodium citrate, and acetate, etc., are used as diluents and buffering agents for commercial products (Bexxar™, Zevalin™, Ontak™) [341]. Developing high-concentration ADCs will have a lower risk of aggregation, because as the concentration increases, the proximity effect retards solubility with colloidal association. As the marketed IV bags are diluted before administration, this causes a decrease in stabilizer levels. The development of necessary administration techniques will eliminate these problems [342]. The excipient-mediated approach still needs to be significantly developed for ADCs, as only limited research is available. Modified excipients can exhibit enhanced physical stability and prevent conformational and colloidal instability [343]. Linker-specific stabilization can be employed for maintaining the linker drug and mAbs association in vivo (Table 5). The stability considerations with mAbs may also apply to ADCs because of their structural similarities, which were emphasized in the previous sections. Hence, there is a need for developing formulation-related approaches specific to ADCs to expedite the stability of ADCs.
Figure 11.
Strategies to improve the stability of ADCs.
3. Conclusions
Advanced biotherapeutics stand out as revolutionary developments, constantly changing the healthcare environment. Many diseases that were previously believed to have few treatment options may now be potentially treated with these therapies. Stability considerations for advanced biotherapeutics are complex and require phase-appropriate and matrix stability strategies owing to the limited batch sizes. A broad range of analytical tests and characterizations are crucial to determine and demonstrate the product quality profile of these biotherapeutics throughout their shelf life. Storage and handling, particularly for cells and vectors, requires additional efforts to maintain viability and activity. The presence of residual impurities such as host cell proteins and DNA affect the stability of the complex proteins (vectors and mAbs), requiring the optimization of purification strategies to enable long-term storage. Cell and gene therapy products are more sensitive to stress conditions than conventional biologics such as mAbs, making scaling up and manufacturing difficult. The growing number of approvals and the increased use of existing therapies are promising for the development of novel, advanced biotherapeutic products. Overall, considering the developments in the last decade and ongoing efforts, these advanced biotherapeutics can treat various diseases in a safe and effective way to address the patient’s unmet needs. This review highlights the major stability challenges and potential solutions for these therapeutics. While significant progress has been made, future research should focus on developing more robust delivery systems and stabilizing agents to further enhance shelf life and therapeutic performance. For instance, employing nanocarriers and advanced excipients could improve the stability of mRNA and protein-based therapies.
Author Contributions
S.R.P. and S.B. contributed by writing the original draft of sections including Antibody Drug Conjugates, Monoclonal Antibodies, and Fusion Proteins, and editing the figures; S.S. contributed by writing the original draft of sections including Introduction, Cell Therapy, and Conclusion; V.V. contributed by writing the original draft of sections including Gene Therapy and editing the figures; A.T. contributed by writing the original draft of sections including mRNA-based Therapies and editing the figures; P.K.B., P.K.N. and A.B. contributed to the conceptualization, supervision, editing, and reviewing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
Author Vartika Verma is employed by the company Lifecare Innovations Private Limited, and Sandeep Bolla is an employee of Fortrea. This affiliation did not influence the design, execution, or interpretation of the study.
Abbreviations
The following abbreviations are used in this manuscript:
| 3′ UTRs | 3′ untranslated regions of mRNA |
| AAV | Adeno-associated virus vectors |
| ADCs | Antibody drug conjugates |
| CAR-T | Chimeric antigen receptor T cell therapy |
| CH2 | Second domain of heavy chain constant regions of mAbs |
| DNA | Deoxyribonucleic acid |
| DAR | Drug to antibody ratio |
| Fc | Fragment crystallizable |
| FFAs | Free fatty acids |
| HPβCD | 2-hydroxypropyl-β-cyclodextrin |
| HSV | Herpes simplex virus vectors |
| IgG | Immunoglobulin G |
| LNPs | Lipid nanoparticles |
| LTRs | Long terminal repeats |
| mAbs | Monoclonal antibodies |
| MMAE | Monomethyl auristatin E |
| PEG | Polyethylene glycol |
| RBPs | RNA-binding proteins |
| RNA | Ribonucleic acid |
| mRNA | Messenger ribonucleic acid |
References
- Phyo, P.; Zhao, X.; Templeton, A.C.; Xu, W.; Cheung, J.K.; Su, Y. Understanding Molecular Mechanisms of Biologics Drug Delivery and Stability from NMR Spectroscopy. Adv. Drug. Deliv. Rev. 2021, 174, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.L. Biologic Therapies: What and When? J. Clin. Pathol. 2007, 60, 8–17. [Google Scholar] [CrossRef]
- FDA. What Are “Biologics” Questions and Answers. Available online: https://www.fda.gov/about-fda/center-biologics-evaluation-and-research-cber/what-are-biologics-questions-and-answers (accessed on 8 April 2025).
- Akbarian, M.; Chen, S.-H. Instability Challenges and Stabilization Strategies of Pharmaceutical Proteins. Pharmaceutics 2022, 14, 2533. [Google Scholar] [CrossRef] [PubMed]
- IDBS. Challenges in Formulating Biologics. Available online: https://www.idbs.com/2019/07/challenges-in-formulating-biologics/ (accessed on 12 February 2025).
- Zu, H.; Gao, D. Non-Viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021, 23, 78. [Google Scholar] [CrossRef]
- El-Kadiry, A.E.-H.; Rafei, M.; Shammaa, R. Cell Therapy: Types, Regulation, and Clinical Benefits. Front. Med. 2021, 8, 756029. [Google Scholar] [CrossRef]
- Meneghel, J.; Kilbride, P.; Morris, G.J. Cryopreservation as a Key Element in the Successful Delivery of Cell-Based Therapies—A Review. Front. Med. 2020, 7, 592242. [Google Scholar] [CrossRef]
- Madhav, V. Advanced Drug Delivery Systems for Biologics. Am. J. Adv. Drug Deliv. 2024, 12, 231–261. [Google Scholar]
- Nature. Advances in the Stability and Delivery of mRNA Therapeutics. Available online: https://www.nature.com/articles/d42473-020-00426-z (accessed on 12 February 2025).
- Schuster, J.; Mahler, H.-C.; Joerg, S.; Kamuju, V.; Huwyler, J.; Mathaes, R. Stability of Monoclonal Antibodies after Simulated Subcutaneous Administration. J. Pharm. Sci. 2021, 110, 2386–2394. [Google Scholar] [CrossRef]
- Shastry, M.; Gupta, A.; Chandarlapaty, S.; Young, M.; Powles, T.; Hamilton, E. Rise of Antibody-Drug Conjugates: The Present and Future. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e390094. [Google Scholar] [CrossRef]
- Adem, Y.T. Physical Stability Studies of Antibody-Drug Conjugates (ADCs) Under Stressed Conditions. Antib.-Drug Conjug. Methods Protoc. 2020, 2078, 301–311. [Google Scholar] [CrossRef]
- Nejadmoghaddam, M.-R.; Minai-Tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.-H. Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna J. Med. Biotechnol. 2019, 11, 3–23. [Google Scholar] [PubMed]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody-Drug Conjugates: Recent Advances in Linker Chemistry. Acta Pharm. Sin. B 2021, 11, 3889–3907. [Google Scholar] [CrossRef]
- Fusion Protein—An Overview|ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/fusion-protein (accessed on 12 February 2025).
- Kulakova, A.; Indrakumar, S.; Sønderby Tuelung, P.; Mahapatra, S.; Streicher, W.W.; Peters, G.H.J.; Harris, P. Albumin-Neprilysin Fusion Protein: Understanding Stability Using Small Angle X-Ray Scattering and Molecular Dynamic Simulations. Sci. Rep. 2020, 10, 10089. [Google Scholar] [CrossRef] [PubMed]
- Rahban, M.; Ahmad, F.; Piatyszek, M.A.; Haertlé, T.; Saso, L.; Saboury, A.A. Stabilization Challenges and Aggregation in Protein-Based Therapeutics in the Pharmaceutical Industry. RSC Adv. 2023, 13, 35947–35963. [Google Scholar] [CrossRef]
- Friedmann, T.; Roblin, R. Gene Therapy for Human Genetic Disease? Science 1972, 175, 949–955. [Google Scholar] [CrossRef]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of Gene Therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef]
- Hanna, E.; Rémuzat, C.; Auquier, P.; Toumi, M. Gene Therapies Development: Slow Progress and Promising Prospect. J. Mark. Access Health. Policy 2017, 5, 1265293. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Veroniaina, H.; Su, N.; Sha, K.; Jiang, F.; Wu, Z.; Qi, X. Applications and Developments of Gene Therapy Drug Delivery Systems for Genetic Diseases. Asian J. Pharm. Sci. 2021, 16, 687–703. [Google Scholar] [CrossRef]
- Butt, M.H.; Zaman, M.; Ahmad, A.; Khan, R.; Mallhi, T.H.; Hasan, M.M.; Khan, Y.H.; Hafeez, S.; Massoud, E.E.S.; Rahman, M.H.; et al. Appraisal for the Potential of Viral and Nonviral Vectors in Gene Therapy: A Review. Genes 2022, 13, 1370. [Google Scholar] [CrossRef]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V. Gene Therapy Leaves a Vicious Cycle. Front. Oncol. 2019, 9, 297. [Google Scholar] [CrossRef]
- Shahryari, A.; Saghaeian Jazi, M.; Mohammadi, S.; Razavi Nikoo, H.; Nazari, Z.; Hosseini, E.S.; Burtscher, I.; Mowla, S.J.; Lickert, H. Development and Clinical Translation of Approved Gene Therapy Products for Genetic Disorders. Front. Genet. 2019, 10, 868. [Google Scholar] [CrossRef]
- Ma, C.-C.; Wang, Z.-L.; Xu, T.; He, Z.-Y.; Wei, Y.-Q. The Approved Gene Therapy Drugs Worldwide: From 1998 to 2019. Biotechnol. Adv. 2020, 40, 107502. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.J. Development of Optimized Vectors for Gene Therapy. Proc. Natl. Acad. Sci. USA 1999, 96, 324–326. [Google Scholar] [CrossRef]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Brown, A.M.; Jenkins, C.; Campbell, K. Viral Vector Systems for Gene Therapy: A Comprehensive Literature Review of Progress and Biosafety Challenges. Appl. Biosaf. 2020, 25, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.-E.S.; Gram, G.J. Viral vectors for clinical gene therapy. Ugeskr. Laeger. 2002, 164, 4272–4276. [Google Scholar]
- Ehrke-Schulz, E.; Zhang, W.; Schiwon, M.; Bergmann, T.; Solanki, M.; Liu, J.; Boehme, P.; Leitner, T.; Ehrhardt, A. Cloning and Large-Scale Production of High-Capacity Adenoviral Vectors Based on the Human Adenovirus Type 5. J. Vis. Exp. 2016, 107, e52894. [Google Scholar] [CrossRef]
- Tatsis, N.; Ertl, H.C.J. Adenoviruses as Vaccine Vectors. Mol. Ther. 2004, 10, 616–629. [Google Scholar] [CrossRef]
- Afkhami, S.; Yao, Y.; Xing, Z. Methods and Clinical Development of Adenovirus-Vectored Vaccines against Mucosal Pathogens. Mol. Ther. Methods Clin. Dev. 2016, 3, 16030. [Google Scholar] [CrossRef]
- Lee, T.F. Gene Therapy. In Gene Future: The Promise and Perils of the New Biology; Lee, T.F., Ed.; Springer: Boston, MA, USA, 1993; pp. 127–163. ISBN 978-1-4899-2760-6. [Google Scholar]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zheng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, H.; An, Y.; Chen, Z. Construction and Application of Adenoviral Vectors. Mol. Ther.-Nucleic Acids 2023, 34, 102027. [Google Scholar] [CrossRef]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.; Wilson, J.M.; Batshaw, M.L. Fatal Systemic Inflammatory Response Syndrome in a Ornithine Transcarbamylase Deficient Patient Following Adenoviral Gene Transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Tazawa, H.; Kagawa, S.; Fujiwara, T. Advances in Adenovirus-Mediated P53 Cancer Gene Therapy. Expert Opin. Biol. Ther. 2013, 13, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Keiser, M.S.; Davidson, B.L. Viral Vectors for Gene Transfer. Curr. Protoc. Mouse Biol. 2018, 8, e58. [Google Scholar] [CrossRef]
- Uprety, T.; Wang, D.; Li, F. Recent Advances in Rotavirus Reverse Genetics and Its Utilization in Basic Research and Vaccine Development. Arch. Virol. 2021, 166, 2369–2386. [Google Scholar] [CrossRef]
- Biasco, L.; Baricordi, C.; Aiuti, A. Retroviral Integrations in Gene Therapy Trials. Mol. Ther. 2012, 20, 709–716. [Google Scholar] [CrossRef]
- Lundstrom, K. Viral Vectors in Gene Therapy: Where Do We Stand in 2023? Viruses 2023, 15, 698. [Google Scholar] [CrossRef]
- Barquinero, J.; Eixarch, H.; Pérez-Melgosa, M. Retroviral Vectors: New Applications for an Old Tool. Gene Ther. 2004, 11 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Kim, W.-J.; Cho, Y.-H.; Lee, Y.-I.; Lee, H.; Jeong, S.; Cho, E.-S.; Chang, S.-I.; Moon, S.-K.; Kang, B.-S.; et al. Cancer Gene Therapy Using Adeno-Associated Virus Vectors. Front. Biosci. 2008, 13, 2653–2659. [Google Scholar] [CrossRef]
- Viral Vectors and AI Within Gene Therapy. Available online: https://www.formbio.com/blog/viral-vectors-gene-therapy (accessed on 14 February 2025).
