
Overview of Computational Toxicology Methods Applied in Drug and Green Chemical Discovery



Abstract
:1. Introduction
2. Machine Learning and Deep Learning in Computational Toxicology
3. Computational Toxicity In Silico Methods
4. Application of QSAR in Toxicity Prediction During Drug Design
5. QSAR Application to Environmental Toxicology
6. New Insights and Challenges for Computational Toxicity Prediction
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bolt, H.M.; Hengstler, J.G. The rapid development of computational toxicology. Arch. Toxicol. 2020, 94, 1371–1372. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S. Progress in computational toxicology. J. Pharmacol. Toxicol. Methods 2014, 69, 115–140. [Google Scholar] [CrossRef] [PubMed]
- United States Environmental Protection Agency. Available online: https://www.epa.gov/tsca-inventory/how-access-tsca-inventory (accessed on 3 October 2024).
- Melagraki, G.; Afantitis, A. Computational toxicology: From cheminformatics to nanoinformatics. Food Chem. Toxicol. 2018, 112, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.A.; Whelan, M.P.; Worth, A.P. The role of validation in establishing the scientific credibility of predictive toxicology approaches intended for regulatory application. Comput. Toxicol. 2021, 17, 100144. [Google Scholar] [CrossRef]
- Ito, S.; Mukherjee, S.; Erami, K.; Muratani, S.; Mori, A.; Ichikawa, S.; White, W.; Yoshino, K.; Fallacara, D. Proof of concept for quantitative adverse outcome pathway modeling of chronic toxicity in repeated exposure. Sci. Rep. 2024, 14, 4741. [Google Scholar] [CrossRef]
- Ekins, S.; Lane, T.R.; Urbina, F.; Puhl, A.C. In silico ADME/tox comes of age: Twenty years later. Xenobiotica 2023, 54, 352–358. [Google Scholar] [CrossRef]
- Luechtefeld, T.; Rowlands, C.; Hartung, T. Big-data and machine learning to revamp computational toxicology and its use in risk assessment. Toxicol. Res. 2018, 7, 732–744. [Google Scholar] [CrossRef]
- Pérez-Santín, E.; Rodríguez Solana, R.; González García, M.; García-Suárez, M.D.M.; Blanco Díaz, G.D.; Cima-Cabal, M.D.; Moreno Rojas, J.M.; López Sánchez, J.I. Toxicity prediction based on artificial intelligence: A multidisciplinary overview. WIREs Comput. Mol. Sci. 2021, 11, e1516. [Google Scholar] [CrossRef]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef]
- Daxberger, E.; Eric, N.; Allingham, J.U.; Antoran, J.; Hernandez-Lobato, J.M. Bayesian deep learning via subnetwork inference. In Proceedings of the 38th International Conference on Machine Learning, Virtual, 18–24 July 2021; Volume 139, pp. 2510–2521. [Google Scholar]
- Hartung, T. ToxAIcology—The evolving role of artificial intelligence in advancing toxicology and modernizing regulatory science. ALTEX 2023, 40, 559–570. [Google Scholar] [CrossRef]
- Baskin, I.I. Machine learning methods in computational toxicology. Methods Mol. Biol. 2018, 1800, 119–139. [Google Scholar] [PubMed]
- QSARPro. Available online: https://www.vlifesciences.com/products/QSARPro/Product_QSARpro.php (accessed on 8 November 2024).
- MedChem Studio. Available online: https://www.simulations-plus.com/software/admetpredictor/medchem-studio/ (accessed on 8 November 2024).
- Vainio, M.J.; Mark, S.; Johnson, M.S. McQSAR: A multiconformational quantitative structure-activity relationship engine driven by genetic algorithms. J. Chem. Inf. Model. 2005, 45, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-Descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Codessa. Available online: http://www.semichem.com/codessa/default.php (accessed on 8 November 2024).
- CQSAR. Available online: http://www.biobyte.com/bb/prod/cqsar.html (accessed on 8 November 2024).
- MCASE. Available online: https://multicase.com/case-ultra/ (accessed on 8 November 2024).
