
Metformin-Based Combination Approaches for Triple-Negative Breast Cancer



Abstract
1. Introduction
2. Metformin and Its Mechanisms
3. Implications of Metformin and Metformin-Based Combination Approaches for TNBC
3.1. Evidence from In Vitro and In Vivo Studies
3.1.1. Metformin’s Effects on Metabolic Pathways
3.1.2. Metformin’s Effects in Combination Therapy Efficacy
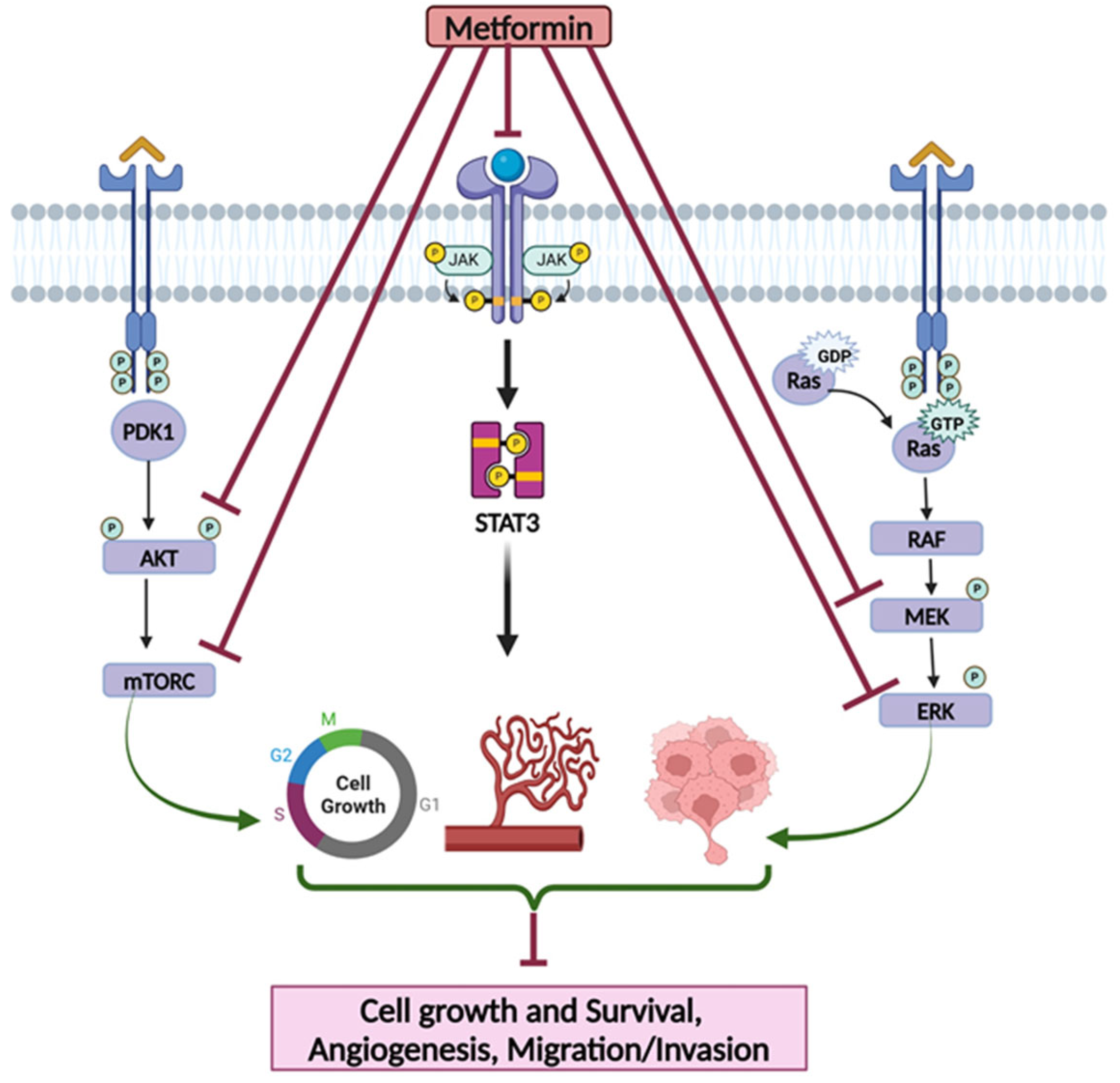
3.1.3. Metformin’s Effects on Targeting Cell Signaling Pathways
3.1.4. Approaches to Enhance Metformin’s Effects via Its Delivery Through Nanoparticles
3.2. Evidence from Clinical Studies
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BC | Breast cancer |
| TNBC | Triple-negative breast cancer |
| ER | Estrogen |
| PR | Progesterone |
| HER2 | Human epidermal growth factor receptor 2 |
| T2D | Type 2 diabetes |
| IGF-1 | Insulin-like growth factor 1 |
| TNF-α | Tumor necrosis factor-α |
| IRS1 | Insulin receptor substrate |
| ROS | Reactive oxygen species |
| IR | Insulin receptor |
| IGFBP-1 | IGF-Binding Protein 1 |
| 2-DG | 2-deoxyglucose |
| GLUT | Glucose transport proteins |
| TNB | Triple-Negative B |
| TNA | Triple-Negative A |
| EMT | Epithelial–mesenchymal transition |
| PI3KI | Phosphatidylinositol-3-kinase inhibitor |
| IGF-1R | Insulin-like growth factor 1 receptor |
| CI | Combination index |
| RPPA | Reverse phase protein lysate microarray |
| MTD | Maximum tolerated dose |
| CSC | Cancer stem cells |
| CMG | Metformin and gefitinib |
| ALDH+ | Aldehyde dehydrogenase |
| HIF | Hypoxia-inducible factor |
| TRAIL | TNF-related apoptosis-inducing ligand |
| XIAP | X-linked inhibitor of apoptosis protein |
| RLN | Relaxin |
| BACH1 | BTB and CNC homology 1 |
| ECAR | Metabolism, extracellular acidification rate |
| OCR | Oxygen consumption rate |
| OCT3 | Organic cation transporter 3 |
| CRP | C-reactive protein |
| SHBG | Sex hormone-binding globulin |
References
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Yeo, S.K.; Guan, J.L. Breast Cancer: Multiple Subtypes within a Tumor? Trends Cancer 2017, 3, 753–760. [Google Scholar] [CrossRef]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast Cancer Development and Progression: Risk Factors, Cancer Stem Cells, Signaling Pathways, Genomics, and Molecular Pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Sirhan, Z.; Thyagarajan, A.; Sahu, R.P. The Efficacy of Tucatinib-Based Therapeutic Approaches for HER2-Positive Breast Cancer. Mil. Med. Res. 2022, 9, 39. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, M.; Sun, Y.; Chen, J.; Chen, C.; Qian, C.; Chen, Y.; Cao, L.; Xu, Q.; Du, X.; et al. Gene Expression Profiling for Diagnosis of Triple-Negative Breast Cancer: A Multicenter, Retrospective Cohort Study. Front. Oncol. 2019, 9, 354. [Google Scholar] [CrossRef]
- Kumar, P.; Aggarwal, R. An Overview of Triple-Negative Breast Cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef]
- Derakhshan, F.; Reis-Filho, J.S. Pathogenesis of Triple-Negative Breast Cancer. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 181–204. [Google Scholar] [CrossRef]
- Baranova, A.; Krasnoselskyi, M.; Starikov, V.; Kartashov, S.; Zhulkevych, I.; Vlasenko, V.; Oleshko, K.; Bilodid, O.; Sadchikova, M.; Vinnyk, Y. Triple-Negative Breast Cancer: Current Treatment Strategies and Factors of Negative Prognosis. J. Med. Life 2022, 2022, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, M.; Bekele, F.; Fekadu, G. Treatment Strategies Against Triple-Negative Breast Cancer: An Updated Review. Breast Cancer Targets Ther. 2022, 14, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.F. Global, Regional, and National Burden and Trend of Diabetes in 195 Countries and Territories: An Analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and Cancer: A Consensus Report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef]
