
Stabilizing Agents for Drug Nanocrystals: Effect on Bioavailability



Abstract
:
1. Introduction
2. Stabilizers
2.1. Poloxamers
2.2. Celluloses
2.3. Vitamin E TPGS
2.4. Soluplus®
2.5. Others
3. Stabilizing Effect
4. Effect of Stabilizers on Cell Layers and Drug Transport
5. Discussion
Author Contributions
Conflicts of Interest
References
- Merisko-Liversidge, E.; Liversidge, G.G. Nanosizing for oral and parenteral drug delivery: A perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv. Drug Deliv. Rev. 2011, 63, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Keck, C.M. Twenty years of drug nanocrystals: Where are we, and where do we go? Eur. J. Pharm. Biopharm. 2012, 80, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Solubility in water and DMSO: Issues and potential solutions. In Pharmaceutical profiling in drug discovery for lead selection; Borchardt, R., Kerns, E., Lipinski, C., Thakker, D., Wang, B., Eds.; AAPS Press: Arlington, TX, USA, 2005; pp. 93–125. [Google Scholar]
- Sigfridsson, K.; Forssén, S.; Holländer, P.; Skantze, U.; de Verdier, J. A formulation comparison, using a solution and different nanosuspensions of a poorly soluble compound. Eur. J. Pharm. Biopharm. 2007, 67, 540–547. [Google Scholar] [CrossRef] [PubMed]
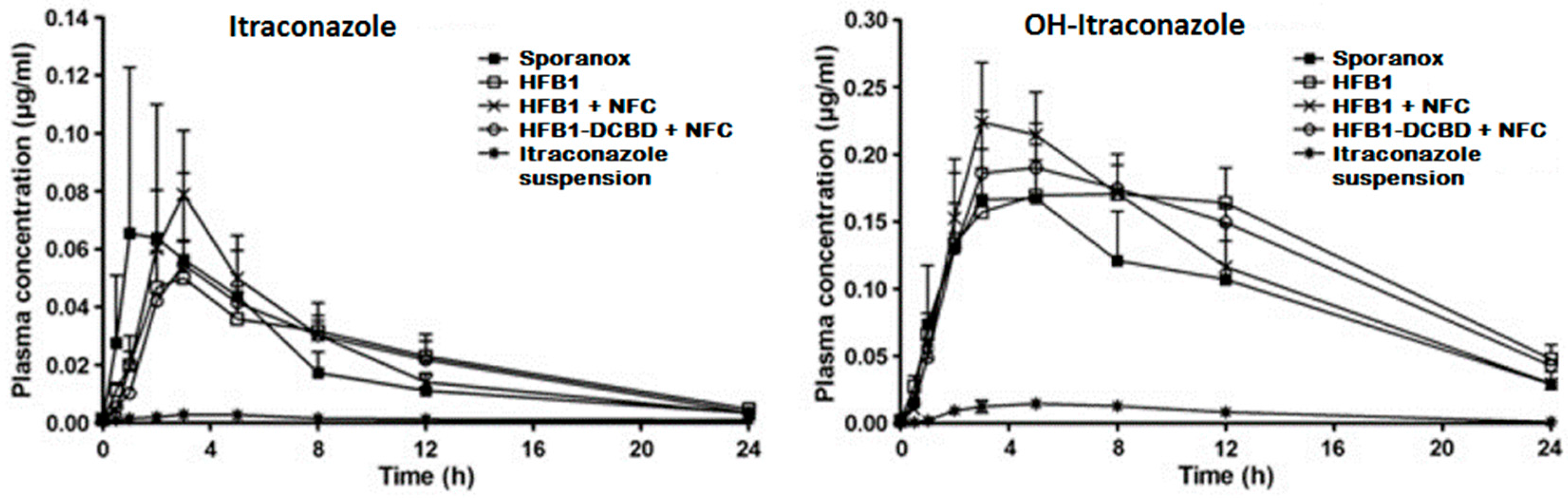
- Sarnes, A.; Kovalainen, M.; Häkkinen, M.R.; Laaksonen, T.; Laru, J.; Kiesvaara, J.; Ilkka, J.; Oksala, O.; Rönkkö, S.; Järvinen, K.; et al. Nanocrystal-based per-oral itraconazole delivery: Superior in vitro dissolution enhancement versus Sporanox® is not realized in in vivo drug absorption. J. Control. Release 2014, 180, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Valo, H.; Kovalainen, M.; Laaksonen, P.; Häkkinen, M.; Auriola, S.; Peltonen, L.; Linder, M.; Järvinen, K.; Hirvonen, J.; Laaksonen, T. Immobilization of protein-coated drug nanoparticles in nanofibrillar cellulose matrices—Enhanced stability and release. J. Control. Release 2011, 156, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, L.; Hirvonen, J. Pharmaceutical nanocrystals by nanomilling: Critical process parameters, particle fracturing and stabilization methods. J. Pharm. Pharmacol. 