- Karwath, A.; De Raedt, L. SMIREP: Predicting chemical activity from SMILES. J. Chem. Inf. Model. 2006, 46, 2432–2444. [Google Scholar] [CrossRef]
- Mauri, A.; Bertola, M. Alvascience: A new software suite for the QSAR workflow applied to the blood–brain barrier permeability. Int. J. Mol. Sci. 2022, 23, 12882. [Google Scholar] [CrossRef]
- Grebner, C.; Malmerberg, E.; Shewmaker, A.; Batista, J.; Nicholls, A.; Sadowski, J. Virtual screening in the cloud: How big is big enough? J. Chem. Inf. Model. 2019, 60, 4274–4282. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—The Konstanz information miner: Version 2.0 and beyond. ACM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Landrum, G. RDKit. Available online: https://www.rdkit.org/ (accessed on 8 February 2024).
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Reactor|ChemAxon. Available online: https://chemaxon.com/products/reactor (accessed on 8 February 2024).
- Ciallella, H.L.; Zhu, H. Advancing computational toxicology in the big data era by artificial intelligence: Data-driven and mechanism-driven modeling for chemical toxicity. Chem. Res. Toxicol. 2019, 32, 536–547. [Google Scholar] [CrossRef]
- Lin, X.; Li, X.; Lin, X. A review on applications of computational methods in drug screening and design. Molecules 2021, 25, 1375. [Google Scholar] [CrossRef]
- Fradera, X.; Babaoglu, K. Overview of methods and strategies for conducting virtual small molecule screening. Curr. Protoc. Chem. Biol. 2017, 9, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol. Inform. 2020, 39, 2000028. [Google Scholar] [CrossRef] [PubMed]
- Khoei, T.; Kaabouch, N. Machine learning: Models, challenges, and research directions. Future Internet 2023, 15, 332. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Schmidhuber, J. Deep learning in neural networks: An overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef]
- Gawehn, E.; Hiss, J.A.; Schneider, G. Deep learning in drug discovery. Mol. Inform. 2016, 35, 3–14. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef]
- Lavecchia, A. Deep learning in drug discovery: Opportunities, challenges and future prospects. Drug Discov. Today 2019, 24, 2017–2032. [Google Scholar] [CrossRef]
- Jeong, J.; Choi, J. Artificial intelligence-based toxicity prediction of environmental chemicals: Future directions for chemical management applications. Environ. Sci. Technol. 2022, 56, 7532–7543. [Google Scholar] [CrossRef]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity prediction using deep learning. Front. Environ. Sci. 2016, 3, 80. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, Y.; Liu, H.; Chang, W.; Xu, Y.; Lin, F. Prediction of hemolytic toxicity for saponins by Machine-Learning methods. Chem. Res. Toxicol. 2019, 32, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Hemmerich, J.; Troger, F.; Füzi, B.; Ecker, G.F. Using machine learning methods and structural alerts for prediction of mitochondrial toxicity. Mol. Inform. 2020, 39, 2000005. [Google Scholar] [CrossRef] [PubMed]
- Koutsoukas, A.; Monaghan, K.J.; Li, X.; Huan, J. Deep-learning: Investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J. Cheminform. 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Kleinstreuer, N.; Hartung, T. Artificial intelligence (AI)—It’s the end of the tox as we know it (and I feel fine). Arch. Toxicol. 2024, 98, 735–754. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Z.; Pang, X.; Cao, H.; Sun, Y.; Wang, L.; Zhou, Z.; Wang, P.; Liang, Y.; Wang, Y. Molecular designing of potential environmentally friendly PFAS based on deep learning and generative models. Sci. Total Environ. 2024, 953, 176095. [Google Scholar] [CrossRef]
- Banerjee, A.; Kar, S.; Roy, K.; Patlewicz, G.; Charest, N.; Benfenati, E.; Cronin, M.T. Molecular similarity in chemical informatics and predictive toxicity modeling: From quantitative read-across (q-RA) to quantitative read-across structure-activity relationship (q-RASAR) with the application of machine learning. Crit. Rev. Toxicol. 2024, 54, 659–684. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.; Ai, H.; Hu, H.; Li, S.; Zhao, J.; Liu, H. Applications of machine learning methods in drug toxicity prediction. Curr. Top. Med. Chem. 2018, 18, 987–997. [Google Scholar] [CrossRef]