- Samuel, S.M.; Varghese, E.; Varghese, S.; Büsselberg, D. Challenges and Perspectives in the Treatment of Diabetes Associated Breast Cancer. Cancer Treat. Rev. 2018, 70, 98–111. [Google Scholar] [CrossRef]
- Min, W.; Wang, B.; Guo, A.; Mao, G.; Zhao, Y.; Zhang, S.; He, R.; Min, Y.; Huang, Y. The Effect of Metformin on the Clinicopathological Features of Breast Cancer with Type 2 Diabetes. World J. Oncol. 2020, 11, 23–32. [Google Scholar] [CrossRef]
- Cejuela, M.; Martin-Castillo, B.; Menendez, J.A.; Pernas, S. Metformin and Breast Cancer: Where Are We Now? Int. J. Mol. Sci. 2022, 23, 2705. [Google Scholar] [CrossRef]
- Corleto, K.A.; Strandmo, J.L.; Giles, E.D. Metformin and Breast Cancer: Current Findings and Future Perspectives from Preclinical and Clinical Studies. Pharmaceuticals 2024, 17, 396. [Google Scholar] [CrossRef]
- Lord, S.R.; Harris, A.L. Is It Still Worth Pursuing the Repurposing of Metformin as a Cancer Therapeutic? Br. J. Cancer 2023, 128, 958–966. [Google Scholar] [CrossRef]
- Wahdan-Alaswad, R.S.; Edgerton, S.M.; Salem, H.S.; Thor, A.D. Metformin Targets Glucose Metabolism in Triple Negative Breast Cancer. J. Oncol. Transl. Res. 2018, 4, 129. [Google Scholar] [CrossRef]
- Jordt, N.; Kjærgaard, K.A.; Thomsen, R.W.; Borgquist, S.; Cronin-Fenton, D. Breast Cancer and Incidence of Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis. Breast Cancer Res. Treat. 2023, 202, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Matou-Nasri, S.; Aldawood, M.; Alanazi, F.; Khan, A.L. Updates on Triple-Negative Breast Cancer in Type 2 Diabetes Mellitus Patients: From Risk Factors to Diagnosis, Biomarkers and Therapy. Diagnostics 2023, 13, 2390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; de Haan-Du, J.; Sidorenkov, G.; Landman, G.W.D.; Jalving, M.; Zhang, Q.; de Bock, G.H. Type 2 Diabetes Mellitus and Clinicopathological Tumor Characteristics in Women Diagnosed with Breast Cancer: A Systematic Review and Meta-Analysis. Cancers 2021, 13, 4992. [Google Scholar] [CrossRef]
- Eketunde, A.O. Diabetes as a Risk Factor for Breast Cancer. Cureus 2020, 12, e8010. [Google Scholar] [CrossRef]
- Bashraheel, S.S.; Kheraldine, H.; Khalaf, S.; Al Moustafa, A.E. Metformin and HER2-Positive Breast Cancer: Mechanisms and Therapeutic Implications. Biomed. Pharmacother. 2023, 162, 114676. [Google Scholar] [CrossRef]
- Christopoulos, P.F.; Msaouel, P.; Koutsilieris, M. The Role of the Insulin-like Growth Factor-1 System in Breast Cancer. Mol. Cancer 2015, 14, 43. [Google Scholar] [CrossRef]
- Durrani, I.A.; Bhatti, A.; John, P. The Prognostic Outcome of ‘Type 2 Diabetes Mellitus and Breast Cancer’ Association Pivots on Hypoxia-Hyperglycemia Axis. Cancer Cell Int. 2021, 21, 351. [Google Scholar] [CrossRef]
- Kurelac, I.; Umesh Ganesh, N.; Iorio, M.; Porcelli, A.M.; Gasparre, G. The Multifaceted Effects of Metformin on Tumor Microenvironment. Semin. Cell Dev. Biol. 2020, 98, 90–97. [Google Scholar] [CrossRef]
- Samuel, S.M.; Varghese, E.; Koklesová, L.; Líšková, A.; Kubatka, P.; Büsselberg, D. Counteracting Chemoresistance with Metformin in Breast Cancers: Targeting Cancer Stem Cells. Cancers 2020, 12, 2482. [Google Scholar] [CrossRef]
- Yee, L.D.; Mortimer, J.E.; Natarajan, R.; Dietze, E.C.; Seewaldt, V.L. Metabolic Health, Insulin, and Breast Cancer: Why Oncologists Should Care About Insulin. Front. Endocrinol. 2020, 11, 58. [Google Scholar] [CrossRef]
- Khan, I.; Kamal, A.; Akhtar, S. Diabetes Driven Oncogenesis and Anticancer Potential of Repurposed Antidiabetic Drug: A Systemic Review. Cell Biochem. Biophys. 2024, 82, 1907–1929. [Google Scholar] [CrossRef] [PubMed]