- Burton, E.A.; Fink, D.J.; Glorioso, J.C. Gene Delivery Using Herpes Simplex Virus Vectors. DNA Cell Biol. 2002, 21, 915–936. [Google Scholar] [CrossRef]
- Jacobs, A.; Breakefield, X.O.; Fraefel, C. HSV-1-Based Vectors for Gene Therapy of Neurological Diseases and Brain Tumors: Part II. Vector Systems and Applications. Neoplasia 1999, 1, 402–416. [Google Scholar] [CrossRef]
- Verlengia, G.; Miyagawa, Y.; Ingusci, S.; Cohen, J.B.; Simonato, M.; Glorioso, J.C. Engineered HSV Vector Achieves Safe Long-Term Transgene Expression in the Central Nervous System. Sci. Rep. 2017, 7, 1507. [Google Scholar] [CrossRef] [PubMed]
- Vickram, A.S.; Manikandan, S.; Richard, T.; Lakshmi, S.V.; Chopra, H. Targeted Gene Therapy: Promises and Challenges in Disease Management. J. Bio-X Res. 2024, 7, 81–89. [Google Scholar] [CrossRef]
- Torchilin, V.P.; Omelyanenko, V.G.; Lukyanov, A.N. Temperature-Dependent Aggregation of pH-Sensitive Phosphatidyl Ethanolamine-Oleic Acid-Cholesterol Liposomes as Measured by Fluorescent Spectroscopy. Anal. Biochem. 1992, 207, 109–113. [Google Scholar] [CrossRef]
- Ogris, M.; Steinlein, P.; Kursa, M.; Mechtler, K.; Kircheis, R.; Wagner, E. The Size of DNA/Transferrin-PEI Complexes Is an Important Factor for Gene Expression in Cultured Cells. Gene Ther. 1998, 5, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.L.; Gøtzsche, C.R.; Woldbye, D.P.D. Current and Future Prospects for Gene Therapy for Rare Genetic Diseases Affecting the Brain and Spinal Cord. Front. Mol. Neurosci. 2021, 14, 695937. [Google Scholar] [CrossRef]
- Jang, M.; Yeom, K.; Han, J.; Fagan, E.; Park, J.-H. Inhalable mRNA Nanoparticle with Enhanced Nebulization Stability and Pulmonary Microenvironment Infiltration. ACS Nano 2024, 18, 24204–24218. [Google Scholar] [CrossRef] [PubMed]
- Pasika, S.; Bulusu, R.; Balaga, V.K.R.; Kommineni, N.; Bolla, P.K.; Kala, S.G.; Godugu, C. Nanotechnology for Biomedical Applications. In Nanomaterials: Advances and Applications; Springer Nature: Singapore, 2023; pp. 297–327. ISBN 978-981-19796-2-0. [Google Scholar]
- Dimov, N.; Kastner, E.; Hussain, M.; Perrie, Y.; Szita, N. Formation and Purification of Tailored Liposomes for Drug Delivery Using a Module-Based Micro Continuous-Flow System. Sci. Rep. 2017, 7, 12045. [Google Scholar] [CrossRef]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Grit, M.; Underberg, W.J.M.; Crommelin, D.J.A. Hydrolysis of saturated soybean phosphatidylcholine in aqueous liposome dispersions. J. Pharm. Sci. 1993, 82, 362–366. [Google Scholar] [CrossRef]
- Giles, A.R.; Sims, J.J.; Turner, K.B.; Govindasamy, L.; Alvira, M.R.; Lock, M.; Wilson, J.M. Deamidation of Amino Acids on the Surface of Adeno-Associated Virus Capsids Leads to Charge Heterogeneity and Altered Vector Function. Mol. Ther. 2018, 26, 2848–2862. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.B.; Chen, M.; Chen, C.-K.; Pfeifer, B.A.; Jones, C.H. Overcoming Gene-Delivery Hurdles: Physiological Considerations for Nonviral Vectors. Trends Biotechnol. 2016, 34, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Chen, Y.Y.; Spencer, M.J. Successes and Challenges in Clinical Gene Therapy. Gene Ther. 2023, 30, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Anchordoquy, T.J.; Girouard, L.G.; Carpenter, J.F.; Kroll, D.J. Stability of Lipid/DNA Complexes during Agitation and Freeze-Thawing. J. Pharm. Sci. 1998, 87, 1046–1051. [Google Scholar] [CrossRef]
- Bee, J.S.; Zhang, Y.; Finkner, S.; O’Berry, K.; Kaushal, A.; Phillippi, M.K.; DePaz, R.A.; Webber, K.; Marshall, T. Mechanistic Studies and Formulation Mitigations of Adeno-Associated Virus Capsid Rupture During Freezing and Thawing: Mechanisms of Freeze/Thaw Induced AAV Rupture. J. Pharm. Sci. 2022, 111, 1868–1878. [Google Scholar] [CrossRef]
- CD Creative Diagnostics. AAV Gene Therapy Manufacturing Process. Available online: https://www.creative-diagnostics.com/aav-gene-therapy-manufacturing-process.htm (accessed on 15 February 2025).
- Trevisan, B.; Morsi, A.; Aleman, J.; Rodriguez, M.; Shields, J.; Meares, D.; Farland, A.M.; Doering, C.B.; Spencer, H.T.; Atala, A.; et al. Effects of Shear Stress on Production of FVIII and vWF in a Cell-Based Therapeutic for Hemophilia A. Front. Bioeng. Biotechnol. 2021, 9, 639070. [Google Scholar] [CrossRef]
- Lengler, J.; Gavrila, M.; Brandis, J.; Palavra, K.; Dieringer, F.; Unterthurner, S.; Fuchsberger, F.; Kraus, B.; Bort, J.A.H. Crucial Aspects for Maintaining rAAV Stability. Sci. Rep. 2024, 14, 27685. [Google Scholar] [CrossRef]
- Nagy, A.; Chakrabarti, L.; Kurasawa, J.; Mulagapati, S.H.R.; Devine, P.; Therres, J.; Chen, Z.; Schmelzer, A.E. Engineered CHO Cells as a Novel AAV Production Platform for Gene Therapy Delivery. Sci. Rep. 2023, 13, 19210. [Google Scholar] [CrossRef]
- Gruntman, A.M.; Su, L.; Su, Q.; Gao, G.; Mueller, C.; Flotte, T.R. Stability and Compatibility of Recombinant Adeno-Associated Virus Under Conditions Commonly Encountered in Human Gene Therapy Trials. Hum. Gene Ther. Methods 2015, 26, 71–76. [Google Scholar] [CrossRef]
- Saunders, M.; Taylor, K.M.G.; Craig, D.Q.M.; Palin, K.; Robson, H. High Sensitivity Differential Scanning Calorimetry Study of DNA-Cationic Liposome Complexes. Pharm. Res. 2007, 24, 1954–1961. [Google Scholar] [CrossRef]
- Bennett, J.I.; Boit, M.O.; Gregorio, N.E.; Zhang, F.; Kibler, R.D.; Hoye, J.W.; Prado, O.; Rapp, P.B.; Murry, C.E.; Stevens, K.R.; et al. Genetically Encoded XTEN-Based Hydrogels with Tunable Viscoelasticity and Biodegradability for Injectable Cell Therapies. Adv. Sci. 2024, 11, 2301708. [Google Scholar] [CrossRef]
- Terradas, G.; Manzano-Alvarez, J.; Vanalli, C.; Werling, K.; Cattadori, I.M.; Rasgon, J.L. Temperature Affects Viral Kinetics and Vectorial Capacity of Aedes Aegypti Mosquitoes Co-Infected with Mayaro and Dengue Viruses. Parasites Vectors 2024, 17, 73. [Google Scholar] [CrossRef] [PubMed]
- Ionova, Y.; Wilson, L. Biologic Excipients: Importance of Clinical Awareness of Inactive Ingredients. PLoS ONE 2020, 15, e0235076. [Google Scholar] [CrossRef] [PubMed]
- Eilts, F.; Harsy, Y.M.J.; Lothert, K.; Pagallies, F.; Amann, R.; Wolff, M.W. An Investigation of Excipients for a Stable Orf Viral Vector Formulation. Virus Res. 2023, 336, 199213. [Google Scholar] [CrossRef] [PubMed]
- Kumru, O.S.; Saleh-Birdjandi, S.; Antunez, L.R.; Sayeed, E.; Robinson, D.; van den Worm, S.; Diemer, G.S.; Perez, W.; Caposio, P.; Früh, K.; et al. Stabilization and Formulation of a Recombinant Human Cytomegalovirus Vector for Use as a Candidate HIV-1 Vaccine. Vaccine 2019, 37, 6696–6706. [Google Scholar] [CrossRef]
- Chen, Y.; Liao, Q.; Chen, T.; Zhang, Y.; Yuan, W.; Xu, J.; Zhang, X. Freeze-Drying Formulations Increased the Adenovirus and Poxvirus Vaccine Storage Times and Antigen Stabilities. Virol. Sin. 2021, 36, 365–372. [Google Scholar] [CrossRef]
- Remes, A.; Basha, D.I.; Puehler, T.; Borowski, C.; Hille, S.; Kummer, L.; Wagner, A.H.; Hecker, M.; Soethoff, J.; Lutter, G.; et al. Alginate Hydrogel Polymers Enable Efficient Delivery of a Vascular-Targeted AAV Vector into Aortic Tissue. Mol. Ther. Methods Clin. Dev. 2021, 21, 83–93. [Google Scholar] [CrossRef]
- Santos-Carballal, B.; Fernández Fernández, E.; Goycoolea, F.M. Chitosan in Non-Viral Gene Delivery: Role of Structure, Characterization Methods, and Insights in Cancer and Rare Diseases Therapies. Polymers 2018, 10, 444. [Google Scholar] [CrossRef]
- Jang, J.-H.; Schaffer, D.V.; Shea, L.D. Engineering Biomaterial Systems to Enhance Viral Vector Gene Delivery. Mol. Ther. 2011, 19, 1407–1415. [Google Scholar] [CrossRef]
- Yu, Y.; Gao, Y.; He, L.; Fang, B.; Ge, W.; Yang, P.; Ju, Y.; Xie, X.; Lei, L. Biomaterial-Based Gene Therapy. MedComm 2023, 4, e259. [Google Scholar] [CrossRef]
- Zhi, L.; Chen, Y.; Lai, K.-Y.N.; Wert, J.; Li, S.; Wang, X.; Tang, X.C.; Shameem, M.; Liu, D. Lyophilization as an Effective Tool to Develop AAV8 Gene Therapy Products for Refrigerated Storage. Int. J. Pharm. 2023, 648, 123564. [Google Scholar] [CrossRef] [PubMed]
- Fenske, D.B.; MacLachlan, I.; Cullis, P.R. Stabilized Plasmid-Lipid Particles: A Systemic Gene Therapy Vector. Methods Enzymol. 2002, 346, 36–71. [Google Scholar] [CrossRef] [PubMed]
- Pikal, M.J. Mechanisms of Protein Stabilization During Freeze-Drying Storage: The Relative Importance of Thermodynamic Stabilization and Glassy State Relaxation Dynamics. In Freeze-Drying/Lyophilization of Pharmaceutical and Biological Products; CRC Press: Boca Raton, FL, USA, 2010; ISBN 978-0-429-15185-9. [Google Scholar]
- Porcari, A.; Maurer, M.S.; Gillmore, J.D. Treatment of ATTR Amyloidosis: From Stabilizers to Gene Editing. In Cardiac Amyloidosis: Diagnosis and Treatment; Emdin, M., Vergaro, G., Aimo, A., Fontana, M., Eds.; Springer Nature: Cham, Switzerland, 2024; pp. 261–278. ISBN 978-3-031-51757-0. [Google Scholar]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Kang, D.D.; Dong, Y. Biomaterials for in Situ Cell Therapy. BMEMat 2023, 1, e12039. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Golchin, A.; Seyedjafari, E.; Ardeshirylajimi, A. Mesenchymal Stem Cell Therapy for COVID-19: Present or Future. Stem Cell Rev. Rep. 2020, 16, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.-T.; Nguyen, T.T.; Tien, N.L.B.; Tran, D.-K.; Jeong, J.-H.; Anh, P.G.; Thanh, V.V.; Truong, D.T.; Dinh, T.C. Recent Progress of Stem Cell Therapy in Cancer Treatment: Molecular Mechanisms and Potential Applications. Cells 2020, 9, 563. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, G.; Wan, X. Challenges and New Technologies in Adoptive Cell Therapy. J. Hematol. Oncol. 2023, 16, 97. [Google Scholar] [CrossRef]
- Temple, S. Advancing Cell Therapy for Neurodegenerative Diseases. Cell Stem Cell 2023, 30, 512–529. [Google Scholar] [CrossRef]
- Pinto, I.S.; Cordeiro, R.A.; Faneca, H. Polymer- and Lipid-Based Gene Delivery Technology for CAR T Cell Therapy. J. Control. Release 2023, 353, 196–215. [Google Scholar] [CrossRef]
- Sarvepalli, S.; Pasika, S.R.; Vadarevu, S.; Bolla, S.; Bolla, P.K. A Comprehensive Review on Injectable Hydrogels for Cell Therapy. J. Drug Deliv. Sci. Technol. 2025, 105, 106648. [Google Scholar] [CrossRef]
- Atouf, F. Cell-Based Therapies Formulations: Unintended Components. AAPS J. 2016, 18, 844–848. [Google Scholar] [CrossRef]
- Whaley, D.; Damyar, K.; Witek, R.P.; Mendoza, A.; Alexander, M.; Lakey, J.R. Cryopreservation: An Overview of Principles and Cell-Specific Considerations. Cell Transplant. 2021, 30, 963689721999617. [Google Scholar] [CrossRef] [PubMed]