- Schmeisser, S.; Miccoli, A.; von Bergen, M.; Berggren, E.; Braeuning, A.; Busch, W.; Desaintes, C.; Gourmelon, A.; Grafström, R.; Harrill, J.; et al. New approach methodologies in human regulatory toxicology—Not if, but how and when! Environ. Int. 2023, 178, 108082. [Google Scholar] [CrossRef]
- Usmani, S.M.; Bremer-Hoffmann, S.; Cheyns, K.; Cubadda, F.; Dumit, V.I.; Escher, S.E.; Fessard, V.; Gutleb, A.C.; Léger, T.; Liu, Y.C.; et al. Review of new approach methodologies for application in risk assessment of nanoparticles in the food and feed sector: Status and challenges. EFSA Support. Publ. 2024, 21, 8826E. [Google Scholar]
- Kleinstreuer, N.C.; Tong, W.; Tetko, I.V. Computational toxicology. Chem. Res. Toxicol. 2020, 33, 687–688. [Google Scholar] [CrossRef]
- Roy, K.; Kar, S.; Das, R.N. A Primer on QSAR/QSPR Modeling: Fundamental Concepts; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Tonk, R.K.; Yadav, V.; Goyal, R.K. Repurposing of immunomodulators for the treatment of cancer with QSAR approaches. In Drug Repurposing for Emerging Infectious Diseases and Cancer, 1st ed.; Sobti, R.C., Lal, S.K., Goyal, R.K., Eds.; Springer: Singapore, 2023; Chapter 13; pp. 283–297. [Google Scholar]
- Suay-Garcia, B.; Falcó, A.; Bueso-Bordils, J.I.; Anton-Fos, G.M.; Pérez-Gracia, M.T.; Alemán-López, P.A. Tree-based QSAR model for drug repurposing in the discovery of new antibacterial compounds against Escherichia coli. Pharmaceuticals 2020, 13, 431. [Google Scholar] [CrossRef] [PubMed]
- Tejera, E.; Munteanu, C.R.; López-Cortés, A.; Cabrera-Andrade, A.; Pérez-Castillo, Y. Drugs repurposing using QSAR, docking and molecular dynamics for possible inhibitors of the SARS-CoV-2 Mpro protease. Molecules 2020, 25, 5172. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Kumar, V.; Kar, S.; Roy, K.; Leszczynski, J. Repurposing FDA approved drugs as possible anti-SARS-CoV-2 medications using ligand-based computational approaches: Sum of ranking difference-based model selection. Struct. Chem. 2022, 33, 1741–1753. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Scotti, M.T.; Speck-Planche, A. Computational drug repurposing for antituberculosis therapy: Discovery of multi-strain inhibitors. Antibiotics 2021, 10, 1005. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, Y.; He, Y.; Zhu, H.; Gao, Y.; Li, Z.; Zhu, J.; Sun, X.; Fang, F.; Wen, H.; et al. Combined pharmacophore modeling, 3D-QSAR and docking studies to identify novel HDAC inhibitors using drug repurposing. J. Biomol. Struct. Dyn. 2020, 38, 533–547. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; Wiley VCH Verlag GmbH: Weinheim, Germany, 2009; Volume 41. [Google Scholar]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef]
- Gálvez, J.; deJulián-Ortiz, J.V.; García-Doménech, R. Diseño y desarrollo de nuevos fármacos contra la malaria. Enf. Emerg. 2005, 7, 44–51. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Huggins, D.J.; Venkitaraman, A.R.; Spring, D.R. Rational methods for the selection of diverse screening compounds. ACS Chem. Biol. 2011, 6, 208–217. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Hitchcock, S.A.; Pennington, L.D. Structure-brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Sun, H.; Chaudhury, S.; Abdulhameed, M.D.; Wallqvist, A.; Tawa, G. A physicochemical descriptor-based scoring scheme for effective and rapid filtering of kinase-like chemical space. J. Cheminform. 2012, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Committee on Acute Exposure Guideline Levels & National Research Council. Acute Exposure Guideline Levels for Selected Airborne Chemicals: Volume 14; National Academies Press: Washington, DC, USA, 2013; pp. 115–138. [Google Scholar]
- European Medicines Agency. ICH Guideline S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use. 2012. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-s2-r1-genotoxicity-testing-and-data-interpretation-pharmaceuticals-intended-human-use-step-5_en.pdf (accessed on 19 November 2024).