- García-Estévez, L.; Cortés, J.; Pérez, S.; Calvo, I.; Gallegos, I.; Moreno-Bueno, G. Obesity and Breast Cancer: A Paradoxical and Controversial Relationship Influenced by Menopausal Status. Front. Oncol. 2021, 11, 705911. [Google Scholar] [CrossRef]
- Sahu, P.; Camarillo, I.G.; Sundararajan, R. Efficacy of Metformin and Electrical Pulses in Breast Cancer MDA-MB-231 Cells. Explor. Target. Antitumor Ther. 2024, 5, 54–73. [Google Scholar] [CrossRef] [PubMed]
- Mallik, R.; Chowdhury, T.A. Metformin in Cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419. [Google Scholar] [CrossRef]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, Regional, and National Prevalence of Overweight and Obesity in Children and Adults during 1980-2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Stambolic, V. Impact of the Obesity Epidemic on Cancer. Annu. Rev. Med. 2015, 66, 281–296. [Google Scholar] [CrossRef]
- Song, J.; Du, J.; Han, L.; Lin, X.; Fan, C.; Chen, G. The Effect of Metformin on Triple-Negative Breast Cancer Cells and Nude Mice ORIGINAL RESEARCH. Altern. Ther. 2023, 29, 389–395. [Google Scholar]
- Lyu, X.; Zhang, Q.; Fares, H.M.; Wang, Y.; Han, Y.; Sun, L. Contribution of Adipocytes in the Tumor Microenvironment to Breast Cancer Metabolism. Cancer Lett. 2022, 534, 215616. [Google Scholar] [CrossRef]
- Verras, G.I.; Tchabashvili, L.; Chlorogiannis, D.D.; Mulita, F.; Argentou, M.I. Updated Clinical Evidence on the Role of Adipokines and Breast Cancer: A Review. Cancers 2023, 15, 1572. [Google Scholar] [CrossRef]
- Georgiou, G.P.; Provatopoulou, X.; Kalogera, E.; Siasos, G.; Menenakos, E.; Zografos, G.C.; Gounaris, A. Serum Resistin Is Inversely Related to Breast Cancer Risk in Premenopausal Women. Breast 2016, 29, 163–169. [Google Scholar] [CrossRef]
- Niu, J.; Jiang, L.; Guo, W.; Shao, L.; Liu, Y.; Wang, L. The Association between Leptin Level and Breast Cancer: A Meta-Analysis. PLoS ONE 2013, 8, e67349. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shukla, S.; Sinha, S.; Meeran, S.M. Role of Adipokines and Cytokines in Obesity-Associated Breast Cancer: Therapeutic Targets. Cytokine Growth Factor Rev. 2013, 24, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Koprivčić, I.; Marjanović, K.; Matić, A.; Levak, M.T.; Lovrić, I.; Pauzar, B.; Erić, I.; Wertheimer, V. SERUM LEPTIN LEVEL IN BREAST CANCER. Acta Clin. Croat. 2022, 61, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Samson, S.L.; Vellanki, P.; Blonde, L.; Christofides, E.A.; Galindo, R.J.; Hirsch, I.B.; Isaacs, S.D.; Izuora, K.E.; Low Wang, C.C.; Twining, C.L.; et al. American Association of Clinical Endocrinology Consensus Statement: Comprehensive Type 2 Diabetes Management Algorithm—2023 Update. Endocr. Pract. 2023, 29, 305–340. [Google Scholar] [CrossRef]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Mahmoudi, G.; Ehteshaminia, Y.; Kokhaei, P.; Jalali, S.F.; Jadidi-Niaragh, F.; Pagheh, A.S.; Enderami, S.E.; Kenari, S.A.; Hassannia, H. Enhancement of Targeted Therapy in Combination with Metformin on Human Breast Cancer Cell Lines. Cell Commun. Signal. 2024, 22, 10. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on Mechanisms of Action and Repurposing Potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin Inhibits Breast Cancer Cell Growth, Colony Formation and Induces Cell Cycle Arrest in Vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef]
- Stoian, A.M.P.; Rizzo, M. Metformin; IntechOpen: Rijeka, Croatia, 2020; ISBN 978-1-83880-428-2. [Google Scholar]
- Deng, X.S.; Wang, S.; Deng, A.; Liu, B.; Edgerton, S.M.; Lind, S.E.; Wahdan-Alaswad, R.; Thor, A.D. Metformin Targets Stat3 to Inhibit Cell Growth and Induce Apoptosis in Triple-Negative Breast Cancers. Cell Cycle 2012, 11, 367–376. [Google Scholar] [CrossRef]