2010, 62, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Beirowski, J.; Inghelbrecht, S.; Arien, A.; Gieseler, H. Freeze-drying of nanosuspensions, 1: Freezing rate versus formulation design as critical factors to preserve the original particle size distribution. J. Pharm. Sci. 2011, 100, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Verma, A.; Pandey, G.; Mittapelly, N.; Mishra, P.R. Investigating the role of pluronic-g-cationic polyelectrolyte as functional stabilizer for nanocrystals: Impact on paclitaxel oral bioavailability and tumor growth. Acta Biomater. 2015, 26, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Mahesh, K.V.; Singh, S.K.; Gulati, M. A comparative study of top-down and bottom-up approaches for the preparation of nanosuspensions of glipizide. Powder Technol. 2014, 256, 436–449. [Google Scholar] [CrossRef]
- Danhier, F.; Ucakar, B.; Vanderhaegen, M.-L.; Brewster, M.E.; Arien, T.; Préat, V. Nanosuspension for the delivery of a poorly soluble anti-cancer kinase inhibitor. Eur. J. Pharm Biopharm. 2014, 88, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Valo, H.; Arola, S.; Laaksonen, P.; Torkkeli, M.; Peltonen, L.; Linder, M.B.; Serimaa, R.; Kuga, S.; Hirvonen, J.; Laaksonen, T. Drug release from nanoparticles embedded in four different nanofibrillar cellulose aerogels. Eur. J. Pharm. Sci. 2013, 50, 69–77. [Google Scholar] [CrossRef] [PubMed]
- George, M.; Ghosh, I. Identifying the correlation between drug/stabilizer properties and critical quality attributes (CQAs) of nanosuspension formulation prepared by wet media milling technology. Eur. J. Pharm. Sci. 2013, 48, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, Z.-H.; Li, T.; McNally, H.; Park, K.; Sturek, M. Development and evaluation of transferrin-stabilized paclitaxel nanocrystal formulation. J. Control. Release 2014, 176, 76–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makhlof, A.; Miyazaki, Y.; Tozuka, Y.; Takeuchi, H. Cyclodextrins as stabilizers for the preparation of drug nanocrystals by the emulsion solvent diffusion method. Int. J. Pharm. 2008, 357, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Laaksonen, T.; Liu, P.; Rahikkala, A.; Peltonen, L.; Kauppinen, E.I.; Hirvonen, J.; Järvinen, K.; Raula, J. Intact nanoparticulate indomethacin in fast-dissolving carrier particles by combined wet milling and aerosol flow reactor methods. Pharm. Res. 2011, 28, 2403–2411. [Google Scholar] [CrossRef] [PubMed]