- Shehata, M.; Durner, J.; Eldenez, A.; Van Landuyt, K.; Styllou, P.; Rothmund, L.; Hickel, R.; Scherthan, H.; Geurtsen, W.; Kana, B.; et al. Cytotoxicity and induction of DNA double-strand breaks by components leached from dental composites in primary human gingival fibroblasts. Dent. Mater. 2013, 29, 971–979. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, L.; Yuan, X.; Luo, F.; Pu, H. Development and validation of a UPLC–MS/MS method for ultra-trace level determination of acyl chloride potential genotoxic impurity in mezlocillin. J. Chromatogr. Sci. 2022, 60, 732–740. [Google Scholar] [CrossRef]
- Limban, C.; Nuţă, D.C.; Chiriţă, C.; Negreș, S.; Arsene, A.L.; Goumenou, M.; Karakitsios, S.P.; Tsatsakis, A.M.; Sarigiannis, D.A. The use of structural alerts to avoid the toxicity of pharmaceuticals. Toxicol. Rep. 2018, 5, 943–953. [Google Scholar] [CrossRef]
- Duart, M.J.; Antón-Fos, G.M.; Alemán, P.A.; Gay-Roig, J.B.; González-Rosende, M.E.; Gálvez, J.; García-Domenech, R. New potential antihistaminic compounds. Virtual combinatorial chemistry, computational screening, real synthesis, and pharmacological evaluation. J. Med. Chem. 2005, 48, 1260–1264. [Google Scholar] [CrossRef]
- Duart, M.J.; García-Doménech, R.; Gálvez, J.; Alemán, P.A.; Martín-Algarra, R.V.; Antón-Fos, G.M. Application of a mathematical topological pattern of antihistaminic activity for the selection of new drug candidates and pharmacology assays. J. Med. Chem. 2006, 49, 3667–3673. [Google Scholar] [CrossRef]
- Wermuth, C.G. Selective optimization of side activities: Another way to drug discovery. J. Med. Chem. 2004, 47, 1303–1314. [Google Scholar] [CrossRef]
- Wang, M.Y.; Liang, J.W.; Olounfeh, K.M.; Sun, Q.; Zhao, N.; Meng, F.H. A comprehensive in silico method to study the QSTR of the aconitine alkaloids for designing novel drugs. Molecules 2018, 23, 2385. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N. Simultaneous virtual prediction of anti-Escherichia coli activities and ADMET profiles: A chemoinformatic complementary approach for high-throughput screening. ACS Comb. Sci. 2014, 16, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.N. Computer-aided discovery in antimicrobial research: In silico model for virtual screening of potent and safe antipseudomonas agents. Comb. Chem. High Throughput Screen. 2015, 18, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.V.; Bansod, G.; Mahajan, M.; Dietrich, P.; Singh, S.P.; Rav, K.; Thissen, A.; Bharde, A.M.; Rothenstein, D.; Kulkarni, S.; et al. Digital transformation in toxicology: Improving communication and efficiency in risk assessment. ACS Omega 2023, 8, 21377–21390. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.S.; Zhao, J.C.; Yang, Y.N.; Zhao, C.X.; Yan, J.; Liu, S.; Hu, Q.N.; Xu, Q.S.; Liang, Y.Z. In silico toxicity prediction by support vector machine and SMILES representation-based string kernel. SAR QSAR Environ. Res. 2012, 23, 141–153. [Google Scholar] [CrossRef]
- Wu, K.; Wei, G.W. Quantitative toxicity prediction using topology based multitask deep neural networks. J. Chem. Inf. Model. 2018, 58, 520–531. [Google Scholar] [CrossRef]
- Huang, X.; Tang, F.; Hua, Y.; Li, X. In silico prediction of drug-induced ototoxicity using machine learning and deep learning methods. Chem. Biol. Drug Des. 2021, 98, 248–257. [Google Scholar] [CrossRef]
- Yu, X. Support vector machine-based model for toxicity of organic compounds against fish. Regul. Toxicol. Pharmacol. 2021, 123, 104942. [Google Scholar] [CrossRef]
- Muster, W.; Breidenbach, A.; Fischer, H.; Kirchner, S.; Müller, L.; Pähler, A. Computational toxicology in drug development. Drug Discov. Today 2008, 13, 303–310. [Google Scholar] [CrossRef]
- Saifi, I.; Bhat, B.A.; Hamdani, S.S.; Bhat, U.Y.; Lobato-Tapia, C.A.; Mir, M.A.; Dar, T.U.H.; Ganie, S.A. Artificial intelligence and cheminformatics tools: A contribution to the drug development and chemical science. J. Biomol. Struct. Dyn. 2024, 42, 6523–6541. [Google Scholar] [CrossRef]