- Amaral, I.; Silva, C.; Correia-Branco, A.; Martel, F. Effect of Metformin on Estrogen and Progesterone Receptor-Positive (MCF-7) and Triple-Negative (MDA-MB-231) Breast Cancer Cells. Biomed. Pharmacother. 2018, 102, 94–101. [Google Scholar] [CrossRef]
- Cingir Koker, S.; Yalcin, B.; Dogan Turacli, I. Metformin Resistant MDA-MB-468 Cells Exhibit EMT-like Phenotype and Increased Migration Capacity. Mol. Biol. Rep. 2022, 49, 5973–5984. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Q.; Yang, J.; He, W.; Jiang, Y.; Chen, Y.; Wang, Y. Metformin Combined with Glucose Starvation Synergistically Suppress Triple-Negative Breast Cancer by Enhanced Unfolded Protein Response. Biochem. Biophys. Res. Commun. 2023, 675, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.M.; Varghese, E.; Satheesh, N.J.; Triggle, C.R.; Büsselberg, D. Metabolic Heterogeneity in TNBCs: A Potential Determinant of Therapeutic Efficacy of 2-Deoxyglucose and Metformin Combinatory Therapy. Biomed. Pharmacother. 2023, 164, 114911. [Google Scholar] [CrossRef] [PubMed]
- Rico, M.; Baglioni, M.; Bondarenko, M.; Laluce, N.C.; Rozados, V.; André, N.; Carré, M.; Scharovsky, O.G.; Márquez, M.M. Metformin and Propranolol Combination Prevents Cancer Progression and Metastasis in Different Breast Cancer Models. Oncotarget 2017, 8, 2874–2889. [Google Scholar] [CrossRef]
- Anselmino, L.E.; Baglioni, M.V.; Malizia, F.; Laluce, N.C.; Etichetti, C.B.; Marignac, V.L.M.; Rozados, V.; Scharovsky, O.G.; Girardini, J.; Rico, M.J.; et al. Repositioning Metformin and Propranolol for Colorectal and Triple Negative Breast Cancers Treatment. Sci. Rep. 2021, 11, 8091. [Google Scholar] [CrossRef]
- Repas, J.; Zupin, M.; Vodlan, M.; Veranič, P.; Gole, B.; Potočnik, U.; Pavlin, M. Dual Effect of Combined Metformin and 2-Deoxy-D-Glucose Treatment on Mitochondrial Biogenesis and PD-L1 Expression in Triple-Negative Breast Cancer Cells. Cancers 2022, 14, 1343. [Google Scholar] [CrossRef]
- Tan, X.; Li, Y.; Hou, Z.; Zhang, M.; Li, L.; Wei, J. Combination Therapy with PD-1 Inhibition plus Rapamycin and Metformin Enhances Anti-Tumor Efficacy in Triple Negative Breast Cancer. Exp. Cell Res. 2023, 429, 113647. [Google Scholar] [CrossRef]
- Xue, L.; Chen, F.; Yue, F.; Camacho, L.; Kothapalli, S.; Wei, G.; Huang, S.; Mo, Q.; Ma, F.; Li, Y.; et al. Metformin and an Insulin/IGF-1 Receptor Inhibitor Are Synergistic in Blocking Growth of Triple-Negative Breast Cancer. Breast Cancer Res. Treat. 2021, 185, 73–84. [Google Scholar] [CrossRef]
- Cheng, T.; Wang, C.; Lu, Q.; Cao, Y.; Yu, W.; Li, W.; Liu, B.; Gao, X.; Lü, J.; Pan, X. Metformin Inhibits the Tumor-Promoting Effect of Low-Dose Resveratrol, and Enhances the Anti-Tumor Activity of High-Dose Resveratrol by Increasing Its Reducibility in Triple Negative Breast Cancer. Free Radic. Biol. Med. 2022, 180, 108–120. [Google Scholar] [CrossRef]
- Lee, J.O.; Kang, M.J.; Byun, W.S.; Kim, S.A.; Seo, I.H.; Han, J.A.; Moon, J.W.; Kim, J.H.; Kim, S.J.; Lee, E.J.; et al. Metformin Overcomes Resistance to Cisplatin in Triple-Negative Breast Cancer (TNBC) Cells by Targeting RAD51. Breast Cancer Res. 2019, 21, 115. [Google Scholar] [CrossRef]
- Sahu, P.; Camarillo, I.G.; Sundararajan, R. Enhanced Antiproliferation Potency of Electrical Pulse-Mediated Metformin and Cisplatin Combination Therapy on MDA-MB-231 Cells. Appl. Biochem. Biotechnol. 2022, 194, 18–36. [Google Scholar] [CrossRef] [PubMed]
- Babak, M.V.; Chong, K.R.; Rapta, P.; Zannikou, M.; Tang, H.M.; Reichert, L.; Chang, M.R.; Kushnarev, V.; Heffeter, P.; Meier-Menches, S.M.; et al. Interfering with Metabolic Profile of Triple-Negative Breast Cancers Using Rationally Designed Metformin Prodrugs. Angew. Chem.—Int. Ed. 2021, 60, 13405–13413. [Google Scholar] [CrossRef] [PubMed]