- Raula, J.; Rahikkala, A.; Halkola, T.; Pessi, J.; Peltonen, L.; Hirvonen, J.; Järvinen, K.; Laaksonen, T.; Kauppinen, E.I. Coated particle assemblies for the concomitant pulmonary administration of budesonide and salbutamol sulphate. Int. J. Pharm. 2013, 221, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Viitala, T.; Kartal-Hodzic, A.; Liang, H.; Laaksonen, T.; Hirvonen, J.; Peltonen, L. Interaction studies between indomethacin nanocrystals and PEO/PPO copolymer stabilizers. Pharm. Res. 2015, 32, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Rachmawati, H.; Al Shaal, L.; Müller, R.H.; Keck, C.M. Development of curcumin nanocrystal: Physical aspects. J. Pharm. Sci. 2013, 102, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Quan, P.; Piao, H.; Sun, S.; Yin, Y.; Cui, F. Preparation of stable nitrendipine nanosuspensions using the precipitation-ultrasonication method for enhancement of dissolution and oral bioavailability. Eur. J. Pharm. Sci. 2010, 40, 325–334. [Google Scholar] [CrossRef] [PubMed]
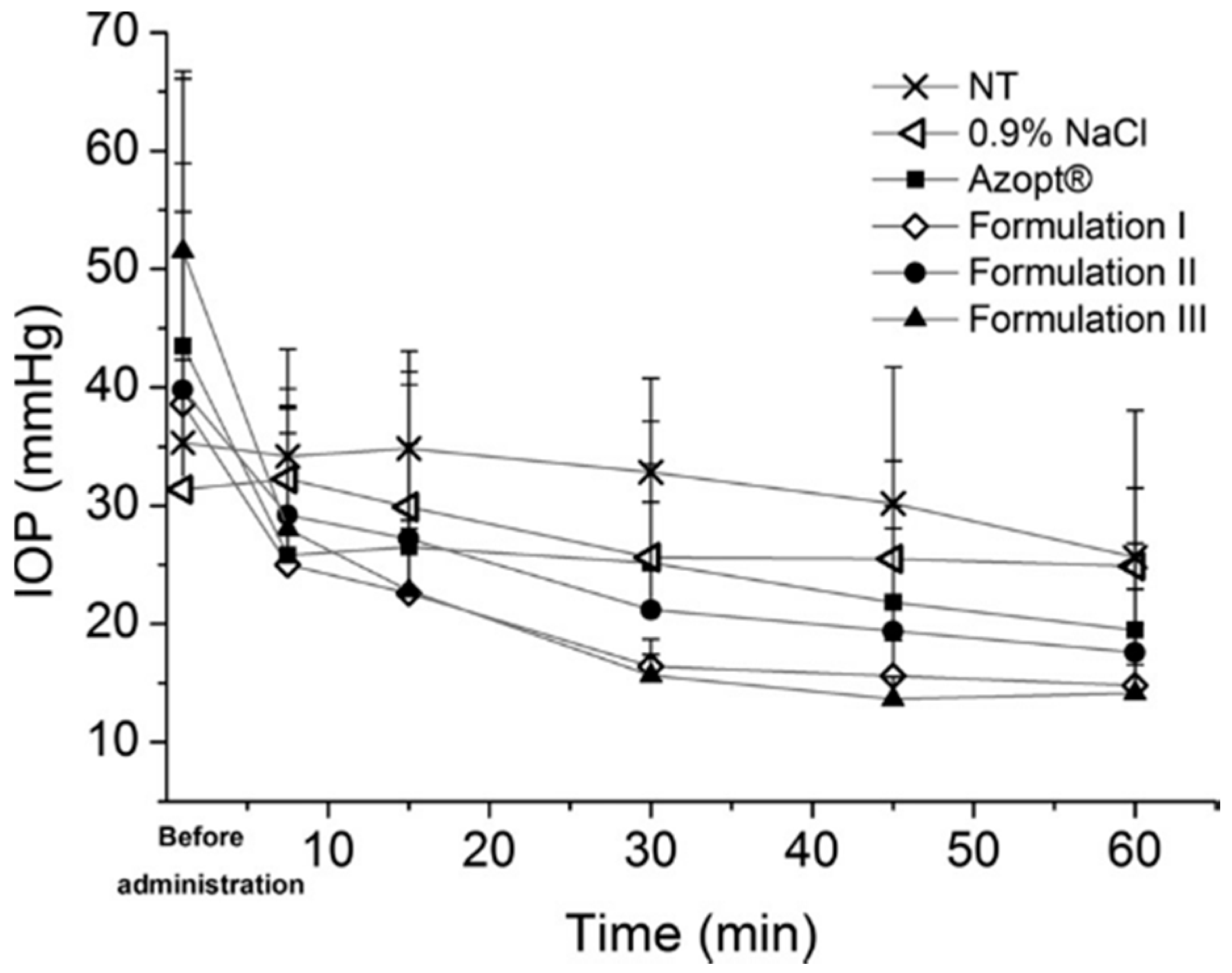
- Tuomela, A.; Liu, P.; Puranen, J.; Rönkkö, S.; Laaksonen, T.; Kalesnykas, G.; Oksala, O.; Ilkka, J.; Laru, J.; Järvinen, K.; et al. Brinzolamide nanocrystal formulations for ophthalmic delivery: Reduction of elevated intraocular pressure in vivo. Int. J. Pharm. 2014, 467, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Teng, F.; Wang, P.; Tian, B.; Lin, X.; Hu, X.; Zhang, L.; Zhang, K.; Zhang, Y.; Tang, X. Investigation of a nanosuspension stabilized by Soluplus® to improve bioavailability. Int. J. Pharm. 2014, 477, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Vermant, J.; Martens, J.A.; Froyen, L.; Van Humbeeck, J.; Augustijns, P.; Van den Mooter, G. A screening study of surface stabilization during the production of drug nanocrystals. J. Pharm. Sci. 2009, 98, 2091–2103. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Sun, J.; Zhang, D.; Li, M.; Wang, Y.; Ling, G.; Liu, X.; Sun, Y.; Sui, X.; Luo, C.; et al. Nimodipine nanocrystals for oral bioavailability improvement: Preparation, characterization and pharmacokinetic studies. Colloids Surf. B 2013, 109, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Rong, X.; Laru, J.; van Veen, B.; Kiesvaara, J.; Hirvonen, J.; Laaksonen, T.; Peltonen, L. Nanosuspensions of poorly soluble drugs: Preparation and development by wet milling. Int. J. Pharm. 2011, 411, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.S.; Saraswathi, R.; Kumar, K.V.; Jha, S.K.; Venkates, D.P.; Dhanaraj, S.A. Development and characterization of lecithin stabilized glibenclamide nanocrystals for enhanced solubility and drug delivery. Drug Deliv. 