- Schür, C.; Gasser, L.; Perez-Cruz, F.; Schirmer, K.; Baity-Jesi, M. A benchmark dataset for machine learning in ecotoxicology. Sci. Data 2023, 10, 718. [Google Scholar] [CrossRef]
- Manzetti, S. Ecotoxicity of polycyclic aromatic hydrocarbons, aromatic amines, and nitroarenes through molecular properties. Environ. Chem. Lett. 2012, 10, 349–361. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, V.; Podder, T.; Ojha, P.K. First report on ecotoxicological QSTR and I-QSTR modeling for the prediction of acute ecotoxicity of diverse organic chemicals against three protozoan species. Chemosphere 2023, 335, 139066. [Google Scholar] [CrossRef] [PubMed]
- Gita, S.; Shukla, S.; Saharan, N.; Prakash, C.; Deshmukhe, G. Toxic effects of selected textile dyes on elemental composition, photosynthetic pigments, protein content and growth of a freshwater chlorophycean alga Chlorella vulgaris. Bull. Environ. Contam. Toxicol. 2019, 102, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Grote, M.; Schüürmann, G.; Altenburger, R. Modeling photoinduced algal toxicity of polycyclic aromatic hydrocarbons. Environ. Sci. Technol. 2005, 39, 4141–4149. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Hu, Y.; Zheng, S.; Lee, J.; Chen, J.; Bi, S.; Xu, Z. Machine learning for mixture toxicity analysis based on high-throughput printing technology. Talanta 2020, 207, 120299. [Google Scholar] [CrossRef]
- Chen, X.; Dang, L.; Yang, H.; Huang, X.; Yu, X. Machine learning-based prediction of toxicity of organic compounds towards fathead minnow. RSC Adv. 2020, 10, 36174–36180. [Google Scholar] [CrossRef]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory–design and description. J. Comput. Aided Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef]
- Hao, Y.; Sun, G.; Fan, T.; Sun, X.; Liu, Y.; Zhang, N.; Zhao, L.; Zhong, R.; Peng, Y. Prediction on the mutagenicity of nitroaromatic compounds using quantum chemistry descriptors based QSAR and machine learning derived classification methods. Ecotoxicol. Environ. Saf. 2019, 186, 109822. [Google Scholar] [CrossRef]
- Michalaki, A.; Kakavas, D.; Giannouli, M.; Grintzalis, K. Toxicity of “green solvents”-The impact of butyl methylimidazolium ionic liquids on daphnids. J. Ion. Liq. 2023, 3, 100059. [Google Scholar] [CrossRef]
- Egorova, K.S.; Ananikov, V.P. Toxicity of ionic liquids: Eco(cyto)activity as complicated, but unavoidable parameter for task-specific optimization. Chem. Sus. Chem. 2014, 7, 336–360. [Google Scholar] [CrossRef]
- Das, R.N.; Roy, K. Development of classification and regression models for Vibrio fischeri toxicity of ionic liquids: Green solvents for the future. Toxicol. Res. 2012, 1, 186–195. [Google Scholar] [CrossRef]
- Wang, C.; Wei, Z.; Wang, L.; Sun, P.; Wang, Z. Assessment of bromide-based ionic liquid toxicity toward aquatic organisms and QSAR analysis. Ecotoxicol. Environ. Saf. 2015, 115, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Das, R.N. QSTR with extended topochemical atom (ETA) indices. 16. Development of predictive classification and regression models for toxicity of ionic liquids towards Daphnia magna. J. Hazard. Mater. 2012, 254, 166–178. [Google Scholar] [CrossRef]
- An, T.; Gao, Y.; Li, G.; Kamat, P.V.; Peller, J.; Joyce, M.V. Kinetics and mechanism of •OH mediated degradation of dimethyl phthalate in aqueous solution: Experimental and theoretical studies. Environ. Sci. Technol. 2014, 48, 641–648. [Google Scholar] [CrossRef]
- Sorell, T.L. Approaches to the development of human health toxicity values for active pharmaceutical ingredients in the environment. AAPS J. 2016, 18, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Ball, N.; Bars, R.; Botham, P.A.; Cuciureanu, A.; Cronin, M.T.; Doe, J.E.; Dudzina, T.; Gant, T.W.; Leist, M.; van Ravenzwaay, B. A framework for chemical safety assessment incorporating new approach methodologies within REACH. Arch. Toxicol. 2022, 96, 743–766. [Google Scholar] [CrossRef] [PubMed]
- REACH. Available online: https://single-market-economy.ec.europa.eu/sectors/chemicals/reach/non-animal-testing_en (accessed on 18 November 2024).