- Brookshaw, T.; Fuller, B.; Erro, E.; Islam, T.; Chandel, S.; Zotova, E.; Selden, C. Cryobiological Aspects of Upscaling Cryopreservation for Encapsulated Liver Cell Therapies. Cryobiology 2024, 117, 105155. [Google Scholar] [CrossRef]
- Śledź, M.; Wojciechowska, A.; Zagożdżon, R.; Kaleta, B. In Situ Programming of CAR-T Cells: A Pressing Need in Modern Immunotherapy. Arch. Immunol. Ther. Exp. 2023, 71, 18. [Google Scholar] [CrossRef] [PubMed]
- Rurik, J.G.; Tombácz, I.; Yadegari, A.; Méndez Fernández, P.O.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T Cells Produced in Vivo to Treat Cardiac Injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef]
- Melocchi, A.; Schmittlein, B.; Sadhu, S.; Nayak, S.; Lares, A.; Uboldi, M.; Zema, L.; di Robilant, B.N.; Feldman, S.A.; Esensten, J.H. Automated Manufacturing of Cell Therapies. J. Control. Release 2025, 381, 113561. [Google Scholar] [CrossRef]
- Abdeen, A.A.; Saha, K. Manufacturing Cell Therapies Using Engineered Biomaterials. Trends Biotechnol. 2017, 35, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Zynda, E. Addressing Cell Therapy Challenges Through a Modular, Closed, and Automated Manufacturing System. BioPharm Int. 2023, 36, 26–29. [Google Scholar]
- Challener, C. Challenges to Cell Expansion for Allogenic Cell Therapies. Biopharm. Int. 2023, 36, 22–25. [Google Scholar]
- CELL THERAPIES—The Challenges & Possible Solutions for Transferring Cell Therapy from the Bench to the Industry. Available online: https://drug-dev.com/cell-therapies-the-challenges-possible-solutions-for-transferring-cell-therapy-from-the-bench-to-the-industry/ (accessed on 14 February 2025).
- Sethi, D.; Cunningham, A. De-Risking the Final Formulation, Fill and Finish Step in Cell Therapy Manufacturing: Considerations for an Automated Solution. Cell Gene Ther. Insights 2020, 6, 1513–1519. [Google Scholar] [CrossRef]
- Thermo Fischer Scientific. Overcoming the Challenges of Cell Therapy Manufacturing. Available online: https://www.bioprocessonline.com/doc/overcoming-the-challenges-of-cell-therapy-manufacturing-0001 (accessed on 15 February 2025).
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The Current Landscape of Nucleic Acid Therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the Potency of siRNA Lipid Nanoparticles for Hepatic Gene Silencing In Vivo. Angew. Chem. Int. Ed. 2012, 51, 8529–8533. [Google Scholar] [CrossRef]
- Meng, C.; Chen, Z.; Li, G.; Welte, T.; Shen, H. Nanoplatforms for mRNA Therapeutics. Adv. Ther. 2021, 4, 2000099. [Google Scholar] [CrossRef]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid Nanoparticles for mRNA Delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA Delivery by Lipid Nanoparticles Is Limited by Endocytic Recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down Barriers: Advances in siRNA Delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-Based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Cote, T.; Chen, Y. Challenges and Advances of the Stability of mRNA Delivery Therapeutics. Nucleic Acid Insights 2024, 1, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Oude Blenke, E.; Örnskov, E.; Schöneich, C.; Nilsson, G.A.; Volkin, D.B.; Mastrobattista, E.; Almarsson, Ö.; Crommelin, D.J.A. The Storage and In-Use Stability of mRNA Vaccines and Therapeutics: Not A Cold Case. J. Pharm. Sci. 2023, 112, 386–403. [Google Scholar] [CrossRef]
- Yang, W.; Chen, P.; Boonstra, E.; Hong, T.; Cabral, H. Polymeric Micelles with pH-Responsive Cross-Linked Core Enhance In Vivo mRNA Delivery. Pharmaceutics 2022, 14, 1205. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Evans, A.C.; Rasley, A.; Bourguet, F.; Peters, S.; Kamrud, K.I.; Wang, N.; Hubby, B.; Felderman, M.; Gouvis, H.; et al. Cationic HDL Mimetics Enhance in Vivo Delivery of Self-Replicating mRNA. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102154. [Google Scholar] [CrossRef]
- Aldosari, B.N.; Alfagih, I.M.; Almurshedi, A.S. Lipid Nanoparticles as Delivery Systems for RNA-Based Vaccines. Pharmaceutics 2021, 13, 206. [Google Scholar] [CrossRef]
- Granot, Y.; Peer, D. Delivering the Right Message: Challenges and Opportunities in Lipid Nanoparticles-Mediated Modified mRNA Therapeutics—An Innate Immune System Standpoint. Semin. Immunol. 2017, 34, 68–77. [Google Scholar] [CrossRef]
- Kayushin, A.L.; Antonov, K.V.; Dorofeeva, E.V.; Berzina, M.Y.; Arnautova, A.O.; Prohorenko, I.A.; Miroshnikov, A.I.; Konstantinova, I.D. Erratum to: A New Approach to the Synthesis of Anti-Reverse Cap Analog (ARCA) 2mGpppG. Russ. J. Bioorganic Chem. 2024, 50, 625. [Google Scholar] [CrossRef]
- Wojtczak, B.A.; Sikorski, P.J.; Fac-Dabrowska, K.; Nowicka, A.; Warminski, M.; Kubacka, D.; Nowak, E.; Nowotny, M.; Kowalska, J.; Jemielity, J. 5′-Phosphorothiolate Dinucleotide Cap Analogues: Reagents for Messenger RNA Modification and Potent Small-Molecular Inhibitors of Decapping Enzymes. J. Am. Chem. Soc. 2018, 140, 5987–5999. [Google Scholar] [CrossRef]
- Strenkowska, M.; Grzela, R.; Majewski, M.; Wnek, K.; Kowalska, J.; Lukaszewicz, M.; Zuberek, J.; Darzynkiewicz, E.; Kuhn, A.N.; Sahin, U.; et al. Cap Analogs Modified with 1,2-Dithiodiphosphate Moiety Protect mRNA from Decapping and Enhance Its Translational Potential. Nucleic Acids Res. 2016, 44, 9578–9590. [Google Scholar] [CrossRef]
- Ziemniak, M.; Strenkowska, M.; Kowalska, J.; Jemielity, J. Potential Therapeutic Applications of RNA Cap Analogs. Future Med. Chem. 2013, 5, 1141–1172. [Google Scholar] [CrossRef]
- Kim, S.C.; Sekhon, S.S.; Shin, W.-R.; Ahn, G.; Cho, B.-K.; Ahn, J.-Y.; Kim, Y.-H. Modifications of mRNA Vaccine Structural Elements for Improving mRNA Stability and Translation Efficiency. Mol. Cell. Toxicol. 2022, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Biziaev, N.; Shuvalov, A.; Salman, A.; Egorova, T.; Shuvalova, E.; Alkalaeva, E. The Impact of mRNA Poly(A) Tail Length on Eukaryotic Translation Stages. Nucleic Acids Res. 2024, 52, 7792–7808. [Google Scholar] [CrossRef]
- Munroe, D.; Jacobson, A. mRNA Poly(A) Tail, a 3′ Enhancer of Translational Initiation. Mol. Cell. Biol. 1990, 10, 3441–3455. [Google Scholar] [CrossRef] [PubMed]
- Gillian-Daniel, D.L.; Gray, N.K.; Aström, J.; Barkoff, A.; Wickens, M. Modifications of the 5′ Cap of mRNAs during Xenopus Oocyte Maturation: Independence from Changes in Poly(A) Length and Impact on Translation. Mol. Cell. Biol. 1998, 18, 6152–6163. [Google Scholar] [CrossRef]
- Xiang, K.; Bartel, D.P. The Molecular Basis of Coupling between Poly(A)-Tail Length and Translational Efficiency. eLife 2021, 10, e66493. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nie, H.; Sun, R.; Wang, J.; Lu, F. Enhancement of Synthetic mRNA Translation Efficiency through Engineered Poly(A) Tails. bioRxiv 2021. [Google Scholar] [CrossRef]
- Beilharz, T.H.; Preiss, T. Widespread Use of Poly(A) Tail Length Control to Accentuate Expression of the Yeast Transcriptome. RNA 2007, 13, 982–997. [Google Scholar] [CrossRef]
- Li, Y.; Yi, Y.; Gao, X.; Wang, X.; Zhao, D.; Wang, R.; Zhang, L.-S.; Gao, B.; Zhang, Y.; Zhang, L.; et al. 2′-O-Methylation at Internal Sites on mRNA Promotes mRNA Stability. Mol. Cell 2024, 84, 2320–2336.e6. [Google Scholar] [CrossRef]
- Pallan, P.S.; Lybrand, T.P.; Schlegel, M.K.; Harp, J.M.; Jahns, H.; Manoharan, M.; Egli, M. Incorporating a Thiophosphate Modification into a Common RNA Tetraloop Motif Causes an Unanticipated Stability Boost. Biochemistry 2020, 59, 4627–4637. [Google Scholar] [CrossRef]
- Leppek, K.; Byeon, G.W.; Kladwang, W.; Wayment-Steele, H.K.; Kerr, C.H.; Xu, A.F.; Kim, D.S.; Topkar, V.V.; Choe, C.; Rothschild, D.; et al. Combinatorial Optimization of mRNA Structure, Stability, and Translation for RNA-Based Therapeutics. Nat. Commun. 2022, 13, 1536. [Google Scholar] [CrossRef] [PubMed]
- Wayment-Steele, H.K.; Kim, D.S.; Choe, C.A.; Nicol, J.J.; Wellington-Oguri, R.; Watkins, A.M.; Sperberg, R.A.P.; Huang, P.-S.; Participants, E.; Das, R. Theoretical Basis for Stabilizing Messenger RNA through Secondary Structure Design. bioRxiv 2020. [Google Scholar] [CrossRef]
- Baptissart, M.; Papas, B.N.; Chi, R.-P.A.; Li, Y.; Lee, D.; Puviindran, B.; Morgan, M. A Unique Poly(A) Tail Profile Uncovers the Stability and Translational Activation of TOP Transcripts during Neuronal Differentiation. iScience 2023, 26, 107511. [Google Scholar] [CrossRef] [PubMed]
- Mauger, D.M.; Cabral, B.J.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. mRNA Structure Regulates Protein Expression through Changes in Functional Half-Life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar] [CrossRef]
- Xian, H.; Zhang, Y.; Yu, C.; Wang, Y. Nanobiotechnology-Enabled mRNA Stabilization. Pharmaceutics 2023, 15, 620. [Google Scholar] [CrossRef]
- Grandi, C.; Emmaneel, M.; Nelissen, F.H.; Roosenboom, L.W.; Petrova, Y.; Elzokla, O.; Hansen, M.M. Decoupled Degradation and Translation Enables Noise-Modulation by Poly(A)-Tails. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kierzek, E.; Malgowska, M.; Lisowiec, J.; Turner, D.H.; Gdaniec, Z.; Kierzek, R. The Contribution of Pseudouridine to Stabilities and Structure of RNAs. Nucleic Acids Res. 2014, 42, 3492–3501. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine into mRNA Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Dutta, N.; Deb, I.; Sarzynska, J.; Lahiri, A. Structural and Thermodynamic Consequences of Base Pairs Containing Pseudouridine and N1-Methylpseudouridine in RNA Duplexes. bioRxiv 2023. [Google Scholar] [CrossRef]
- Nelson, J.; Sorensen, E.W.; Mintri, S.; Rabideau, A.E.; Zheng, W.; Besin, G.; Khatwani, N.; Su, S.V.; Miracco, E.J.; Issa, W.J.; et al. Impact of mRNA Chemistry and Manufacturing Process on Innate Immune Activation. Sci. Adv. 2020, 6, eaaz6893. [Google Scholar] [CrossRef]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N1-Methylpseudouridine-Incorporated mRNA Outperforms Pseudouridine-Incorporated mRNA by Providing Enhanced Protein Expression and Reduced Immunogenicity in Mammalian Cell Lines and Mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef]
- Svitkin, Y.V.; Cheng, Y.M.; Chakraborty, T.; Presnyak, V.; John, M.; Sonenberg, N. N1-Methyl-Pseudouridine in mRNA Enhances Translation through eIF2α-Dependent and Independent Mechanisms by Increasing Ribosome Density. Nucleic Acids Res. 2017, 45, 6023–6036. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Xiao, S.; Burrows, C.J. Pseudouridine and N1-Methylpseudouridine Display pH-Independent Reaction Rates with Bisulfite Yielding Ribose Adducts. Org. Lett. 2022, 24, 6182–6185. [Google Scholar] [CrossRef] [PubMed]