2014, 21, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lorenzo, C.; Rey-Rico, A.; Sosnik, A.; Taboada, P.; Concheiro, A. Poloxamine-based nanomaterials for drug delivery. Front. Biosci. (Elite ed) 2010, 2, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lorenzo, C.; Sosnik, A.; Concheiro, A. PEO-PPO block copolymers for passive micellar targeting and overcoming multidrug resistance in cancer therapy. Curr. Drug Targets 2011, 12, 1112–1130. [Google Scholar] [CrossRef] [PubMed]
- Chiappetta, D.A.; Sosnik, A. Poly(ethylene oxide)-poly(propylene oxide) block copolymer micelles as drug delivery agents: Improved hydrosolubility, stability and bioavailability of drugs. Eur. J. Pharm. Biopharm. 2007, 66, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, J.Y.; Park, C.H. Characteristics of polymers enabling nano-comminution of water-insoluble drugs. Int. J. Pharm. 2008, 355, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Kim, S.; Ahn, C.H.; Lee, J. Hydrophilic and hydrophobic amino acid copolymers for nano-comminution of poorly soluble drugs. Int. J. Pharm. 2010, 384, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Tuomela, A.; Laaksonen, T.; Laru, J.; Antikainen, O.; Kiesvaara, J.; Ilkka, J.; Oksala, O.; Rönkkö, S.; Järvinen, K.; Hirvonen, J.; et al. Solid formulations by a nanocrystal approach: Critical process parameters regarding scale-ability of nanocrystals for tableting applications. Int. J. Pharm. 2015, 485, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Tian, W.; Zhang, Y.; He, J.; Mao, S.; Fang, L. Effect of novel stabilizers—Cationic polymers on the particle size and physical stability of poorly soluble drug nanocrystals. Nanomedicine 2012, 8, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2001, 48, 139–157. [Google Scholar] [CrossRef]
- El-Sousi, S.; Nácher, A.; Mura, C.; Catalán-Latorre, A.; Merino, V.; Merino-Sanjuán, M.; Díez-Sales, O. Hydroxypropylmethylcellulose films for the ophthalmic delivery of diclofenac sodium. J. Pharm. Pharmacol. 2013, 65, 193–200. [Google Scholar] [CrossRef] [PubMed]
- FDA, US Food and Drug Administration: Inactive Ingredients for Approved Drug Products. Available online: http://www.accessdata.fda.gov/scripts/cder/iig/getiigWEB.cfm (accessed on 17 May 2016).
