

Preparation of Gypsum–Urea with Enhanced Sustainability from Flue Gas Desulfurization Gypsum in Saturated Urea Solution

 ,
,
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Preparation Methods
2.3. Characterization
3. Results and Discussion
3.1. Analysis of Initial FGD Gypsum
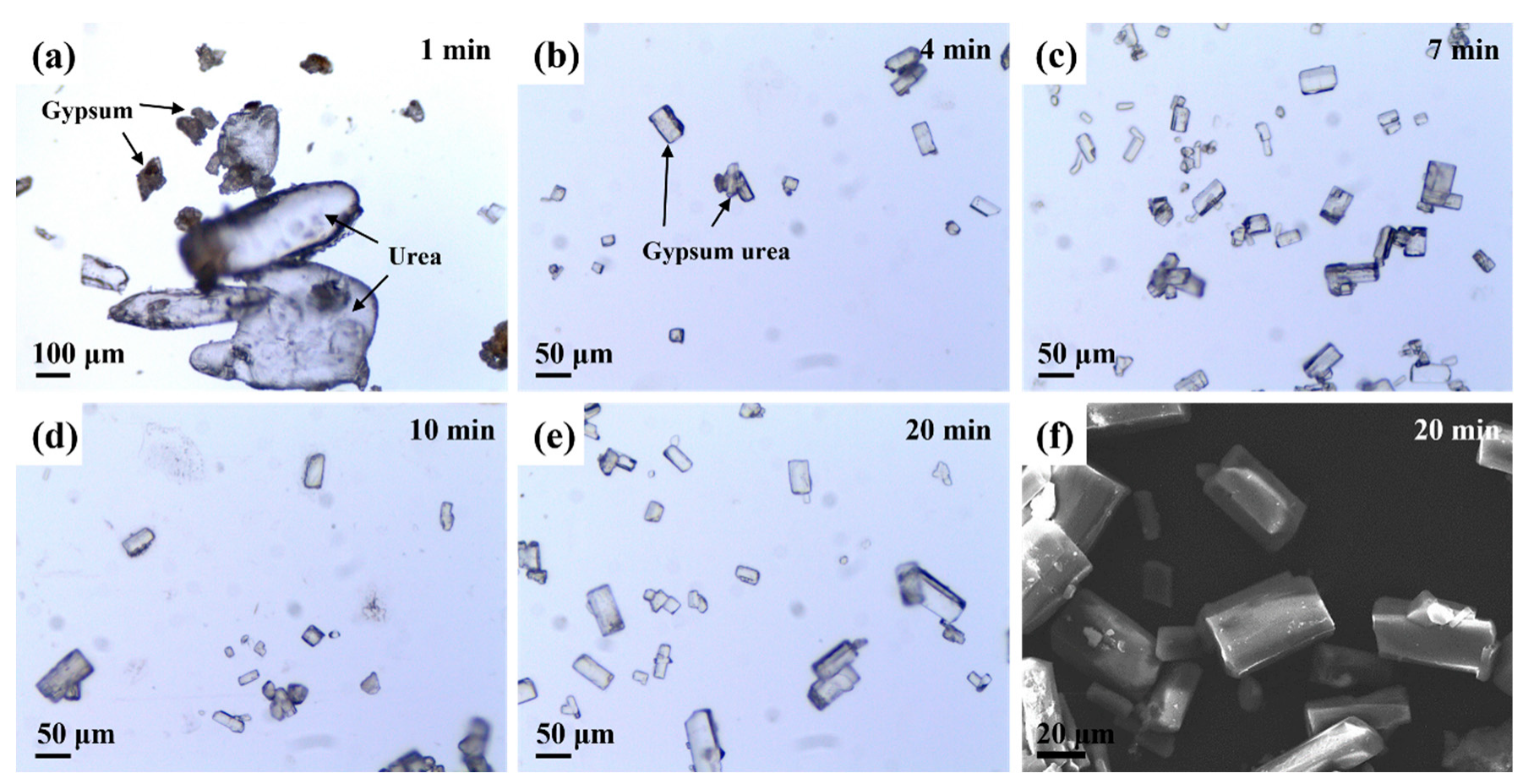
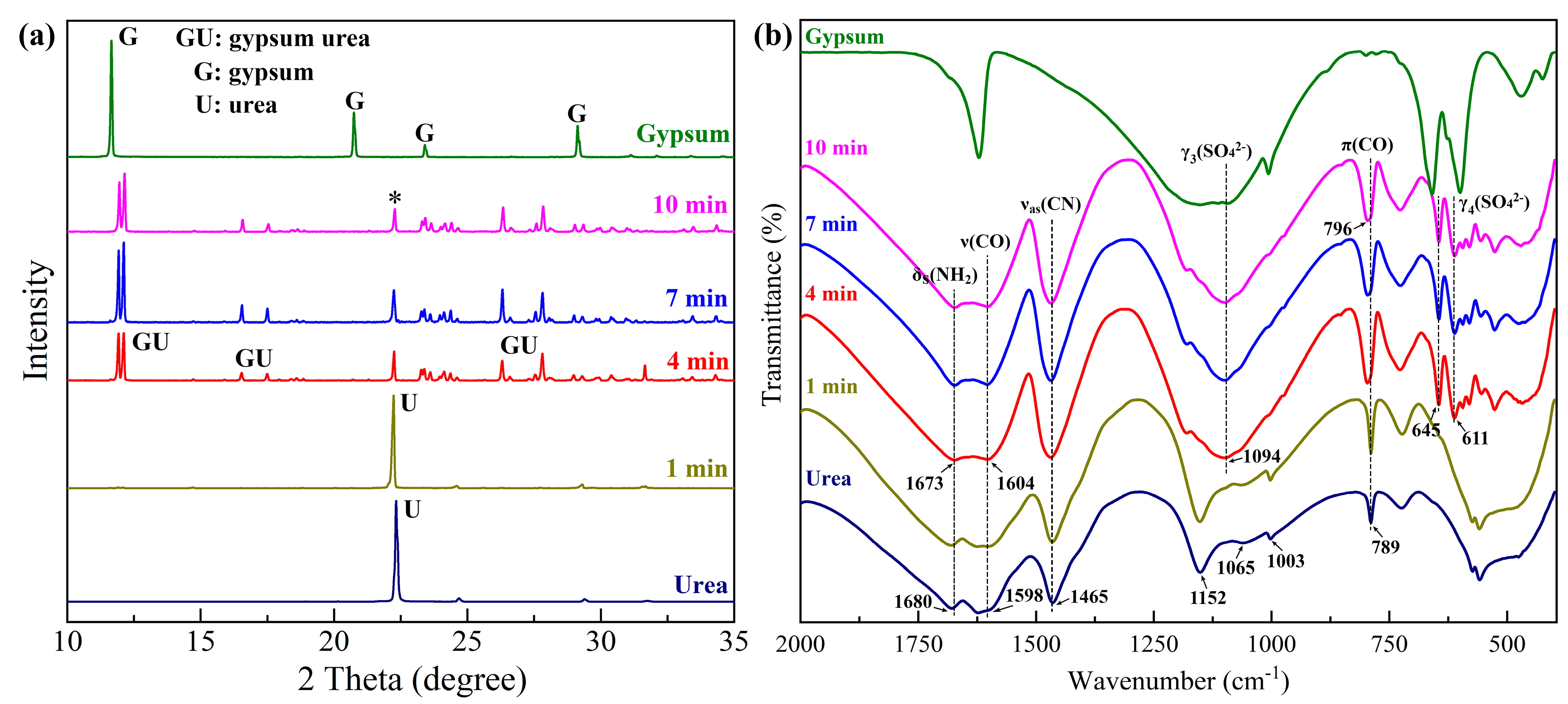
3.2. Preparation of Gypsum–Urea
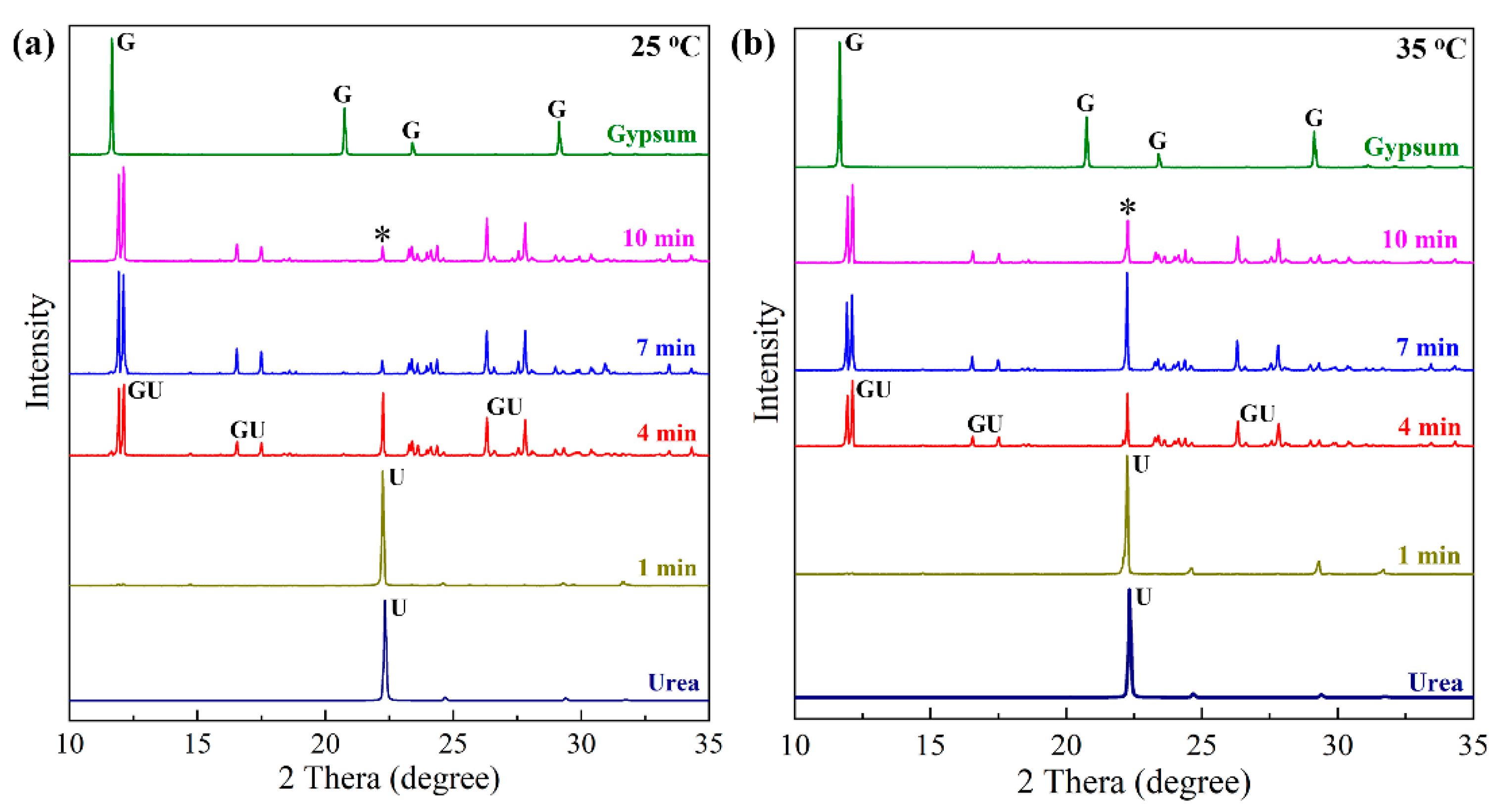
3.3. Effect of Temperature on Preparation Process and Morphology Evolution
3.4. Hygroscopicity and Nitrogen Release Efficiency of Gypsum–Urea
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bremner, J.M.I.S. Recent research on problems in the use of urea as a nitrogen fertilizer. Fertil. Res. 1995, 42, 321–329. [Google Scholar] [CrossRef]
- Canfield, D.E.; Glazer, A.N.; Falkowski, P.G. The evolution and future of Earth’s nitrogen cycle. Science 2010, 330, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Duce, R.A.; Laroche, J.; Altieri, K.; Arrigo, K.R.; Baker, A.R.; Capone, D.G.; Cprnell, S.; Dentener, F.; Galloway, J.; Ganeshram, R.S.; et al. Impacts of atmospheric anthropogenic nitrogen on the open