- Nance, K.D.; Meier, J.L. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines. ACS Central Sci. 2021, 7, 748–756. [Google Scholar] [CrossRef]
- Hoehn, S.J.; Krul, S.E.; Skory, B.J.; Crespo-Hernández, C.E. Increased Photostability of the Integral mRNA Vaccine Component N1-Methylpseudouridine Compared to Uridine. Chemistry 2022, 28, e202103667. [Google Scholar] [CrossRef]
- Gao, Y.; Fang, J. RNA 5-Methylcytosine Modification and Its Emerging Role as an Epitranscriptomic Mark. RNA Biol. 2021, 18, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, L.; Lin, A.; Xu, C.; Li, Z.; Liu, K.; Liu, B.; Ma, X.; Zhao, F.; Jiang, H.; et al. Algorithm for Optimized mRNA Design Improves Stability and Immunogenicity. Nature 2023, 621, 396–403. [Google Scholar] [CrossRef]
- Hanson, G.; Coller, J. Codon Optimality, Bias and Usage in Translation and mRNA Decay. Nat. Rev. Mol. Cell Biol. 2018, 19, 20–30. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, L.; Lin, A.; Xu, C.; Li, Z.; Liu, K.; Liu, B.; Ma, X.; Zhao, F.; Yao, W. Lineardesign: Efficient Algorithms for Optimized Mrna Sequence Design. arXiv 2021, arXiv:2004.10177. [Google Scholar] [CrossRef]
- Luna-Cerralbo, D.; Blasco-Machín, I.; Adame-Pérez, S.; Lampaya, V.; Larraga, A.; Alejo, T.; Martínez-Oliván, J.; Broset, E.; Bruscolini, P. A Statistical-Physics Approach for Codon Usage Optimisation. Comput. Struct. Biotechnol. J. 2024, 23, 3050–3064. [Google Scholar] [CrossRef]
- Nelson, E.V.; Ross, S.J.; Olejnik, J.; Hume, A.J.; Deeney, D.J.; King, E.; Grimins, A.O.; Lyons, S.M.; Cifuentes, D.; Mühlberger, E. The 3′ Untranslated Regions of Ebola Virus mRNAs Contain AU-Rich Elements Involved in Posttranscriptional Stabilization and Decay. J. Infect. Dis. 2023, 228, S488–S497. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-Y.; Wang, Y.-L.; Hsieh, Y.-T.; Chang, Y.-C.; Yang, C.-H.; Kang, Y.; Huang, Y.-T.; Lin, C.-L. Multiplexed Assays of Human Disease-Relevant Mutations Reveal UTR Dinucleotide Composition as a Major Determinant of RNA Stability. eLife 2025, 13, RP97682. [Google Scholar] [CrossRef] [PubMed]
- Meganck, R.M.; Ogurlu, R.; Liu, J.; Moller-Tank, S.; Tse, V.; Blondel, L.O.; Rosales, A.; Hall, A.C.; Vincent, H.A.; Moorman, N.J.; et al. Sub-Genomic Flaviviral RNA Elements Increase the Stability and Abundance of Recombinant AAV Vector Transcripts. J. Virol. 2024, 98, e0009524. [Google Scholar] [CrossRef] [PubMed]
- Awah, C.U.; Glemaud, Y.; Levine, F.; Yang, K.; Ansary, A.; Dong, F.; Ash, L.; Zhang, J.; Ogunwobi, O.O. Destabilized 3′UTR Elements Therapeutically Degrade ERBB2 mRNA in Drug-Resistant ERBB2+ Cancer Models. Front. Genet. 2023, 14. [Google Scholar] [CrossRef]
- Wu, S.; Lin, L.; Shi, L.; Liu, S. An Overview of Lipid Constituents in Lipid Nanoparticle mRNA Delivery Systems. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2024, 16, e1978. [Google Scholar] [CrossRef]
- Jung, O.; Jung, H.-Y.; Thuy, L.T.; Choi, M.; Kim, S.; Jeon, H.-G.; Yang, J.; Kim, S.-M.; Kim, T.-D.; Lee, E.; et al. Modulating Lipid Nanoparticles with Histidinamide-Conjugated Cholesterol for Improved Intracellular Delivery of mRNA. Adv. Heal. Mater. 2024, 13, e2303857. [Google Scholar] [CrossRef]
- Ongun, M.; Lokras, A.G.; Baghel, S.; Shi, Z.; Schmidt, S.T.; Franzyk, H.; Rades, T.; Sebastiani, F.; Thakur, A.; Foged, C. Lipid Nanoparticles for Local Delivery of mRNA to the Respiratory Tract: Effect of PEG-Lipid Content and Administration Route. Eur. J. Pharm. Biopharm. 2024, 198, 114266. [Google Scholar] [CrossRef]
- Gilbert, J.; Sebastiani, F.; Arteta, M.Y.; Terry, A.; Fornell, A.; Russell, R.; Mahmoudi, N.; Nylander, T. Evolution of the Structure of Lipid Nanoparticles for Nucleic Acid Delivery: From in Situ Studies of Formulation to Colloidal Stability. J. Colloid Interface Sci. 2024, 660, 66–76. [Google Scholar] [CrossRef]
- Liu, S.; Wen, Y.; Shan, X.; Ma, X.; Yang, C.; Cheng, X.; Zhao, Y.; Li, J.; Mi, S.; Huo, H.; et al. Charge-Assisted Stabilization of Lipid Nanoparticles Enables Inhaled mRNA Delivery for Mucosal Vaccination. Nat. Commun. 2024, 15, 9471. [Google Scholar] [CrossRef]
- Hashiba, K.; Taguchi, M.; Sakamoto, S.; Otsu, A.; Maeda, Y.; Ebe, H.; Okazaki, A.; Harashima, H.; Sato, Y. Overcoming Thermostability Challenges in mRNA–Lipid Nanoparticle Systems with Piperidine-Based Ionizable Lipids. Commun. Biol. 2024, 7, 556. [Google Scholar] [CrossRef]
- Friis, K.P.; Gracin, S.; Oag, S.; Leijon, A.; Sand, E.; Lindberg, B.; Lázaro-Ibáñez, E.; Lindqvist, J.; Whitehead, K.A.; Bak, A. Spray Dried Lipid Nanoparticle Formulations Enable Intratracheal Delivery of mRNA. J. Control. Release 2023, 363, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.; Zhang, L.; Ang, M.J.Y.; Ng, J.Y.; Babu, C.V.S.; Schneider, S.; Gudihal, R.; Bae, K.H.; Yang, Y.Y. Quantitative Analysis of mRNA-Lipid Nanoparticle Stability in Human Plasma and Serum by Size-Exclusion Chromatography Coupled with Dual-Angle Light Scattering. Nanomed. Nanotechnol. Biol. Med. 2024, 58, 102745. [Google Scholar] [CrossRef]
- Kamiya, M.; Matsumoto, M.; Yamashita, K.; Izumi, T.; Kawaguchi, M.; Mizukami, S.; Tsurumaru, M.; Mukai, H.; Kawakami, S. Stability Study of mRNA-Lipid Nanoparticles Exposed to Various Conditions Based on the Evaluation between Physicochemical Properties and Their Relation with Protein Expression Ability. Pharmaceutics 2022, 14, 2357. [Google Scholar] [CrossRef] [PubMed]
- Kafetzis, K.N.; Papalamprou, N.; McNulty, E.; Thong, K.X.; Sato, Y.; Mironov, A.; Purohit, A.; Welsby, P.J.; Harashima, H.; Yu-Wai-Man, C.; et al. The Effect of Cryoprotectants and Storage Conditions on the Transfection Efficiency, Stability, and Safety of Lipid-Based Nanoparticles for mRNA and DNA Delivery. Adv. Healthc. Mater. 2023, 12, e2203022. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.A.; Brinkman, H.M.; Lee, J.S.; Murphy, W.L. Optimized Biomimetic Minerals Maintain Activity of mRNA Complexes after Long Term Storage. Acta Biomater. 2024, 174, 428–436. [Google Scholar] [CrossRef]
- He, Y.; Johnston, A.P.R.; Pouton, C.W. Therapeutic Applications of Cell Engineering Using mRNA Technology. Trends Biotechnol. 2025, 43, 83–97. [Google Scholar] [CrossRef]
- Shirane, D.; Tanaka, H.; Sakurai, Y.; Taneichi, S.; Nakai, Y.; Tange, K.; Ishii, I.; Akita, H. Development of an Alcohol Dilution-Lyophilization Method for the Preparation of mRNA-LNPs with Improved Storage Stability. Pharmaceutics 2023, 15, 1819. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, X.; Liu, X.; Liu, Y.; Bui-Le, L.; Blakney, A.K.; Yeow, J.; Zhu, Y.; Stevens, M.M.; Shattock, R.J.; et al. Thermally Robust Solvent-Free Liquid Polyplexes for Heat-Shock Protection and Long-Term Room Temperature Storage of Therapeutic Nucleic Acids. Biomacromolecules 2024, 25, 2965–2972. [Google Scholar] [CrossRef]
- Li, M.; Jia, L.; Xie, Y.; Ma, W.; Yan, Z.; Liu, F.; Deng, J.; Zhu, A.; Siwei, X.; Su, W.; et al. Lyophilization Process Optimization and Molecular Dynamics Simulation of mRNA-LNPs for SARS-CoV-2 Vaccine. NPJ Vaccines 2023, 8, 153. [Google Scholar] [CrossRef]
- Meulewaeter, S.; Nuytten, G.; Cheng, M.H.Y.; De Smedt, S.C.; Cullis, P.R.; De Beer, T.; Lentacker, I.; Verbeke, R. Continuous Freeze-Drying of Messenger RNA Lipid Nanoparticles Enables Storage at Higher Temperatures. J. Control. Release 2023, 357, 149–160. [Google Scholar] [CrossRef]
- Lamoot, A.; Lammens, J.; De Lombaerde, E.; Zhong, Z.; Gontsarik, M.; Chen, Y.; De Beer, T.R.M.; De Geest, B.G. Successful Batch and Continuous Lyophilization of mRNA LNP Formulations Depend on Cryoprotectants and Ionizable Lipids. Biomater. Sci. 2023, 11, 4327–4334. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Li, Y.; Zhou, L.; Yao, W.; Zhang, H.; Hu, Z.; Han, J.; Wang, W.; Wu, J.; Xu, P.; et al. Lyophilized mRNA-Lipid Nanoparticle Vaccines with Long-Term Stability and High Antigenicity against SARS-CoV-2. Cell Discov. 2023, 9, 9. [Google Scholar] [CrossRef]
- Muramatsu, H.; Lam, K.; Bajusz, C.; Laczkó, D.; Karikó, K.; Schreiner, P.; Martin, A.; Lutwyche, P.; Heyes, J.; Pardi, N. Lyophilization Provides Long-Term Stability for a Lipid Nanoparticle-Formulated, Nucleoside-Modified mRNA Vaccine. Mol. Ther. 2022, 30, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; He, Z.; Chen, Q.; He, X.; Su, L.; Yu, W.; Zhang, M.; Yang, H.; Huang, X.; Li, J. Helper-Polymer Based Five-Element Nanoparticles (FNPs) for Lung-Specific mRNA Delivery with Long-Term Stability after Lyophilization. Nano Lett. 2022, 22, 6580–6589. [Google Scholar] [CrossRef]
- Pontes, A.P.; van der Wal, S.; Roelofs, K.; Grobbink, A.; Creemers, L.B.; Engbersen, J.F.J.; Rip, J. A Poly(amidoamine)-Based Polymeric Nanoparticle Platform for Efficient in Vivo Delivery of mRNA. Biomater. Adv. 2024, 156, 213713. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Han, H.S.; Yang, K.; Kim, Y.M.; Nam, K.; Park, K.H.; Choi, S.Y.; Park, H.W.; Choi, K.Y.; Roh, Y.H. Nanoengineered Polymeric RNA Nanoparticles for Controlled Biodistribution and Efficient Targeted Cancer Therapy. ACS Nano 2024, 18, 7972–7988. [Google Scholar] [CrossRef]
- Uchida, S.; Lau, C.Y.J.; Oba, M.; Miyata, K. Polyplex Designs for Improving the Stability and Safety of RNA Therapeutics. Adv. Drug Deliv. Rev. 2023, 199, 114972. [Google Scholar] [CrossRef]
- Currie, J.; Dahlberg, J.R.; Lundberg, E.; Thunberg, L.; Eriksson, J.; Schweikart, F.; Nilsson, G.A.; Örnskov, E. Stability Indicating Ion-Pair Reversed-Phase Liquid Chromatography Method for Modified mRNA. J. Pharm. Biomed. Anal. 2024, 245, 116144. [Google Scholar] [CrossRef]
- Zhou, D.-W.; Wang, K.; Zhang, Y.-A.; Ma, K.; Yang, X.-C.; Li, Z.-Y.; Yu, S.-S.; Chen, K.-Z.; Qiao, S.-L. mRNA Therapeutics for Disease Therapy: Principles, Delivery, and Clinical Translation. J. Mater. Chem. B 2023, 11, 3484–3510. [Google Scholar] [CrossRef]
- Han, G.; Noh, D.; Lee, H.; Lee, S.; Kim, S.; Yoon, H.Y.; Lee, S.H. Advances in mRNA Therapeutics for Cancer Immunotherapy: From Modification to Delivery. Adv. Drug Deliv. Rev. 2023, 199, 114973. [Google Scholar] [CrossRef]
- Eralp, Y. Application of mRNA Technology in Cancer Therapeutics. Vaccines 2022, 10, 1262. [Google Scholar] [CrossRef]
- Parhiz, H.; Atochina-Vasserman, E.N.; Weissman, D. mRNA-Based Therapeutics: Looking beyond COVID-19 Vaccines. Lancet 2024, 403, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Liszewski, K. RNA Medicines Address Stability and Deliverability Challenges. Available online: https://www.genengnews.com/topics/omics/rna-medicines-address-stability-and-deliverability-challenges/ (accessed on 14 February 2025).