- Ludwig, A. The use of mucoadhesive polymers in ocular drug delivery. Adv. Drug Deliv. Rev. 2005, 57, 1595–1639. [Google Scholar] [CrossRef] [PubMed]
- Toda, I.; Shinozaki, N.; Tsubota, K. Hydroxypropyl methylcellulose for the treatment of severe dry eye associated with Sjogren’s syndrome. Cornea 1996, 15, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Zuo, B.; Sun, Y.; Li, H.; Liu, X.; Zhai, Y.; Sun, J.; He, Z. Preparation and in vitro/in vivo evaluation of fenofibrate nanocrystals. Int. J. Pharm. 2013, 455, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Douroumis, D.; Fahr, A. Stable carbamazepine colloidal systems using the cosolvent technique. Eur. J. Pharm. Sci. 2007, 30, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Sepassi, S.; Goodwin, D.J.; Drake, A.F.; Holland, S.; Leonard, G.; Martini, L.; Lawrence, M.J. Effect of polymer molecular weight on the production of drug nanoparticles. J. Pharm. Sci. 2007, 96, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Hui, H.; Robinson, J.R. Effect of particle dissolution rate on ocular drug bioavailability. J. Pharm. Sci. 1986, 75, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Kassem, M.A.; Abdel Rahman, A.A.; Ghorab, M.M.; Ahmed, M.B.; Khalil, R.M. Nanosuspension as an ophthalmic delivery system for certain glucocorticoid drugs. Int. J. Pharm. 2007, 340, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Yoo, J.Y.; Kwak, H.S.; Nam, B.U.; Lee, J. Role of polymeric stabilizers for drug nanocrystal dispersions. Curr. Appl. Phys. 2005, 5, 472–474. [Google Scholar] [CrossRef]
- Ghosh, I.; Schenck, D.; Bose, S.; Liu, F.; Motto, M. Identification of critical process parameters and its interplay with nanosuspension formulation prepared by top down media milling technology--a QbD perspective. Pharm. Dev. Technol. 2013, 18, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tan, S.; Feng, S.S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials 2012, 33, 4889–4906. [Google Scholar] [CrossRef] [PubMed]
- Vitamin E TPGS. Available online: isochem.eu/page/vitamin-e-tpgs (accessed on 17 May 2016).
- Feng, S.S.; Mei, L.; Anitha, P.; Gan, C.W.; Zhou, W. Poly(lactide)-vitamin E derivative/montmorillonite nanoparticle formulations for the oral delivery of Docetaxel. Biomaterials 2009, 19, 3297–3306. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Luo, J.; Tan, S.; Otieno, B.O.; Zhang, Z. The applications of vitamin E TPGS in drug delivery. Eur. J. Pharm. Sci. 2013, 49, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Win, K.Y.; Feng, S.S. In vitro and in vivo studies on vitamin E TPGS-emulsified poly(D,L-lactic-co-glycolic acid) nanoparticles for paclitaxel formulation. Biomaterials 2006, 10, 2285–2291. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, M.; Lu, Y.; Ding, L.Y.; Ron, W.T.; Liu, Y.Q.; Song, F.F.; Yu, S.Q. Alpha-tocopheryl polyethylene glycol succinate-emulsified poly(lactic-co-glycolic acid) nanoparticles for reversal of multidrug resistance in vitro. Nanotechnology 2012, 23, 495103. [Google Scholar]
- Ge, Z.-Q.; Du, X.-Y.; Huang, X.-N.; Qiao, B. Enhanced oral bioavailability of ursolic acid nanoparticles via antisolvent presipitation with TPGS1000 as a stabilizer. J. Drug Deliv. Sci. Technol. 2015, 29, 210–217. [Google Scholar] [CrossRef]
- Zhang, K.; Yu, H.; Luo, Q.; Yang, S.; Lin, X.; Zhang, Y.; Tian, B.; Tang, X. Increased dissolution and oral absorption of itraconazole/Soluplus extrudate compared with itraconazole nanosuspension. Eur. J. Pharm. Biopharm. 2013, 85, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Tsinman, O.; Tsinman, K.; Shaukat, A. Excipient update - Soluplus®: An understanding of supersaturation from amorphous solid dispersions. Drug Dev. Deliv. 2015. Available online: http://www.drug-dev.com/Main/Back-Issues/EXCIPIENT-UPDATE-Soluplus-An-Understanding-of-Supe-824.aspx (accessed on 17 May 2016).