- Saeed, H.K.; Sutar, Y.; Patel, P.; Bhat, R.; Mallick, S.; Hatada, A.E.; Koomoa, D.L.T.; Lange, I.; Date, A.A. Synthesis and Characterization of Lipophilic Salts of Metformin to Improve Its Repurposing for Cancer Therapy. ACS Omega 2021, 6, 2626–2637. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; Chambers, J.; Al-Kadi, E.; Phan, A.; Li, L.; Mediratta, K.; Dimitroulakos, J.; Addison, C.; Li, X.; et al. Targeting Hypoxia Sensitizes TNBC to Cisplatin and Promotes Inhibition of Both Bulk and Cancer Stem Cells. Int. J. Mol. Sci. 2020, 21, 5788. [Google Scholar] [CrossRef]
- Aoun, R.; El Hadi, C.; Tahtouh, R.; El Habre, R.; Hilal, G. Microarray Analysis of Breast Cancer Gene Expression Profiling in Response to 2-Deoxyglucose, Metformin, and Glucose Starvation. Cancer Cell Int. 2022, 22, 123. [Google Scholar] [CrossRef]
- Liu, S.; Polsdofer, E.V.; Zhou, L.; Ruan, S.; Lyu, H.; Hou, D.; Liu, H.; Thor, A.D.; He, Z.; Liu, B. Upregulation of Endogenous TRAIL-Elicited Apoptosis Is Essential for Metformin-Mediated Antitumor Activity against TNBC and NSCLC. Mol. Ther. Oncolytics 2021, 21, 303–314. [Google Scholar] [CrossRef]
- Cai, H.; Everett, R.S.; Thakker, D.R. Efficacious Dose of Metformin for Breast Cancer Therapy Is Determined by Cation Transporter Expression in Tumours. Br. J. Pharmacol. 2019, 176, 2724–2735. [Google Scholar] [CrossRef]
- Pateliya, B.; Burade, V.; Goswami, S. Combining Naringenin and Metformin with Doxorubicin Enhances Anticancer Activity against Triple-Negative Breast Cancer in Vitro and in Vivo. Eur. J. Pharmacol. 2021, 891, 173725. [Google Scholar] [CrossRef]
- Strekalova, E.; Malin, D.; Rajanala, H.; Cryns, V.L. Metformin Sensitizes Triple-Negative Breast Cancer to Proapoptotic TRAIL Receptor Agonists by Suppressing XIAP Expression. Breast Cancer Res. Treat. 2017, 163, 435–447. [Google Scholar] [CrossRef]
- Lee, J.; Yesilkanal, A.E.; Wynne, J.P.; Frankenberger, C.; Liu, J.; Yan, J.; Elbaz, M.; Rabe, D.C.; Rustandy, F.D.; Tiwari, P.; et al. Effective Breast Cancer Combination Therapy Targeting BACH1 and Mitochondrial Metabolism. Nature 2019, 568, 254–258. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Zhao, Y.; Shi, F.; Zhou, Q.; Wu, J.; Lyu, S.; Song, Q. Metformin Inducing the Change of Functional and Exhausted Phenotypic Tumor-Infiltrated Lymphocytes and the Correlation with JNK Signal Pathway in Triple-Negative Breast Cancer. Breast Cancer Targets Ther. 2022, 14, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Singh, H.; Trinidad, C.M.; Albarracin, C.T.; Hunt, K.K.; Bedrosian, I. The IsomiR-140-3p-Regulated Mevalonic Acid Pathway as a Potential Target for Prevention of Triple Negative Breast Cancer. Breast Cancer Res. 2018, 20, 150. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cai, H.; Xiong, Y.; Tang, L.; Li, L.; Zhang, L.; Shen, Y.; Yang, Y.; Lin, L.; Huang, J. YAP/TAZ Axis Was Involved in the Effects of Metformin on Breast Cancer. J. Chemother. 2023, 35, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Upadhyay, P.; Ghosh, A.; Bose, A.; Gupta, P.; Chattopadhyay, S.; Chattopadhyay, D.; Adhikary, A. Hyaluronic Acid Engrafted Metformin Loaded Graphene Oxide Nanoparticle as CD44 Targeted Anti-Cancer Therapy for Triple Negative Breast Cancer. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129841. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, L.; Zhao, Y.; Luo, N.; Shi, J.; Xu, S.; Ma, L.; Wang, M.; Gu, M.; Mu, C.; et al. Relaxin-Encapsulated Polymeric Metformin Nanoparticles Remodel Tumor Immune Microenvironment by Reducing CAFs for Efficient Triple-Negative Breast Cancer Immunotherapy. Asian J. Pharm. Sci. 2023, 18, 100796. [Google Scholar] [CrossRef]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef]