- Kovarik, P.; Bestehorn, A.; Fesselet, J. Conceptual Advances in Control of Inflammation by the RNA-Binding Protein Tristetraprolin. Front. Immunol. 2021, 12, 751313. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, X.; Chen, J. RNA-Binding Proteins in Regulating mRNA Stability and Translation: Roles and Mechanisms in Cancer. Semin. Cancer Biol. 2022, 86, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Sinnott, R.W.; Cao, Y.; Dickinson, B.C. Engineered RNA-Binding Proteins: Studying and Controlling RNA Regulation. Isr. J. Chem. 2024, 64, e202300169. [Google Scholar] [CrossRef]
- Hudy, D.; Rzeszowska-Wolny, J. New Features of Micro-RNA Regulation of mRNA Translation and Stability Revealed by Expression of Targeted or Not Targeted Reporter Genes. bioRxiv 2022. [Google Scholar] [CrossRef]
- Vos, P.D.; Leedman, P.J.; Filipovska, A.; Rackham, O. Modulation of miRNA Function by Natural and Synthetic RNA-Binding Proteins in Cancer. Cell. Mol. Life Sci. 2019, 76, 3745–3752. [Google Scholar] [CrossRef]
- Boo, S.H.; Kim, Y.K. The Emerging Role of RNA Modifications in the Regulation of mRNA Stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef]
- Kawata, K.; Akimitsu, N. Regulation of RNA Stability Through RNA Modification. In Epitranscriptomics; Springer: Cham, Switzerland, 2024; pp. 217–246. [Google Scholar] [CrossRef]
- Lyu, X.; Zhao, Q.; Hui, J.; Wang, T.; Lin, M.; Wang, K.; Zhang, J.; Shentu, J.; Dalby, P.A.; Zhang, H.; et al. The Global Landscape of Approved Antibody Therapies. Antib. Ther. 2022, 5, 233–257. [Google Scholar] [CrossRef]
- Strohl, W.R. Structure and Function of Therapeutic Antibodies Approved by the US FDA in 2023. Antib. Ther. 2024, 7, 132–156. [Google Scholar] [CrossRef]
- Bittner, B.; Richter, W.; Schmidt, J. Subcutaneous Administration of Biotherapeutics: An Overview of Current Challenges and Opportunities. BioDrugs 2018, 32, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Pitiot, A.; Heuzé-Vourc’h, N.; Sécher, T. Alternative Routes of Administration for Therapeutic Antibodies—State of the Art. Antibodies 2022, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Matucci, A.; Vultaggio, A.; Danesi, R. The Use of Intravenous versus Subcutaneous Monoclonal Antibodies in the Treatment of Severe Asthma: A Review. Respir. Res. 2018, 19, 154. [Google Scholar] [CrossRef] [PubMed]
- Jackisch, C.; Müller, V.; Maintz, C.; Hell, S.; Ataseven, B. Subcutaneous Administration of Monoclonal Antibodies in Oncology. Geburtshilfe Und Frauenheilkd. 2014, 74, 343. [Google Scholar] [CrossRef]
- Shah, K. Monoclonal Antibody Formulations: Challenges and Developments. Master’s Thesis, University of Nottingham, Nottingham, UK, 2025. Available online: https://eprints.nottingham.ac.uk/67069/1/KeshaShah_MResDissertation2021.pdf (accessed on 14 April 2025).
- Viola, M.; Sequeira, J.; Seiça, R.; Veiga, F.; Serra, J.; Santos, A.C.; Ribeiro, A.J. Subcutaneous Delivery of Monoclonal Antibodies: How Do We Get There? J. Control. Release 2018, 286, 301–314. [Google Scholar] [CrossRef]
- Moussa, E.M.; Panchal, J.P.; Moorthy, B.S.; Blum, J.S.; Joubert, M.K.; Narhi, L.O.; Topp, E.M. Immunogenicity of Therapeutic Protein Aggregates. J. Pharm. Sci. 2016, 105, 417–430. [Google Scholar] [CrossRef]
- Bodier-Montagutelli, E.; Respaud, R.; Watier, H.; Guillon-Munos, A. MAbDelivery: Administration Routes for Antibody Therapy Third LabEx MAbImprove Industrial Workshop, July 2, 2015 Tours, France. mAbs 2017, 9, 579–585. [Google Scholar] [CrossRef]
- Thiagarajan, G.; Semple, A.; James, J.K.; Cheung, J.K.; Shameem, M. A Comparison of Biophysical Characterization Techniques in Predicting Monoclonal Antibody Stability. mAbs 2016, 8, 1088–1097. [Google Scholar] [CrossRef]
- Kopp, M.R.G.; Capasso Palmiero, U.; Arosio, P. A Nanoparticle-Based Assay To Evaluate Surface-Induced Antibody Instability. Mol. Pharm. 2020, 17, 909–918. [Google Scholar] [CrossRef]
- Smith, C.; Li, Z.; Holman, R.; Pan, F.; Campbell, R.A.; Campana, M.; Li, P.; Webster, J.R.P.; Bishop, S.; Narwal, R.; et al. Antibody Adsorption on the Surface of Water Studied by Neutron Reflection. mAbs 2017, 9, 466–475. [Google Scholar] [CrossRef]
- van Haaren, C.; Byrne, B.; Kazarian, S.G. Study of Monoclonal Antibody Aggregation at the Air–Liquid Interface under Flow by ATR-FTIR Spectroscopic Imaging. Langmuir 2024, 40, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Tawab, M.; Banerjee, S.; Kirchner, R.; Wellenhofer, T.; Hahn, L.; Meinel, L.; Holzgrabe, U.; Schubert-Zsilavecz, M.; Seidl, A.; Stadler, F. An Exploratory Study on the Effect of Mechanical Stress on Particle Formation in Monoclonal Antibody Infusions. Arch. Pharm. 2023, 356, e2300101. [Google Scholar] [CrossRef]
- Tathe, U.; Khopkar, S.; Rasam, P.; Kancherla, A.; Dandekar, P.; Jain, R. Impact of Stirring Material on Formation of Submicron and Subvisible Aggregates in mAbs by Quantitative Laser Diffraction, Dynamic Light Scattering and Background Membrane Imaging. Int. J. Pharm. 2024, 660, 124321. [Google Scholar] [CrossRef] [PubMed]
- Wälchli, R.; Vermeire, P.-J.; Massant, J.; Arosio, P. Accelerated Aggregation Studies of Monoclonal Antibodies: Considerations for Storage Stability. J. Pharm. Sci. 2020, 109, 595–602. [Google Scholar] [CrossRef]
- Jin, M.-J.; Ge, X.-Z.; Huang, Q.; Liu, J.-W.; Ingle, R.G.; Gao, D.; Fang, W.-J. The Effects of Excipients on Freeze-Dried Monoclonal Antibody Formulation Degradation and Sub-Visible Particle Formation during Shaking. Pharm. Res. 2024, 41, 321–334. [Google Scholar] [CrossRef]
- Malani, H.; Shrivastava, A.; Nupur, N.; Rathore, A.S. LC-MS Characterization and Stability Assessment Elucidate Correlation Between Charge Variant Composition and Degradation of Monoclonal Antibody Therapeutics. AAPS J. 2024, 26, 42. [Google Scholar] [CrossRef]
- Kannan, A.; Shieh, I.C.; Fuller, G.G. Linking Aggregation and Interfacial Properties in Monoclonal Antibody-Surfactant Formulations. J. Colloid Interface Sci. 2019, 550, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Brudar, S.; Hribar-Lee, B. Effect of Buffer on Protein Stability in Aqueous Solutions: A Simple Protein Aggregation Model. J. Phys. Chem. B 2021, 125, 2504–2512. [Google Scholar] [CrossRef]
- Kayser, V.; Chennamsetty, N.; Voynov, V.; Helk, B.; Trout, B.L. Conformational Stability and Aggregation of Therapeutic Monoclonal Antibodies Studied with ANS and Thioflavin T Binding. mAbs 2011, 3, 408–411. [Google Scholar] [CrossRef]
- Roberts, C.J. Therapeutic Protein Aggregation: Mechanisms, Design, and Control. Trends Biotechnol. 2014, 32, 372–380. [Google Scholar] [CrossRef]
- Nowak, C.; K Cheung, J.; M Dellatore, S.; Katiyar, A.; Bhat, R.; Sun, J.; Ponniah, G.; Neill, A.; Mason, B.; Beck, A.; et al. Forced Degradation of Recombinant Monoclonal Antibodies: A Practical Guide. mAbs 2017, 9, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.E.; Sahin, E. Chemical and Physical Instabilities in Manufacturing and Storage of Therapeutic Proteins. Curr. Opin. Biotechnol. 2019, 60, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.E.; Slaney, T.R.; Wu, L.; Das, T.K.; Kar, S.; Barnett, G.V.; Leone, A.; Tessier, P.M. Unique Impacts of Methionine Oxidation, Tryptophan Oxidation, and Asparagine Deamidation on Antibody Stability and Aggregation. J. Pharm. Sci. 2020, 109, 656–669. [Google Scholar] [CrossRef]
- Fischer, P.; Merkel, O.M.; Siedler, M.; Huelsmeyer, M. Development of a High Throughput Oxidation Profiling Strategy for Monoclonal Antibody Products. Eur. J. Pharm. Biopharm. 2024, 199, 114301. [Google Scholar] [CrossRef]
- Shah, D.D.; Zhang, J.; Hsieh, M.-C.; Sundaram, S.; Maity, H.; Mallela, K.M.G. Effect of Peroxide—Versus Alkoxyl-Induced Chemical Oxidation on the Structure, Stability, Aggregation, and Function of a Therapeutic Monoclonal Antibody. J. Pharm. Sci. 2018, 107, 2789–2803. [Google Scholar] [CrossRef]
- Manikwar, P.; Majumdar, R.; Hickey, J.M.; Thakkar, S.V.; Samra, H.S.; Sathish, H.A.; Bishop, S.M.; Middaugh, C.R.; Weis, D.D.; Volkin, D.B. Correlating Excipient Effects on Conformational and Storage Stability of an IgG1 Monoclonal Antibody with Local Dynamics as Measured by Hydrogen/Deuterium-Exchange Mass Spectrometry. J. Pharm. Sci. 2013, 102, 2136–2151. [Google Scholar] [CrossRef]
- Gregoritza, K.; Theodorou, C.; Heitz, M. Enzymatic Degradation Pattern of Polysorbate 20 Impacts Interfacial Properties of Monoclonal Antibody Formulations. Eur. J. Pharm. Biopharm. 2024, 194, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Carle, S.; Evers, D.-H.; Hagelskamp, E.; Garidel, P.; Buske, J. All-in-One Stability Indicating Polysorbate 20 Degradation Root-Cause Analytics via UPLC-QDa. J. Chromatogr. B 2024, 1232, 123955. [Google Scholar] [CrossRef]