- Soluplus®—The Solid Solution. Available online: www.pharma-ingredients.basf.com/Soluplus/Home.aspx (accessed on 17 May 2016).
- Linn, M.; Collnot, E.M.; Djuric, D.; Hempel, K.; Fabian, E.; Kolter, K.; Lehr, C.M. Soluplus® as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur. J. Pharm. Sci. 2012, 45, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Gadadare, R.; Mandpe, L.; Pokharkar, V. Ultra rapidly dissolving repaglinide nanosized crystals prepared via bottom-up and top-down approach: influence of food on pharmacokinetics behavior. AAPS PharmSciTech 2015, 16, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Valo, H.K.; Laaksonen, P.H.; Peltonen, L.J.; Linder, M.B.; Hirvonen, J.T.; Laaksonen, T.J. Multifunctional hydrophobin: Toward functional coatings for drug nanoparticles. ACS Nano 2010, 4, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Becket, G.; Schep, L.J.; Tan, M.Y. Improvement of the in vitro dissolution of praziquantel by complexation with α-, β-, and γ-cyclodextrins. Int. J. Pharm. 1999, 179, 65–71. [Google Scholar] [CrossRef]
- Lindfors, L.; Forssn, S.; Skantze, P.; Skantze, U.; Zackrisson, A.; Olsson, U. Amorphous drug nanosuspensions. 2. Experimental determination of bulk monomer concentrations. Langmuir 2006, 22, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Kurakula, M.; El-Helw, A.M.; Sobahi, T.R.; Abdelaal, M.Y. Chitosan based atorvastatin nanocrystals: Effect of cationic charge on particle size, formulation stability, and in vivo efficacy. Int. J. Nanomed. 2015, 10, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Lestari, M.L.A.D.; Müller, R.H.; Möschwitzer, J.P. Systematic screening of different surface modifiers for the production of physically stable nanosuspensions. J. Pharm. Sci. 2015, 104, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Gauniya, A.; Mazumder, R.; Pathak, K. Formulation, optimization and characterization of ziprasidone nanocrystals prepared by media milling technique. Int. J. Pharm. Pharm. Sci. 2015, 7, 146–150. [Google Scholar]
- Pokharkar, V.B.; Malhi, T.; Mandpe, L. Bicalutamide nanocrystals with improved oral bioavailability: In vitro and in vivo evaluation. Pharm. Dev. Technol. 2013, 18, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, B.K.; Jena, S.K.; Paidi, S.K.; Bagri, S.; Suresh, S. Formulation, optimization and in vitro-in vivo evaluation of febuxostat nanosuspension. Int. J. Pharm. 2015, 478, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Tu, L.; Hu, K.; Wu, W.; Feng, J. The construction of puerarin nanocrystals and its pharmacokinetic and in vivo-in vitro correlation (IVIVC) studies on beagle dog. Colloids Surf. B 2015, 133, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Susarla, R.; Afolabi, A.; Patel, D.; Bilgili, E.; Davé, R.N. Novel use of superdisintegrants as viscosity enhancing agents in biocompatible polymer films containing griseofulvin nanoparticles. Powder Tech. 2015, 285, 25–33. [Google Scholar] [CrossRef]
- Bhakay, A.; Merwade, M.; Bilgili, E.; Dave, R.N. Novel aspects of wet milling for the production of microsuspensions and nanosuspensions of poorly water-soluble drugs. Drug Dev. Ind. Pharm. 2011, 37, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J. Effective polymeric dispersants for vacuum, convection and freeze drying of drug nanosuspensions. Int. J. Pharm. 2010, 397, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Huang, L.; Liu, F. Understanding the structure and stability of paclitaxel nanocrystals. Int. J. Pharm. 