ocean. Science 2008, 320, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Burger, M.; Doane, T.A.; Horwath, W.R. Ammonia oxidation pathways and nitrifier denitrification are significant sources of N2O and NO under low oxygen availability. Proc. Natl. Acad. Sci. USA 2013, 110, 6328–6333. [Google Scholar] [CrossRef] [PubMed]
- Tilman, D.; Fargione, J.; Wolff, B.; D’Antonio, C.; Dobson, A.; Howarth, R.; Schindler, D.; Schesinger, W.H.; Simberloff, D.; Swackhamer, D. Forecasting agriculturally driven global environmental change. Science 2001, 292, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Conley, D.J.; Paerl, H.W.; Howarth, R.W.; Boesch, D.F.; Seitzinger, S.P.; Havens, K.E.; Lancelot, C.; Likens, G.E. Controlling eutrophication: Nitrogen and phosphorus. Science 2009, 323, 1014–1015. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, W.; Yin, J.; Chadwick, D.; Norse, D.; Lu, Y.; Liu, X.; Chen, X.; Zhang, F.; Powlson, D.; et al. Enhanced-efficiency fertilizers are not a panacea for resolving the nitrogen problem. Glob. Chang. Biol. 2018, 24, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Naz, M.Y.; Sulaiman, S.A. Slow release coating remedy