- Oyama, H.; Koga, H.; Tadokoro, T.; Maenaka, K.; Shiota, A.; Yokoyama, M.; Noda, M.; Torisu, T.; Uchiyama, S. Relation of Colloidal and Conformational Stabilities to Aggregate Formation in a Monoclonal Antibody. J. Pharm. Sci. 2020, 109, 308–315. [Google Scholar] [CrossRef]
- Saurabh, S.; Zhang, Q. Seddon Unraveling the Microscopic Mechanism of Molecular Ion Interaction with Monoclonal Antibodies: Impact on Protein Aggregation. Mol. Pharm. 2024, 21, 1285–1299. [Google Scholar] [CrossRef]
- Rott, E.; Leppin, C.; Diederichs, T.; Garidel, P.; Johannsmann, D. Protein–Protein Interactions in Solutions of Monoclonal Antibodies Probed by the Dependence of the High-Frequency Viscosity on Temperature and Concentration. Analyst 2023, 148, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Kannan, A.; Shieh, I.C.; Hristov, P.; Fuller, G.G. In-Use Interfacial Stability of Monoclonal Antibody Formulations Diluted in Saline i.v. Bags. J. Pharm. Sci. 2021, 110, 1687–1692. [Google Scholar] [CrossRef]
- Zapadka, K.L.; Becher, F.J.; Gomes dos Santos, A.L.; Jackson, S.E. Factors Affecting the Physical Stability (Aggregation) of Peptide Therapeutics. Interface Focus 2017, 7, 20170030. [Google Scholar] [CrossRef]
- Roberts, D.; Keeling, R.; Tracka, M.; van der Walle, C.F.; Uddin, S.; Warwicker, J.; Curtis, R. Specific Ion and Buffer Effects on Protein-Protein Interactions of a Monoclonal Antibody. Mol. Pharm. 2015, 12, 179–193. [Google Scholar] [CrossRef]
- Schermeyer, M.-T.; Wöll, A.K.; Kokke, B.; Eppink, M.; Hubbuch, J. Characterization of Highly Concentrated Antibody Solution—A Toolbox for the Description of Protein Long-Term Solution Stability. mAbs 2017, 9, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- Bramham, J.E.; Davies, S.A.; Podmore, A.; Golovanov, A.P. Stability of a High-Concentration Monoclonal Antibody Solution Produced by Liquid-Liquid Phase Separation. mAbs 2021, 13, 1940666. [Google Scholar] [CrossRef] [PubMed]
- Meza, N.P.; Hardy, C.A.; Morin, K.H.; Huang, C.; Raghava, S.; Song, J.; Zhang, J.; Wang, Y. Predicting Colloidal Stability of High-Concentration Monoclonal Antibody Formulations in Common Pharmaceutical Buffers Using Improved Polyethylene Glycol Induced Protein Precipitation Assay. Mol. Pharm. 2023, 20, 5842–5855. [Google Scholar] [CrossRef]
- Shah, D.D.; Zhang, J.; Maity, H.; Mallela, K.M.G. Effect of Photo-Degradation on the Structure, Stability, Aggregation, and Function of an IgG1 Monoclonal Antibody. Int. J. Pharm. 2018, 547, 438–449. [Google Scholar] [CrossRef]
- Du, C.; Barnett, G.; Borwankar, A.; Lewandowski, A.; Singh, N.; Ghose, S.; Borys, M.; Li, Z.J. Protection of Therapeutic Antibodies from Visible Light Induced Degradation: Use Safe Light in Manufacturing and Storage. Eur. J. Pharm. Biopharm. 2018, 127, 37–43. [Google Scholar] [CrossRef]
- Sreedhara, A.; Yin, J.; Joyce, M.; Lau, K.; Wecksler, A.T.; Deperalta, G.; Yi, L.; John Wang, Y.; Kabakoff, B.; Kishore, R.S.K. Effect of Ambient Light on IgG1 Monoclonal Antibodies during Drug Product Processing and Development. Eur. J. Pharm. Biopharm. 2016, 100, 38–46. [Google Scholar] [CrossRef]
- Gupta, S.; Jiskoot, W.; Schöneich, C.; Rathore, A.S. Oxidation and Deamidation of Monoclonal Antibody Products: Potential Impact on Stability, Biological Activity, and Efficacy. J. Pharm. Sci. 2022, 111, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Zbacnik, T.J.; Holcomb, R.E.; Katayama, D.S.; Murphy, B.M.; Payne, R.W.; Coccaro, R.C.; Evans, G.J.; Matsuura, J.E.; Henry, C.S.; Manning, M.C. Role of Buffers in Protein Formulations. J. Pharm. Sci. 2017, 106, 713–733. [Google Scholar] [CrossRef]
- Lebar, B.; Zidar, M.; Mravljak, J.; Šink, R.; Žula, A.; Pajk, S. Alternative Buffer Systems in Biopharmaceutical Formulations and Their Effect on Protein Stability. Acta Pharm. 2024, 74, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Ren, S. Effects of Arginine in Therapeutic Protein Formulations: A Decade Review and Perspectives. Antib. Ther. 2023, 6, 265–276. [Google Scholar] [CrossRef]
- Saito, S.; Hasegawa, J.; Kobayashi, N.; Tomitsuka, T.; Uchiyama, S.; Fukui, K. Effects of Ionic Strength and Sugars on the Aggregation Propensity of Monoclonal Antibodies: Influence of Colloidal and Conformational Stabilities. Pharm. Res. 2013, 30, 1263–1280. [Google Scholar] [CrossRef] [PubMed]
- Bahrenburg, S.; Karow, A.R.; Garidel, P. Buffer-Free Therapeutic Antibody Preparations Provide a Viable Alternative to Conventionally Buffered Solutions: From Protein Buffer Capacity Prediction to Bioprocess Applications. Biotechnol. J. 2015, 10, 610–622. [Google Scholar] [CrossRef]
- Chavez, B.K.; Agarabi, C.D.; Read, E.K.; Boyne, M.T.; Khan, M.A.; Brorson, K.A. Improved Stability of a Model IgG3 by DoE-Based Evaluation of Buffer Formulations. BioMed Res. Int. 2016, 2016, 2074149. [Google Scholar] [CrossRef]
- Salvi, G.; De Los Rios, P.; Vendruscolo, M. Effective Interactions between Chaotropic Agents and Proteins. Proteins 2005, 61, 492–499. [Google Scholar] [CrossRef]
- Kheddo, P.; Tracka, M.; Armer, J.; Dearman, R.J.; Uddin, S.; van der Walle, C.F.; Golovanov, A.P. The Effect of Arginine Glutamate on the Stability of Monoclonal Antibodies in Solution. Int. J. Pharm. 2014, 473, 126–133. [Google Scholar] [CrossRef]
- Olgenblum, G.I.; Carmon, N.; Harries, D. Not Always Sticky: Specificity of Protein Stabilization by Sugars Is Conferred by Protein–Water Hydrogen Bonds. J. Am. Chem. Soc. 2023, 145, 23308–23320. [Google Scholar] [CrossRef]
- Lv, J.-Y.; Ingle, R.G.; Wu, H.; Liu, C.; Fang, W.-J. Histidine as a Versatile Excipient in the Protein-Based Biopharmaceutical Formulations. Int. J. Pharm. 2024, 662, 124472. [Google Scholar] [CrossRef] [PubMed]
- Brovč, E.V.; Mravljak, J.; Šink, R.; Pajk, S. Degradation of Polysorbates 20 and 80 Catalysed by Histidine Chloride Buffer. Eur. J. Pharm. Biopharm. 2020, 154, 236–245. [Google Scholar] [CrossRef]
- Shen, K.; Hu, X.; Li, Z.; Liao, M.; Zhuang, Z.; Ruane, S.; Wang, Z.; Li, P.; Micciulla, S.; Kasinathan, N.; et al. Competitive Adsorption of a Monoclonal Antibody and Nonionic Surfactant at the PDMS/Water Interface. Mol. Pharm. 2023, 20, 2502–2512. [Google Scholar] [CrossRef]
- Kanthe, A.D.; Carnovale, M.R.; Katz, J.S.; Jordan, S.; Krause, M.E.; Zheng, S.; Ilott, A.; Ying, W.; Bu, W.; Bera, M.K.; et al. Differential Surface Adsorption Phenomena for Conventional and Novel Surfactants Correlates with Changes in Interfacial mAb Stabilization. Mol. Pharm. 2022, 19, 3100–3113. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.L.; McAuley, A.; McGuire, J. Protein Effects on Surfactant Adsorption Suggest the Dominant Mode of Surfactant-Mediated Stabilization of Protein. J. Pharm. Sci. 2014, 103, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Kannan, A.; Shieh, I.C.; Negulescu, P.G.; Chandran Suja, V.; Fuller, G.G. Adsorption and Aggregation of Monoclonal Antibodies at Silicone Oil-Water Interfaces. Mol. Pharm. 2021, 18, 1656–1665. [Google Scholar] [CrossRef]
- Grapentin, C.; Müller, C.; Kishore, R.S.K.; Adler, M.; ElBialy, I.; Friess, W.; Huwyler, J.; Khan, T.A. Protein-Polydimethylsiloxane Particles in Liquid Vial Monoclonal Antibody Formulations Containing Poloxamer 188. J. Pharm. Sci. 2020, 109, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Kanthe, A.D.; Krause, M.; Zheng, S.; Ilott, A.; Li, J.; Bu, W.; Bera, M.K.; Lin, B.; Maldarelli, C.; Tu, R.S. Armoring the Interface with Surfactants to Prevent the Adsorption of Monoclonal Antibodies. ACS Appl. Mater. Interfaces 2020, 12, 9977–9988. [Google Scholar] [CrossRef]
- Gerhardt, A.; Mcumber, A.C.; Nguyen, B.H.; Lewus, R.; Schwartz, D.K.; Carpenter, J.F.; Randolph, T.W. Surfactant Effects on Particle Generation in Antibody Formulations in Pre-Filled Syringes. J. Pharm. Sci. 2015, 104, 4056–4064. [Google Scholar] [CrossRef]
- Katz, J.S.; Chou, D.K.; Christian, T.R.; Das, T.K.; Patel, M.; Singh, S.N.; Wen, Y. Emerging Challenges and Innovations in Surfactant-Mediated Stabilization of Biologic Formulations. J. Pharm. Sci. 2022, 111, 919–932. [Google Scholar] [CrossRef]
- Armstrong, G.B.; Shah, V.; Patel, M.; Casey, R.; Jamieson, C.J.; Burley, G.A.; Lewis, W.J.; Rattray, Z. Enhancing Viscosity Control in Antibody Formulations: A Framework for the Biophysical Screening of Mutations Targeting Solvent-Accessible Hydrophobic and Electrostatic Patches. bioRxiv 2024. [Google Scholar] [CrossRef]