2010, 390, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, G.; Ma, J.; Wang, X.; Wang, F.; Wang, H.; Sun, J. Paclitaxel nanosuspension coated with P-gp inhibitory surfactants: II. Ability to reverse the drug-resistance of H460 human lung cancer cells. Colloids Surf. B 2014, 117, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.J.; Fu, Y.H.; Meng, Q.H.; Li, M.; Ying, X.Y.; Han, M.; He, Q.J.; Yang, B.; Zeng, S.; Hu, Y.Z.; et al. Evaluation of pluronic nanosuspensions loading a novel insoluble anticancer drug both in vitro and in vivo. Int. J. Pharm. 2013, 456, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, J.; Chen, Q.; Gao, Y.; Li, L.; Li, H.; Leng, D.; Wang, Y.; Sun, Y.; Jing, Y.; et al. Star-shape copolymer of lysine-linked dl-tocopherol polyethylene glycol 2000 succinate for doxorubicin delivery with reversal of multidrug resistance. Biomaterials 2012, 33, 6877–6888. [Google Scholar] [CrossRef] [PubMed]
- Darville, N.; Saarinen, J.; Isomäki, A.; Khriachtchev, L.; Cleeren, D.; Sterkens, P.; van Heerden, M.; Annaert, P.; Peltonen, L.; Santos, H.A.; et al. Multimodal non-linear optical imaging for the investigation of drug nano-/microcrystal-cell interactions. Eur. J. Pharm. Biopharm. 2015, 96, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Li, B.; Zhang, D.; Fang, M.; Shao, J.; Guo, M.; Guo, Z.; Li, M.; Sun, J.; Zhai, Y. Comparative studies of the in vitro dissolution and in vivo pharmacokinetics for different formulation strategies (solid dispersion, micronization, and nanocrystals) for poorly water-soluble drugs: A case study for lacidipine. Colloids Surf. B 2015, 132, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Abu-Diak, O.A.; Jones, D.S.; Andrews, G.P. An investigation into the dissolution properties of celecoxibmelt extrudates: Understanding the role of polymer type and concentration in stabilizing supersaturated drug concentrations. Mol. Pharm. 2011, 8, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Ochi, M.; Kawachi, T.; Toita, E.; Hashimoto, I.; Yuminoki, K.; Onoue, S.; Hashimoto, N. Development of nanocrystal formulation of meloxicam with improved dissolution and pharmacokinetic behaviors. Int. J. Pharm. 2014, 474, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.Y.; Yoo, S.D.; Lee, S.H.; Kim, K.H.; Yoon, D.S.; Lee, K.H. Enhanced solubility and dissolution rate of itraconazole by a solid dispersion technique. Int. J. Pharm. 1999, 187, 209–218. [Google Scholar] [CrossRef]
- Surwase, S.A.; Itkonen, L.; Aaltonen, J.; Saville, D.; Rades, T.; Peltonen, L.; Strachan, C.J. Polymer incorporation method affects the physical stability of amorphous indomethacin in aqueous suspension. Eur. J. Pharm. Biopharm. 2015, 96, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.J.; Westedt, U.; Rosenblatt, K.M.; Hölig, P.; Rosenberg, J.; Mägerlein, M.; Fricker, G.; Brandl, M. What is the mechanism behind increased permeation rate of a poorly soluble drug from aqueous dispersions of an amorphous solid dispersion? J. Pharm. Sci. 2014, 103, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Fu, Q.; Wu, C.; Guo, Z.; Li, M.; Sun, J.; He, Z.; Yang, L. Rod shaped nanocrystals exhibit superios in vitro dissolution and in vivo bioavailability over spherical like nanocrystals: A case study of lovastatin. Colloids Surf. B 2015, 128, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Higashino, H.; Hasegawa, T.; Yamamoto, M.; Matsui, R.; Masaoka, Y.; Kataoka, M.; Sakuma, S.; Yamashita, S. In vitro-in vivo correlation of the effect of supersaturation on the intestinal absorption of BCS class 2 drugs. Mol. Pharm. 