- European Chemicals Agency. Available online: https://echa.europa.eu/guidance-documents/guidance-on-reach (accessed on 18 November 2024).
- European Chemicals Agency. How to Use and Report (Q)SARs. Practical Guide 5; European Chemicals Agency: Helsinki, Finland, 2016.
- Choi, S.M.; Roh, T.H.; Lim, D.S.; Kacew, S.; Kim, H.S.; Lee, B.M. Risk assessment of benzalkonium chloride in cosmetic products. J. Toxicol. Environ. Health B Crit. Rev. 2018, 21, 8–23. [Google Scholar] [CrossRef]
- Goldstein, B.D.; Brooks, B.W.; Cohen, S.D.; Gates, A.E.; Honeycutt, M.E.; Morris, J.B.; Orme-Zavaleta, J.; Penning, T.M.; Snawder, J. The role of toxicological science in meeting the challenges and opportunities of hydraulic fracturing. Toxicol. Sci. 2014, 139, 271–283. [Google Scholar] [CrossRef]
- Daood, N.J.; Russo, D.P.; Chung, E.; Qin, X.; Zhu, H. Predicting chemical immunotoxicity through data-driven QSAR modeling of aryl hydrocarbon receptor agonism and related toxicity mechanisms. Environ. Health 2024, 2, 474–485. [Google Scholar] [CrossRef]
- Konkel, M.E.; Tilly, K. Temperature-regulated expression of bacterial virulence genes. Microbes Infect. 2000, 2, 157–166. [Google Scholar] [CrossRef]
- Taszlow, P.; Vertyporokh, L.; Wojda, I. Humoral immune response of Galleria mellonella after repeated infection with Bacillus thuringiensis. J. Invertebr. Pathol. 2017, 149, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Beard, R.L. Species-specificity of toxicants as related to route of administration. J. Econ. Entomol. 1949, 42, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Jander, G.; Rahme, L.G.; Ausubel, F.M. Positive correlation between virulence of Pseudomonas aeruginosa mutants in mice and insects. J. Bacteriol. 2000, 182, 3843–3845. [Google Scholar] [CrossRef]
- Brennan, M.; Thomas, D.Y.; Whitewat, M.; Kavanagh, K. Correlation between virulence of Candida albicans mutants in mice and Galleria mellonella larvae. FEMS Immunol. Med. Microbiol. 2002, 34, 153–157. [Google Scholar] [CrossRef]
- Fuchs, B.B.; O’Brien, E.; Khoury, J.B.; Mylonakis, E. Methods for using Galleria mellonella as a model host to study fungal pathogenesis. Virulence 2010, 1, 475–482. [Google Scholar] [CrossRef]
- Merkling, S.H.; Lambrechts, L. Taking insect immunity to the single-cell level. Trends Immunol. 2020, 41, 190–199. [Google Scholar] [CrossRef]
- Champion, O.L.; Wagley, S.; Titball, R.W. Galleria mellonella as a model host for microbiological and toxin research. Virulence 2016, 7, 840–845. [Google Scholar] [CrossRef]
- Ignasiak, K.; Maxwell, A. Galleria mellonella (greater wax moth) larvae as a model for antibiotic susceptibility testing and acute toxicity trials. BMC Res. Notes 2017, 10, 428. [Google Scholar] [CrossRef]
- Allegra, E.; Titball, R.W.; Carter, J.; Champion, O.L. Galleria mellonella larvae allow the discrimination of toxic and non-toxic chemicals. Chemosphere 2018, 198, 469–472. [Google Scholar] [CrossRef]
- Maguire, R.; Duggan, O.; Kavanagh, K. Evaluation of Galleria mellonella larvae as an in vivo model for assessing the relative toxicity of food preservative agents. Cell Biol. Toxicol. 2016, 32, 209–216. [Google Scholar] [CrossRef]
- Maguire, R.; Kunc, M.; Hyrsl, P.; Kavanagh, K. Caffeine administration alters the behavior and development of Galleria mellonella larvae. Neurotox. Teratol. 2017, 64, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Megaw, J.; Thompson, T.P.; Lafferty, R.A.; Gilmore, B.F. Galleria mellonella as a novel in vivo model for assessment of the toxicity of 1-alkyl-3-methylimidazolium chloride ionic liquids. Chemosphere 2015, 139, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Suay-García, B.; Alemán-López, P.A.; Bueso-Bordils, J.I.; Falcó, A.; Antón-Fos, G.; Pérez-Gracia, M.T. New solvent options for in vivo assays in the Galleria mellonella larvae model. Virulence 2019, 10, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.M.; McArthur, J.D. Developing Galleria mellonella as a model host for human pathogens. Virulence 2013, 4, 350–353. [Google Scholar] [CrossRef]