- Zidar, M.; Rozman, P.; Belko-Parkel, K.; Ravnik, M. Control of Viscosity in Biopharmaceutical Protein Formulations. J. Colloid Interface Sci. 2020, 580, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Prašnikar, M.; Proj, M.; Bjelošević Žiberna, M.; Lebar, B.; Knez, B.; Kržišnik, N.; Roškar, R.; Gobec, S.; Grabnar, I.; Žula, A.; et al. The Search for Novel Proline Analogs for Viscosity Reduction and Stabilization of Highly Concentrated Monoclonal Antibody Solutions. Int. J. Pharm. 2024, 655, 124055. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.; Tanenbaum, L.M.; Thirumangalathu, R.; Somani, S.; Zhang, K.; Kumar, V.; Amin, K.; Thakkar, S.V. Product-Specific Impact of Viscosity Modulating Formulation Excipients During Ultra-High Concentration Biotherapeutics Drug Product Development. J. Pharm. Sci. 2021, 110, 1077–1082. [Google Scholar] [CrossRef]
- Dear, B.J.; Hung, J.J.; Truskett, T.M.; Johnston, K.P. Contrasting the Influence of Cationic Amino Acids on the Viscosity and Stability of a Highly Concentrated Monoclonal Antibody. Pharm. Res. 2017, 34, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Friesner, R.A.; Berne, B.J. Competition of Electrostatic and Hydrophobic Interactions between Small Hydrophobes and Model Enclosures. J. Phys. Chem. B 2010, 114, 7294–7301. [Google Scholar] [CrossRef]
- Minton, A.P. Influence of Macromolecular Crowding upon the Stability and State of Association of Proteins: Predictions and Observations. J. Pharm. Sci. 2005, 94, 1668–1675. [Google Scholar] [CrossRef]
- Zeng, Y.; Tran, T.; Wuthrich, P.; Naik, S.; Davagnino, J.; Greene, D.G.; Mahoney, R.P.; Soane, D.S. Caffeine as a Viscosity Reducer for Highly Concentrated Monoclonal Antibody Solutions. J. Pharm. Sci. 2021, 110, 3594–3604. [Google Scholar] [CrossRef]
- Hu, Y.; Toth, R.T.; Joshi, S.B.; Esfandiary, R.; Middaugh, C.R.; Volkin, D.B.; Weis, D.D. Characterization of Excipient Effects on Reversible Self-Association, Backbone Flexibility, and Solution Properties of an IgG1 Monoclonal Antibody at High Concentrations: Part 2. J. Pharm. Sci. 2020, 109, 353–363. [Google Scholar] [CrossRef]
- Soeda, K.; Fukuda, M.; Takahashi, M.; Imai, H.; Arai, K.; Saitoh, S.; Kishore, R.S.K.; Oltra, N.S.; Duboeuf, J.; Hashimoto, D.; et al. Impact of Poloxamer 188 Material Attributes on Proteinaceous Visible Particle Formation in Liquid Monoclonal Antibody Formulations. J. Pharm. Sci. 2022, 111, 2191–2200. [Google Scholar] [CrossRef]
- Zürcher, D.; Caduff, S.; Aurand, L.; Capasso Palmiero, U.; Wuchner, K.; Arosio, P. Comparison of the Protective Effect of Polysorbates, Poloxamer and Brij on Antibody Stability Against Different Interfaces. J. Pharm. Sci. 2023, 112, 2853–2862. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, A.; Dagallier, C.; Huille, S.; Tribet, C. Poly(Glutamic Acid)-Based Viscosity Reducers for Concentrated Formulations of a Monoclonal IgG Antibody. Mol. Pharm. 2024, 21, 982–991. [Google Scholar] [CrossRef]
- Alkhawaja, B.; Al-Akayleh, F.; Al-Rubaye, Z.; AlDabet, G.; Bustami, M.; Smairat, M.; Agha, A.S.A.A.; Nasereddin, J.; Qinna, N.; Michael, A.; et al. Dissecting the Stability of Atezolizumab with Renewable Amino Acid-Based Ionic Liquids: Colloidal Stability and Anticancer Activity under Thermal Stress. Int. J. Biol. Macromol. 2024, 270, 132208. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hong, S.; Goh, L.Y.H.; Zhang, H.; Peng, T.; Chow, K.T.; Gokhale, R.; Tuliani, V. Investigation on the Combined Effect of Hydroxypropyl Beta-Cyclodextrin (HPβCD) and Polysorbate in Monoclonal Antibody Formulation. Pharmaceuticals 2024, 17, 528. [Google Scholar] [CrossRef]
- Messina, K.M.M.; Woys, A.M. Random Heteropolymer Excipients Improve the Colloidal Stability of a Monoclonal Antibody for Subcutaneous Administration. Pharm. Res. 2023, 40, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasan, S.; Rathore, A.S. Taurine, a Naturally Occurring Amino Acid, as a Physical Stability Enhancer of Different Monoclonal Antibodies. AAPS J. 2024, 26, 25. [Google Scholar] [CrossRef]
- Klich, J.H.; Kasse, C.M.; Mann, J.L.; Huang, Y.; d’Aquino, A.I.; Grosskopf, A.K.; Baillet, J.; Fuller, G.G.; Appel, E.A. Stable High-Concentration Monoclonal Antibody Formulations Enabled by an Amphiphilic Copolymer Excipient. Adv. Ther. 2023, 6, 2200102. [Google Scholar] [CrossRef]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor Regression in Cancer Patients by Very Low Doses of a T Cell-Engaging Antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef]
- Contag, C.H.; Bachmann, M.H. Advances in in Vivo Bioluminescence Imaging of Gene Expression. Annu. Rev. Biomed. Eng. 2002, 4, 235–260. [Google Scholar] [CrossRef]
- Smith, D.B.; Johnson, K.S. Single-Step Purification of Polypeptides Expressed in Escherichia Coli as Fusions with Glutathione S-Transferase. Gene 1988, 67, 31–40. [Google Scholar] [CrossRef]
- Mohler, K.M.; Torrance, D.S.; Smith, C.A.; Goodwin, R.G.; Stremler, K.E.; Fung, V.P.; Madani, H.; Widmer, M.B. Soluble Tumor Necrosis Factor (TNF) Receptors Are Effective Therapeutic Agents in Lethal Endotoxemia and Function Simultaneously as Both TNF Carriers and TNF Antagonists. J. Immunol. 1993, 151, 1548–1561. [Google Scholar] [CrossRef]
- Tsien, R.Y. The Green Fluorescent Protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef] [PubMed]
- Bornhorst, J.A.; Falke, J.J. Purification of Proteins Using Polyhistidine Affinity Tags. Methods Enzymol. 2000, 326, 245–254. [Google Scholar] [CrossRef]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. VEGF-Trap: A VEGF Blocker with Potent Antitumor Effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Challener, C. Fusion Proteins Pose Manufacturability Challenges. BioPharm Int. 2017, 30, 30–31, 37. [Google Scholar]
- Unverdorben, F.; Richter, F.; Hutt, M.; Seifert, O.; Malinge, P.; Fischer, N.; Kontermann, R.E. Pharmacokinetic Properties of IgG and Various Fc Fusion Proteins in Mice. mAbs 2016, 8, 120–128. [Google Scholar] [CrossRef]
- Demelenne, A.; Ben Yahia, A.; Lempereur, D.; Crommen, J.; Servais, A.-C.; Fradi, I.; Fillet, M. Comparison of Three Complementary Analytical Techniques for the Evaluation of the Biosimilar Comparability of a Monoclonal Antibody and an Fc-Fusion Protein. Front. Chem. 2021, 9, 782099. [Google Scholar] [CrossRef]
- Tang, S.; Tao, J.; Li, Y. Challenges and Solutions for the Downstream Purification of Therapeutic Proteins. Antib. Ther. 2023, 7, 1–12. [Google Scholar] [CrossRef]
- Berger, S.; Lowe, P.; Tesar, M. Fusion Protein Technologies for Biopharmaceuticals: Applications and Challenges. mAbs 2015, 7, 456–460. [Google Scholar] [CrossRef]
- Strand, J.; Huang, C.-T.; Xu, J. Characterization of Fc-Fusion Protein Aggregates Derived from Extracellular Domain Disulfide Bond Rearrangements. J. Pharm. Sci. 2013, 102, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Ballow, M. Monoclonal Antibodies and Fusion Proteins and Their Complications: Targeting B Cells in Autoimmune Diseases. J. Allergy Clin. Immunol. 2010, 125, 814–820. [Google Scholar] [CrossRef]
- Ueno, N.; Kashiwagi, M.; Kanekatsu, M.; Marubashi, W.; Yamada, T. Accumulation of Protein Aggregates Induces Autolytic Programmed Cell Death in Hybrid Tobacco Cells Expressing Hybrid Lethality. Sci. Rep. 2019, 9, 10223. [Google Scholar] [CrossRef]
- Ilyinskii, P.O.; Meriin, A.B.; Gabai, V.L.; Usachev, E.V.; Prilipov, A.G.; Thoidis, G.; Shneider, A.M. The Proteosomal Degradation of Fusion Proteins Cannot Be Predicted from the Proteosome Susceptibility of Their Individual Components. Protein Sci. 2008, 17, 1077–1085. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chakrabarti, S.; Barrow, C.J.; Kanwar, R.K.; Ramana, V.; Kanwar, J.R. Studies to Prevent Degradation of Recombinant Fc-Fusion Protein Expressed in Mammalian Cell Line and Protein Characterization. Int. J. Mol. Sci. 2016, 17, 913. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted Protein Degradation: Mechanisms, Strategies and Application. Signal Transduct. Target. Ther. 2022, 7, 113. [Google Scholar] [CrossRef]
- Gonzalez, K.J.; Huang, J.; Criado, M.F.; Banerjee, A.; Tompkins, S.; Mousa, J.J.; Strauch, E.-M. A General Computational Design Strategy for Stabilizing Viral Class I Fusion Proteins. bioRxiv 2023. [Google Scholar] [CrossRef]
- Liu, K.; Li, M.; Li, Y.; Li, Y.; Chen, Z.; Tang, Y.; Yang, M.; Deng, G.; Liu, H. A Review of the Clinical Efficacy of FDA-Approved Antibody‒drug Conjugates in Human Cancers. Mol. Cancer 2024, 23, 62. [Google Scholar] [CrossRef] [PubMed]
- ADC Approval up to 2023|BroadPharm. Available online: https://broadpharm.com/blog/ADC-Approval-up-to-2023 (accessed on 16 February 2025).