2014, 11, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F.; Elsadek, B. Clinical impact of serum proteins on drug delivery. J. Control. Release 2012, 161, 429–445. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhao, W.; Liu, H.; Marquez, R.; Huang, Y.; Zhang, Y.; Li, J.; Xie, W.; Venkataramanan, R.; Xu, L.; et al. An improved D-α-tocopherol-based nanocarrier for targeted delivery of doxorubicin with reversal of multidrug resistance. J. Control. Release 2014, 196, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.; Panchagnula, R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: Effect on solubility and permeability in vitro, in situ and in vivo. Eur. J. Pharm. Sci. 2005, 25, 445–453. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Stabilizer | Process | Ref. |
|---|---|---|---|
| Glibizide | Sodium lauryl sulfate, polyvinyl pyrrolidone K30, Pluronics F68 and F127, Tween 80, hydroxypropyl methylcellulose | Milling, antisolvent precipitation | [10] |
| MTKi-327 | Pluronic F108, Lipid S75 | Milling | [11] |
| Beclomethasone dipropionate | Hydrophobin | Antisolvent precipitation | [12] |
| Naproxen | Vitamin E tocopherol polyethylene glycol succinate, Pluronic F127, sodium lauryl sulfate, di(2-ethylhexyl) sulfosuccinate | Milling | [13] |
| Paclitaxel | Hydroxypropyl methylcellulose, polyvinyl pyrrolidone, polyethylene glycol 400, Pluronics F127 and F68, sodium lauryl sulfate, Tween 20 and 80, transferrin, Immunoglobulin G, Human serum albumin | Antisolvent precipitation + sonication | [14] |
| Indomethacin | α-, β- and γ-cyclodextrins | Emulsion solvent diffusion | [15] |
| Indomethacin | Pluronic F68 | Milling | [16] |
| Budesonide | Lecithin, Pluronic F68 | Milling | [17] |
| Indomethacin | Pluronics F68, 17R4 and L64, Tetronics 908 and 1107 | Milling | [18] |
| Curcumin | Polyvinyl alcohol, polyvinyl pyrrolidone, Vitamin E tocopherol polyethylene glycol succinate, sodium lauryl sulfate, carboxymethylcellulose sodium | High pressure homogenization | [19] |
| Nitrendipine | Polyvinyl alcohol | Antisolvent precipitation–ultrasonication | [20] |
| Brinzolamide | Tween 80, Pluronics F68 and F127, hydroxypropyl methylcellulose | Milling | [21] |
| Fenofibrate | Hydroxypropyl methylcellulose, Soluplus | Milling | [22] |
| Loviridine, itraconazole, cinnarizine, griseofulvin, indomethacin, mebendazole, naproxen, phenylbutazone, phenytoin | Polyvinyl pyrrolidone, polyvinyl alcohol-polyethylene glycol, Pluronic F68, tocopherol polyethylene glycol succinate, hydroxypropyl methylcellulose, hydroxyethyl cellulose, hydroxypropyl cellulose, methylcellulose, carboxymethylcellulose sodium, polyvinyl alcohol, sodium alginate, Tween 80 | Milling | [23] |
| Nimodipine | Pluronic F127, hydroxypropyl methylcellulose | Microprecipitation, high-pressure homogenization | [24] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuomela, A.; Hirvonen, J.; Peltonen, L. Stabilizing Agents for Drug Nanocrystals: Effect on Bioavailability. Pharmaceutics 2016, 8, 16. https://doi.org/10.3390/pharmaceutics8020016
Tuomela A, Hirvonen J, Peltonen L. Stabilizing Agents for Drug Nanocrystals: Effect on Bioavailability. Pharmaceutics. 2016; 8(2):16. https://doi.org/10.3390/pharmaceutics8020016
Chicago/Turabian StyleTuomela, Annika, Jouni Hirvonen, and Leena Peltonen. 2016. "Stabilizing Agents for Drug Nanocrystals: Effect on Bioavailability" Pharmaceutics 8, no. 2: 16. https://doi.org/10.3390/pharmaceutics8020016
APA StyleTuomela, A., Hirvonen, J., & Peltonen, L. (2016). Stabilizing Agents for Drug Nanocrystals: Effect on Bioavailability. Pharmaceutics, 8(2), 16. https://doi.org/10.3390/pharmaceutics8020016