- Attri, K.S.; Park, J.H.; Kaipparettu, B.A. Redox Regulation of Hybrid Metabolic State in Breast Cancer Metastasis. Ann. Transl. Med. 2022, 10, 1032. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells without Mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting Mitochondria Metabolism for Cancer Therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Xu, Q.; Biener-Ramanujan, E.; Yang, W.; Ramanujan, V.K. Targeting Metabolic Plasticity in Breast Cancer Cells via Mitochondrial Complex I Modulation. Breast Cancer Res. Treat. 2015, 150, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Jung, K.H.; Jia, D.; Yang, S.; Attri, K.S.; Ahn, S.; Murthy, D.; Samanta, T.; Dutta, D.; Ghidey, M.; et al. Biguanides Antithetically Regulate Tumor Properties by the Dose-Dependent Mitochondrial Reprogramming-Driven c-Src Pathway. Cell Rep. Med. 2025, 6, 101941. [Google Scholar] [CrossRef] [PubMed]
- Fenn, K.; Maurer, M.; Lee, S.M.; Crew, K.D.; Trivedi, M.S.; Accordino, M.K.; Hershman, D.L.; Kalinsky, K. Phase 1 Study of Erlotinib and Metformin in Metastatic Triple-Negative Breast Cancer. Clin. Breast Cancer 2020, 20, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.E.; Marinac, C.R.; Sears, D.D.; Kerr, J.; Hartman, S.J.; Cadmus-Bertram, L.; Villaseñor, A.; Flatt, S.W.; Godbole, S.; Li, H.; et al. The Effects of Metformin and Weight Loss on Biomarkers Associated with Breast Cancer Outcomes. J. Natl. Cancer Inst. 2018, 110, 1239–1247. [Google Scholar] [CrossRef]
- Chen, H.; Cook, L.S.; Tang, M.T.C.; Hill, D.A.; Wiggins, C.L.; Li, C.I. Relationship between Diabetes and Diabetes Medications and Risk of Different Molecular Subtypes of Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1802–1808. [Google Scholar] [CrossRef]
- Park, Y.M.M.; Bookwalter, D.B.; O’Brien, K.M.; Jackson, C.L.; Weinberg, C.R.; Sandler, D.P. A Prospective Study of Type 2 Diabetes, Metformin Use, and Risk of Breast Cancer. Ann. Oncol. 2021, 32, 351–359. [Google Scholar] [CrossRef]
- Fondazione IRCCS Istituto Nazionale dei Tumori, Milano. Calorie Restriction With or Without Metformin in Triple Negative Breast Cancer (BREAKFAST). In ClinicalTrials.gov; ClinicalTrials.gov Identifier: NCT04248998; National Library of Medicine: Bethesda, MD, USA, 2023. Available online: https://clinicaltrials.gov/study/NCT04248998 (accessed on 18 March 2025).
- Ministry of Health, Saudi Arabia. The Study of Quadruple Therapy Quercetin, Zinc, Metformin, and EGCG as Adjuvant Therapy for Early, Metastatic Breast Cancer and Triple-negative Breast Cancer, a Novel Mechanism. In ClinicalTrials.gov; ClinicalTrials.gov Identifier: NCT05680662; National Library of Medicine: Bethesda, MD, USA, 2023. Available online: https://clinicaltrials.gov/study/NCT05680662 (accessed on 18 March 2025).
- Columbia University. Study of Erlotinib and Metformin in Triple Negative Breast Cancer. In ClinicalTrials.gov; ClinicalTrials.gov Identifier: NCT01650506; National Library of Medicine: Bethesda, MD, USA, 2017. Available online: https://clinicaltrials.gov/study/NCT01650506 (accessed on 18 March 2025).
- The Methodist Hospital Research Institute. Alpelisib/iNOS Inhibitor/Nab-paclitaxel in Patients with HER2 Negative Metaplastic Breast Cancer (MpBC). In ClinicalTrials.gov; ClinicalTrials.gov Identifier: NCT05660083; National Library of Medicine: Bethesda, MD, USA, 2025. Available online: https://clinicaltrials.gov/study/NCT05660083 (accessed on 18 March 2025).
- QuantumLeap Healthcare Collaborative. I-SPY TRIAL: Neoadjuvant and Personalized Adaptive Novel Agents to Treat Breast Cancer (I-SPY). In ClinicalTrials.gov; ClinicalTrials.gov Identifier: NCT01042379; National Library of Medicine: Bethesda, MD, USA, 2024. Available online: https://clinicaltrials.gov/study/NCT01042379 (accessed on 18 March 2025).