- Aneja, B.; Irfan, M.; Kapil, C.; Jairajpuri, M.A.; Maguire, R.; Kavanagh, K.; Rizvi, M.M.A.; Manzoor, N.; Azam, A.; Abid, M. Effect of novel triazole-amino acid hybrids on growth and virulence of Candida species: In vitro and in vivo studies. Org. Biomol. Chem. 2016, 14, 10599. [Google Scholar] [CrossRef]
- Zhang, K.; Ding, W.; Sun, J.; Zhang, B.; Lu, F.; Lai, R.; Zou, Y.; Yedid, G. Antioxidant and antitumor activities of 4-arylcoumarins and 4-aryl-3,4-dihydrocoumarins. Biochimie 2014, 107 Pt B, 203–230. [Google Scholar] [CrossRef]

| Software | Main Features | Ref. |
|---|---|---|
| QSARPro | Performs group-based QSAR approach, establishing a correlation between chemical group variation at different molecular sites of interest and the biological activity. | [14] |
| MedChem Studio | Cheminformatics platform supporting lead identification and prioritization, de novo design, scaffold hopping and lead optimization. | [15] |
| McQSAR | Free program to generate QSAR equations using the genetic function approximation paradigm. | [16] |
| PADEL | Free software to calculate molecular descriptors and fingerprints. | [17] |
| Codessa | Uses quantum mechanics-derived descriptors to develop QSAR/QSPR models. | [18] |
| cQSAR | Program for interactive, visual compound promotion and optimization. It includes PD and PK parameters and can be linked to other modules for physicochemical and ADME. | [19] |
| MCASE | ML approach to automatically evaluate compounds/activity data set and identify the biophores. It then creates organized dictionaries of them and develops ad hoc local QSAR correlations. | [20] |
| SMIREP | System for predicting the structural activity of chemical compounds. | [21] |
| Alvascience | QSAR software package that uses in silico techniques to analyze chemical datasets and evaluate the physico-chemical and ecotoxicological properties of chemicals. | [22] |
| Methodology | Definitions | Model Types | Limitations |
|---|---|---|---|
| Quantitative structure–activity relationships (QSARs) | Use molecular descriptors Predict chemical’s toxicity | Local and global QSAR, SAR, QSTR and QSPR | Requires large database, feature selection |
| Pharmacokinetic (PK), Pharmacodynamic (PD) | PK and PD models evaluate concentration at a given time and calculate effect at a given concentration, respectively | One-compartment models, two-compartment models | PK and PD parameters may be unavailable or inaccurate |
| Structural alerts (SAs), rule-based | Chemical structures associated with toxicity | Human-based rules, induction-based rules, pattern growth | SAs cannot provide insight into the biological pathways of toxicity |
| Read across (RA) | Predict unknown toxicity of chemical using similar chemicals with known toxicity | Analog approach, category approach, qualitative and quantitative RA | Use small datasets, accuracy depending on the number and choice of analogs, similarity metrics |
| Chemical Structure | Group Name | Screening Liability |
|---|---|---|
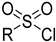 | Sulfonyl chloride | Can metabolize, causing genotoxicity |
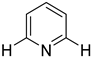 | 2,6-unsubstituted pyridine | Potential interference with cytochrome P450s due to metal ion coordination |
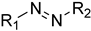 | Azo | Potentially carcinogenic and mutagenic |
 | Acetal | Metabolically unstable due to acetal hydrolysis |