- Chen, H.; Lin, Z.; Arnst, K.E.; Miller, D.D.; Li, W. Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules 2017, 22, 1281. [Google Scholar] [CrossRef]
- Gogia, P.; Ashraf, H.; Bhasin, S.; Xu, Y. Antibody–Drug Conjugates: A Review of Approved Drugs and Their Clinical Level of Evidence. Cancers 2023, 15, 3886. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, Y.; Li, W.; Jeanty, C.; Xiang, G.; Dong, Y. Recent Advances of Antibody Drug Conjugates for Clinical Applications. Acta Pharm. Sin. B 2020, 10, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody-Drug Conjugates-A Tutorial Review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.-H.; Lin, Y.-H.; Luo, H.-L.; Sung, W.-W. Antibody-Drug Conjugates Targeting HER2 for the Treatment of Urothelial Carcinoma: Potential Therapies for HER2-Positive Urothelial Carcinoma. Front. Pharmacol. 2024, 15, 1326296. [Google Scholar] [CrossRef]
- Boylan, N.J.; Zhou, W.; Proos, R.J.; Tolbert, T.J.; Wolfe, J.L.; Laurence, J.S. Conjugation Site Heterogeneity Causes Variable Electrostatic Properties in Fc Conjugates. Bioconjug. Chem. 2013, 24, 1008–1016. [Google Scholar] [CrossRef]
- Tomar, D.S.; Kumar, S.; Singh, S.K.; Goswami, S.; Li, L. Molecular Basis of High Viscosity in Concentrated Antibody Solutions: Strategies for High Concentration Drug Product Development. mAbs 2016, 8, 216–228. [Google Scholar] [CrossRef]
- Mohamed, H.E.; Al-Ghobashy, M.A.; Abbas, S.S.; Boltia, S.A. Stability Assessment of Polatuzumab Vedotin and Brentuximab Vedotin Using Different Analytical Techniques. J. Pharm. Biomed. Anal. 2023, 228, 115249. [Google Scholar] [CrossRef]
- Ebrahimi, S.B.; Hong, X.; Ludlow, J.; Doucet, D.; Thirumangalathu, R. Studying Intermolecular Interactions in an Antibody-Drug Conjugate Through Chemical Screening and Computational Modeling. J. Pharm. Sci. 2023, 112, 2621–2628. [Google Scholar] [CrossRef]
- Gandhi, A.V.; Randolph, T.W.; Carpenter, J.F. Conjugation of Emtansine Onto Trastuzumab Promotes Aggregation of the Antibody–Drug Conjugate by Reducing Repulsive Electrostatic Interactions and Increasing Hydrophobic Interactions. J. Pharm. Sci. 2019, 108, 1973–1983. [Google Scholar] [CrossRef]
- Schumacher, D.; Hackenberger, C.P.R.; Leonhardt, H.; Helma, J. Current Status: Site-Specific Antibody Drug Conjugates. J. Clin. Immunol. 2016, 36 (Suppl. 1), 100–107. [Google Scholar] [CrossRef]
- Ross, P.L.; Wolfe, J.L. Physical and Chemical Stability of Antibody Drug Conjugates: Current Status. J. Pharm. Sci. 2016, 105, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Kumar, S.; Chipley, M.; Marcq, O.; Gupta, D.; Jin, Z.; Tomar, D.S.; Swabowski, C.; Smith, J.; Starkey, J.A.; et al. Characterization and Higher-Order Structure Assessment of an Interchain Cysteine-Based ADC: Impact of Drug Loading and Distribution on the Mechanism of Aggregation. Bioconjug. Chem. 2016, 27, 604–615. [Google Scholar] [CrossRef]
- Buecheler, J.W.; Winzer, M.; Tonillo, J.; Weber, C.; Gieseler, H. Impact of Payload Hydrophobicity on the Stability of Antibody–Drug Conjugates. Mol. Pharm. 2018, 15, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Johann, F.; Wöll, S.; Gieseler, H. “Negative” Impact: The Role of Payload Charge in the Physicochemical Stability of Auristatin Antibody-Drug Conjugates. J. Pharm. Sci. 2024, 113, 2433–2442. [Google Scholar] [CrossRef]
- Mills, B.J.; Kruger, T.; Bruncko, M.; Zhang, X.; Jameel, F. Effect of Linker-Drug Properties and Conjugation Site on the Physical Stability of ADCs. J. Pharm. Sci. 2020, 109, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody-Drug Conjugates. Bioconjug. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef]
- Fujii, T.; Reiling, C.; Quinn, C.; Kliman, M.; Mendelsohn, B.A.; Matsuda, Y. Physical Characteristics Comparison between Maytansinoid-Based and Auristatin-Based Antibody-Drug Conjugates. Explor. Target. Anti-Tumor Ther. 2021, 2, 576–585. [Google Scholar] [CrossRef]
- Buecheler, J.W.; Winzer, M.; Weber, C.; Gieseler, H. Alteration of Physicochemical Properties for Antibody-Drug Conjugates and Their Impact on Stability. J. Pharm. Sci. 2020, 109, 161–168. [Google Scholar] [CrossRef]
- Wuxi App Tec. Drug Conjugate Linkers and Their Effects on Drug Properties—WuXi AppTec DMPK Service. Available online: https://dmpkservice.wuxiapptec.com/articles/15-drug-conjugate-linkers-and-their-effects-on-drug-properties/ (accessed on 16 February 2025).
- Zhao, H.; Rubio, B.; Sapra, P.; Wu, D.; Reddy, P.; Sai, P.; Martinez, A.; Gao, Y.; Lozanguiez, Y.; Longley, C.; et al. Novel Prodrugs of SN38 Using Multiarm Poly(Ethylene Glycol) Linkers. Bioconjug. Chem. 2008, 19, 849–859. [Google Scholar] [CrossRef]
- Biopharma PEG Provides PEG Products Used as ADC Linker. Available online: https://www.clinicalresearchnewsonline.com/news/2023/01/18/biopharma-peg-provides-peg-products-used-as-adc-linker (accessed on 16 February 2025).
- Trodelvy (Sacituzumab Govitecan)|BroadPharm. Available online: https://broadpharm.com/blog/trodelvy-sacituzumab-govitecan-breast-cancer (accessed on 16 February 2025).
- Li, Q.; Li, W.; Xu, K.; Xing, Y.; Shi, H.; Jing, Z.; Li, S.; Hong, Z. PEG Linker Improves Antitumor Efficacy and Safety of Affibody-Based Drug Conjugates. Int. J. Mol. Sci. 2021, 22, 1540. [Google Scholar] [CrossRef]
- Sonzini, S.; Greco, M.L.; Cailleau, T.; Adams, L.; Masterson, L.; Vijayakrishnan, B.; Barry, C.; Howard, P.; Ravn, P.; van der Walle, C.F. Improved Physical Stability of an Antibody-Drug Conjugate Using Host-Guest Chemistry. Bioconjug. Chem. 2020, 31, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Tedeschini, T.; Campara, B.; Grigoletto, A.; Bellini, M.; Salvalaio, M.; Matsuno, Y.; Suzuki, A.; Yoshioka, H.; Pasut, G. Polyethylene Glycol-Based Linkers as Hydrophilicity Reservoir for Antibody-Drug Conjugates. J. Control. Release 2021, 337, 431–447. [Google Scholar] [CrossRef] [PubMed]
- Dian, S.; Donglu, Z. Linker Design Impacts Antibody-Drug Conjugate Pharmacokinetics and Efficacy via Modulating the Stability and Payload Release Efficiency. Front. Pharmacol. 2021, 12, 687926. [Google Scholar] [CrossRef]
- Karunaratne, S.P.; Moussa, E.M.; Mills, B.J.; Weis, D.D. Understanding the Effects of Site-Specific Light Chain Conjugation on Antibody Structure Using Hydrogen Exchange-Mass Spectrometry (HX-MS). J. Pharm. Sci. 2024, 113, 2065–2071. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.-Y.; Hsieh, Y.-C.; Fung, K.-S.; Huang, Y.-C.; Shia, C.-S.; Chiang, M.-F.; Wang, N.-H.; Li, W.-F.; Lai, M.-T. Abstract 3149: Development of a Novel Site-Specific ADC Glycan Platform with Potential for Improved In Vivo Efficacy and Stability of the ADC in Animal Studies. Cancer Res. 2024, 84, 3149. [Google Scholar] [CrossRef]
- Kaempffe, A.; Dickgiesser, S.; Rasche, N.; Paoletti, A.; Bertotti, E.; De Salve, I.; Sirtori, F.R.; Kellner, R.; Könning, D.; Hecht, S.; et al. Effect of Conjugation Site and Technique on the Stability and Pharmacokinetics of Antibody-Drug Conjugates. J. Pharm. Sci. 2021, 110, 3776–3785. [Google Scholar] [CrossRef]
- Gandhi, A.V.; Arlotta, K.J.; Chen, H.-N.; Owen, S.C.; Carpenter, J.F. Biophysical Properties and Heating-Induced Aggregation of Lysine-Conjugated Antibody-Drug Conjugates. J. Pharm. Sci. 2018, 107, 1858–1869. [Google Scholar] [CrossRef]
- Rakotoarinoro, N.; Dyck, Y.F.K.; Krebs, S.K.; Assi, M.-K.; Parr, M.K.; Stech, M. A Disruptive Clickable Antibody Design for the Generation of Antibody-Drug Conjugates. Antib. Ther. 2023, 6, 298–310. [Google Scholar] [CrossRef]
- Dorywalska, M.; Strop, P.; Melton-Witt, J.A.; Hasa-Moreno, A.; Farias, S.E.; Galindo Casas, M.; Delaria, K.; Lui, V.; Poulsen, K.; Loo, C.; et al. Effect of Attachment Site on Stability of Cleavable Antibody Drug Conjugates. Bioconjug. Chem. 2015, 26, 650–659. [Google Scholar] [CrossRef]
- Strop, P.; Liu, S.-H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.-T.; Ho, W.-H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-Selective Modification Strategies in Antibody–Drug Conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef] [PubMed]
- Ferhati, X.; Jiménez-Moreno, E.; Hoyt, E.A.; Salluce, G.; Cabeza-Cabrerizo, M.; Navo, C.D.; Compañón, I.; Akkapeddi, P.; Matos, M.J.; Salaverri, N.; et al. Single Mutation on Trastuzumab Modulates the Stability of Antibody-Drug Conjugates Built Using Acetal-Based Linkers and Thiol-Maleimide Chemistry. J. Am. Chem. Soc. 2022, 144, 5284–5294. [Google Scholar] [CrossRef] [PubMed]
- Dovgan, I.; Ehkirch, A.; Lehot, V.; Kuhn, I.; Koniev, O.; Kolodych, S.; Hentz, A.; Ripoll, M.; Ursuegui, S.; Nothisen, M.; et al. On the Use of DNA as a Linker in Antibody-Drug Conjugates: Synthesis, Stability and in Vitro Potency. Sci. Rep. 2020, 10, 7691. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.J.; Song, D.; Kim, J.; Kim, J.Y.; Eom, J.; Sung, B.; Son, Y.-G.; Kim, Y.M.; Lee, S.H.; You, W.-K.; et al. N-Terminal Selective Conjugation Method Widens the Therapeutic Window of Antibody-Drug Conjugates by Improving Tolerability and Stability. mAbs 2021, 13, 1914885. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, F.; Liu, L.; Xu, X.; Fan, S.; Zhong, W.; Zhou, X. Development of Applicable Thiol-Linked Antibody-Drug Conjugates with Improved Stability and Therapeutic Index. Drug. Deliv. 2022, 29, 754–766. [Google Scholar] [CrossRef]
- Watanabe, T.; Arashida, N.; Fujii, T.; Shikida, N.; Ito, K.; Shimbo, K.; Seki, T.; Iwai, Y.; Hirama, R.; Hatada, N.; et al. Exo-Cleavable Linkers: A Paradigm Shift for Enhanced Stability and Therapeutic Efficacy in Antibody-Drug Conjugates. ChemRxiv 2023. [Google Scholar] [CrossRef]
- Goldberg, S.D.; Satomaa, T.; Aina, O.; Aitio, O.; Burke, K.; Dudkin, V.; Geist, B.; Irrechukwu, O.; Hänninen, A.-L.; Heiskanen, A.; et al. Trastuzumab-MMAU Antibody-Auristatin Conjugates: Valine-Glucoserine Linker with Stabilized Maleimide Conjugation Improves In Vivo Efficacy and Tolerability. Mol. Cancer Ther. 2024, 23, 1530–1543. [Google Scholar] [CrossRef]
- Chuprakov, S.; Ogunkoya, A.O.; Barfield, R.M.; Bauzon, M.; Hickle, C.; Kim, Y.C.; Yeo, D.; Zhang, F.; Rabuka, D.; Drake, P.M. Tandem-Cleavage Linkers Improve the In Vivo Stability and Tolerability of Antibody–Drug Conjugates. Bioconjug. Chem. 2021, 32, 746–754. [Google Scholar] [CrossRef]
- Zheng, Y.; Xu, R.; Cheng, H.; Tai, W. Mono-Amino Acid Linkers Enable Highly Potent Small Molecule-Drug Conjugates by Conditional Release. Mol. Ther. 2024, 32, 1048–1060. [Google Scholar] [CrossRef]
- Xiao, D.; Liu, L.; Xie, F.; Dong, J.; Wang, Y.; Xu, X.; Zhong, W.; Deng, H.; Zhou, X.; Li, S. Azobenzene-Based Linker Strategy for Selective Activation of Antibody-Drug Conjugates. Angew. Chem. Int. Ed. 2024, 63, e202310318. [Google Scholar] [CrossRef]
- Li, X.; Patel, N.; Kalen, J.; Schnermann, M. Alpha-Ammonium Carbamates Undergo Efficient Two-Step Linker Cleavage and Improve the Properties of Antibody Conjugates. ChemRxiv 2024. [Google Scholar] [CrossRef]
- Duerr, C.; Friess, W. Antibody-Drug Conjugates-Stability and Formulation. Eur. J. Pharm. Biopharm. 2019, 139, 168–176. [Google Scholar] [CrossRef]
- Sreedhara, A.; Glover, Z.K.; Piros, N.; Xiao, N.; Patel, A.; Kabakoff, B. Stability of IgG1 Monoclonal Antibodies in Intravenous Infusion Bags under Clinical In-Use Conditions. J. Pharm. Sci. 2012, 101, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Johann, F.; Wöll, S.; Gieseler, H. Evaluating the Potential of Cyclodextrins in Reducing Aggregation of Antibody–Drug Conjugates with Different Payloads. J. Pharm. Sci. 2024, 113, 2443–2453. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).