{kind=link}
| Agents | Target (s) | Parameters | Findings | Refs. |
|---|---|---|---|---|
| Metformin | GLUT1 mRNA expression | Glycemic reuptake, lactate production, cell viability, and culture growth | Glucose uptake decreased after short-term exposure but increased in long-term exposure. Increased formation of lactate was observed. In addition, the cell viability and growth were reduced in a concentration-dependent manner. | [51] |
| Metformin | Expressions of ZEB1, Vimentin MMP9, N-cadherin, MMP2, Slug and Snail | Long-term exposure on the metastatic profile of different BC genomic subtypes | Upregulation of ZEB1, Vimentin, MMP9, N-cadherin, MMP2, Slug and Snail expression, and downregulation of E-cadherin, claudin, and β-catenin were noted by metformin treatment. In addition, combined treatment of metformin and LY294002 reduced cell viability as compared to either one of them alone. | [52] |
| Metformin | UPR | Rate of inhibition of cell proliferation and induction of apoptosis | Metformin + glucose deprivation resulted in higher activation of UPR and cell apoptosis compared to glucose starvation alone. | [53] |
| Metformin and 2-DG | mTOR | Effects on cellular activities and cell signaling pathways | Metformin and 2DG alone inhibited cell proliferation in a dose-dependent manner and significantly higher effects were noticed with combination. Also, the combination in glucose-starved cells resulted in the inhibition of activation of the mTOR pathway and its downstream targets. | [54] |
| Metformin and propranolol | Cell proliferation, clonogenic efficiency, apoptosis, migration, invasion and metabolic potential, and in vivo studies | Drug combination reduced cell proliferation, mitochondrial activity, migration, and invasion, and induced apoptosis. In vivo results validated the reduction in invasion and inhibition of cell proliferation. | [55] | |
| Metformin and propranolol | Metastasis, intravasation, and extravasation | The combination of metformin and propranolol resulted in a significant decline in circulating tumor cell survival. | [56] | |
| Metformin and 2DG | PD-L1 | Mitochondrial biogenesis and protein PD-L1 expression | Metformin + 2DG resulted in a significant increase in mitochondrial mass, and decrease in PD-L1 expression. | [57] |
| Metformin and rapamycin | PD-L1 and p-S6 | PD-1 and PD-L1 inhibition. In vivo studies. | Rapamycin resulted in the downregulation of PD-L1 and p-S6 expression, and mice treated with rapamycin or metformin had a smaller tumor size as compared to those of the control group. | [58] |
| Metformin and BMS-754807 | Cell viability, combination index | Metformin and BMS-754807 had a synergistic effect on 11 out of 13 cell lines. | [59] | |
| Metformin and resveratrol | Cell viability | Metformin in combination with resveratrol inhibited cell proliferation caused by LRes but enhanced the inhibition of cell growth caused by HRes. | [60] | |
| Metformin and cisplatin | Cell viability, proliferation, migration, invasion, and regulation of RAD51 expression. In vivo measurement of mice tumor. | Cisplatin + metformin resulted in reduced cell viability, cell migration, and invasion and exerted a potent antiproliferative effect on cancer cells. The combination resulted in a lower average tumor weight as compared to metformin and cisplatin alone groups, respectively. | [61] | |
| Metformin, cisplatin, and electric pulses | Cell viability, colony-forming ability, oxidative stress, and glucose consumption | Cell viability and colony forming ability were decreased with cisplatin+ metformin subjected to Eps. When cells were treated with metformin and exposed to EP, glucose levels were decreased and ROS level was increased. | [62] | |
| Metformin and phenformin prodrugs | Intracellular accumulation of the compounds and in vivo studies to determine MTD and tumor burden | All complexes were shown to accumulate intracellularly in a dose-dependent manner, and metformin + phenformin prodrugs decreased tumor burden with MTD determined as 15 mg/kg. | [63] | |
| Lipophilic salts of metformin | Solubility and permeation | Metformin docusate exerted greater anticancer effects due to its enhanced lipophilicity and lipid solubility properties. | [64] | |
| Metformin, cisplatin, and gefitinib | Cell viability, hypoxia gene expression, and CSC population were studied through luciferase activity | Greater reduction in cell viability, HIF activity, and CSC population was seen in the cisplatin + metformin + gefitinib (CMG) group as compared to individual or two-agent combination. | [65] | |
| Metformin and 2-DG | Exploring the differential expression of genes with glucose starvation or glucose-lowering agents such as 2-DG or metformin | The glucose starvation environment was most effective in damaging cancerous cells as compared to 2-DG and metformin. | [66] | |
| Metformin | TRAIL | Expression of TRAIL, cell viability | The apoptotic process was triggered by the increased expression of TRAIL. Higher metformin concentrations were associated with increased TRAIL protein levels, and the inhibition of TRAIL led to the attenuation of metformin-induced apoptosis. | [67] |
| Metformin | Metformin and its relationship to metformin transporters named organic cation transporter 3 (OCT3) | Metformin was more efficacious in OCT3-MCF7 cells as compared to MCF-7 cells. | [68] | |
| Metformin and naringenin | Cell viability | The cotreatment with both metformin and naringenin resulted in a significant reduced cell viability. | [69] | |