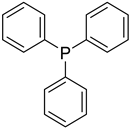 | Triphenylphosphane | Produces DNA double-strand breaks (genotoxic) and human cell death effects (cytotoxic) |
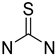 | Thiourea | Metabolically unstable due to flavin oxidation Potential non-specific protein binding |
 | 1,2-dicarbonyl | Metabolically unstable Potential toxicity due to mutagenicity |
 | Nitro | Prone to reduction, yielding reactive species Potential hepatocarcinogen |
 | α,β-unsaturated carbonyl | Prone to reactivity by acting as a Michael acceptor |
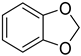 | Methylenedioxy | Metabolically unstable due to acetal hydrolysis Prone to oxidation, yielding reactive quinones |
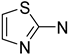 | Aminotiazole | Potential toxicity |
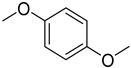 | 1,4-dimethoxybenzene | Very prone to oxidation, yielding reactive quinones |
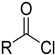 | Chlorocarbonyl | Potential genotoxic impurity |
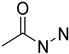 | Acylhidrazide | Metabolically unstable due to acyl hydrolysis |
| Year | Content | Method | Conclusion | Author |
|---|---|---|---|---|
| 2012 | A series of SMILES meant serial kernels and SVM were used to classify chemical toxicity in a toxicity database network (DSSTox) | SVM | The AUC values of DBPCAN data, NCTRER data, EPAFHM data, CPDBAS data and FDAMDD data are 0.950, 0.901, 0.740, 0.823 and 0.840, respectively | Cao et al. [78] |
| 2018 | Four quantitative toxicity data sets were used: LC50, LC50-DM, IGC50 and LD50. DNN, RF and GBDT are used to build the model | DNN RF GBDT a | According to the coefficient r2 of four data sets, the fitting effect of the DNN is the best, and the results obtained are more accurate | Wu et al. [79] |
| 2021 | In a study on drug-induced chemical ototoxicity, 1102 ototoxic drugs and 1705 non-ototoxic drugs were collected. ML and DL algorithms were used to construct individual models and consensus models, and a structural characteristics analysis of ototoxic drugs was conducted | ANN SVM RF XGBoost b TCNN c | The performance of the consensus model on the test set and external verification set is better than that of the single model, and the accuracy rates are 0.95 and 0.90, respectively | Huang et al. [80] |
| 2021 | An SVM and GA model was established on a large data set of 840 organic compounds to explore the toxicity prediction of chemicals to various fish | SVM GA d | The decision coefficient r2 of the SVM model is above 0.70 on both the training set and testing set, which shows good prediction performance | Yu et al. [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bueso-Bordils, J.I.; Antón-Fos, G.M.; Martín-Algarra, R.; Alemán-López, P.A. Overview of Computational Toxicology Methods Applied in Drug and Green Chemical Discovery. J. Xenobiot. 2024, 14, 1901-1918. https://doi.org/10.3390/jox14040101
Bueso-Bordils JI, Antón-Fos GM, Martín-Algarra R, Alemán-López PA. Overview of Computational Toxicology Methods Applied in Drug and Green Chemical Discovery. Journal of Xenobiotics. 2024; 14(4):1901-1918. https://doi.org/10.3390/jox14040101
Chicago/Turabian StyleBueso-Bordils, Jose I., Gerardo M. Antón-Fos, Rafael Martín-Algarra, and Pedro A. Alemán-López. 2024. "Overview of Computational Toxicology Methods Applied in Drug and Green Chemical Discovery" Journal of Xenobiotics 14, no. 4: 1901-1918. https://doi.org/10.3390/jox14040101
APA StyleBueso-Bordils, J. I., Antón-Fos, G. M., Martín-Algarra, R., & Alemán-López, P. A. (2024). Overview of Computational Toxicology Methods Applied in Drug and Green Chemical Discovery. Journal of Xenobiotics, 14(4), 1901-1918. https://doi.org/10.3390/jox14040101