| Metformin and TRAIL receptor agonists | TRAIL, TRAIL-R2 mRNA | TRAIL receptor sensitization to agonists, caspase activation, receptor expression over cell surface, and X-linked inhibitor of apoptosis protein (XIAP) expression and in vivo studies | Metformin sensitized TNBC cell lines to TRAIL agonists via inducing caspase activation. Real-time PCR results showed only a slight increase in TRAIL-R2 mRNA levels. In vitro investigation was consistent with the in vivo testing as the combination group had reduced XIAP protein levels. Combination treatment inhibited lung metastases to a similar degree to that in TRAIL alone. | [70] |
| Metformin and hemin | Metabolism, extracellular acidification rate (ECAR), oxygen consumption rate (OCR) | ECAR, OCR | A notable increase in OCR and ATP levels but decreased ECAR and intermediate compounds of the glycolysis pathway were noted. | [71] |
| Metformin and JNK signal pathway | Tumor-infiltrating lymphocytes, cell viability | Higher concentrations of metformin-suppressed cell viability. | [72] | |
| Metformin, Fluvastatin, and aspirin | Cell-colonizing ability and in vivo studies | The synergistic combination of fluvastatin, metformin, and aspirin resulted in 100% inhibition of cell colonization. | [73] | |
| Metformin | The role of metformin on YAP/TAZ axis regulation and its relation to achieving BC therapeutic effects | Metformin impeded cell growth in a dose-dependent manner and induced apoptosis via significantly arresting the cells in the G1 phase. | [74] | |
| Hyaluronic acid-engrafted metformin-loaded graphene oxide (HA-GO-Met) nanoparticles | Cell viability and cell migration | HA-GO-Met nanoparticles were effective in inducing cell death and attenuating cell migration as compared to GO-Met or metformin alone. | [75] | |
| Metformin and relaxin | Altering the immune microenvironment of tumors by reducing cancer-associated fibroblasts | Coumarins with LPP remained on the edges of the tumor sphere while those with LPPR were efficient in penetrating deep into the tumor, which indicates the efficiency of pRLN in increasing penetration. | [76] |
| Agents | Parameters | Findings | Refs. |
|---|---|---|---|
| Metformin and erlotinib | MTD, DLT, response rate, stable disease rate, and PFS | Absence of sufficient clinical benefits of this combination. | [84] |
| Metformin | Fasting insulin, glucose, CRP, estradiol, testosterone, and SHBG | Fasting insulin, estradiol, testosterone, and SHBG concentrations were improved by metformin. In addition, while metformin resulted in a higher average weight loss, glucose and CRP were unaffected as compared to placebo. | [85] |
| Metformin | Statistical insights | Women using metformin with T2D were at increased risk of developing TNBC. | [86] |
| Metformin | Relationship between T2D, use of metformin, and BC risk | T2D patients treated with metformin were found to have an increased risk of TNBC. | [87] |
| Study Title | Study Phase (Status) | Conditions | Intervention | Primary Outcome Measures | NCT Number | Refs. |
|---|---|---|---|---|---|---|
| Calorie Restriction With or Without Metformin in Triple-Negative Breast Cancer | Phase II (Unknown) | TNBC | Fasting-mimicking diet, Metformin, Preoperative Chemotherapy | Rate of pathologic complete responses. | NCT04248998 | [88] |
| The Study of Quadruple Therapy Quercetin, Zinc, Metformin, and EGCG as Adjuvant Therapy for Early, Metastatic Breast Cancer and Triple-Negative Breast Cancer, a Novel Mechanism | Phase I (Completed) | TNBC | Combination product of Quercetin, EGCG, metformin, zinc | Invasive disease-free survival at 3 years. | NCT05680662 | [89] |
| Study of Erlotinib and Metformin in Triple-Negative Breast Cancer | Phase I (Completed) | TNBC | Erlotinib in combination with metformin | Maximum tolerated dose of metformin in combination with a fixed dose of 150 mg erlotinib daily. | NCT01650506 | [90] |
| Alpelisib/iNOS Inhibitor/Nab-paclitaxel in Patients With HER2 Negative Metaplastic Breast Cancer (MpBC) | Phase II (Recruiting) | HER2-negative BC, MBC, Metaplastic breast carcinoma, TNBC | L-NMMA | Define recommended phase II dose and objective response rate. | NCT05660083 | [91] |
| I-SPY TRIAL: Neoadjuvant and Personalized Adaptive Novel Agents to Treat Breast Cancer | Phase II (Recruiting) | Breast neoplasms, BC, Breast tumors, TNBC, HER2-positive BC, HER2-negative BC, Hormone receptor positive tumor, Hormone receptor negative tumor, early-stage BC, Locally advanced BC | Standard therapy, AMG 386 with or without Trastuzumab, AMG 479 (Ganitumab) plus metformin, and others | Determine whether adding experimental agents to standard neoadjuvant medications increases the probability of pathologic complete response over standard neoadjuvant chemotherapy for each biomarker signature established at trial entry. | NCT01042379 | [92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirhan, Z.; Abu Nada, A.; Anabtawi, N.; Thyagarajan, A.; Sahu, R.P. Metformin-Based Combination Approaches for Triple-Negative Breast Cancer. Pharmaceutics 2025, 17, 558. https://doi.org/10.3390/pharmaceutics17050558
Sirhan Z, Abu Nada A, Anabtawi N, Thyagarajan A, Sahu RP. Metformin-Based Combination Approaches for Triple-Negative Breast Cancer. Pharmaceutics. 2025; 17(5):558. https://doi.org/10.3390/pharmaceutics17050558
Chicago/Turabian StyleSirhan, Zaid, Aya Abu Nada, Nadeen Anabtawi, Anita Thyagarajan, and Ravi P. Sahu. 2025. "Metformin-Based Combination Approaches for Triple-Negative Breast Cancer" Pharmaceutics 17, no. 5: 558. https://doi.org/10.3390/pharmaceutics17050558
APA StyleSirhan, Z., Abu Nada, A., Anabtawi, N., Thyagarajan, A., & Sahu, R. P. (2025). Metformin-Based Combination Approaches for Triple-Negative Breast Cancer. Pharmaceutics, 17(5), 558. https://doi.org/10.3390/pharmaceutics17050558