for nitrogen loss from conventional urea: A review. J. Control. Release 2016, 225, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, M.; Li, Y.; Fan, X.; Geng, Y. Improving the quality of polymer-coated urea with recycled plastic, proper additives, and large tablets. J. Agric. Food Chem. 2012, 60, 11229–11237. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.M.E.; Cabral-Albuquerque, E.C.M.; Alves, T.L.M.; Pinto, J.C.; Fialho, R.L. Use of polyhydroxybutyrate and ethyl cellulose for coating of urea granules. J. Agric. Food Chem. 2013, 61, 9984–9991. [Google Scholar] [CrossRef]
- Cantarella, H.; Otto, R.; Soares, J.R.; Silva, A.G.D.B. Agronomic efficiency of NBPT as a urease inhibitor: A review. J. Adv. Res. 2018, 13, 19–27. [Google Scholar] [CrossRef]
- Chien, S.H.; Prochnow, L.I.; Cantarella, H. Recent developments of fertilizer production and use to improve nutrient efficiency and minimize environmental impacts. Adv. Agron. 2009, 102, 267–322. [Google Scholar]
- Liu, L.; Shen, T.; Yang, Y.; Gao, B.; Li, Y.C.; Xie, J.; Tang, Y.; Zhang, S.; Wang, Z.; Chen, J. Bio-based large tablet controlled-release urea: Synthesis, characterization, and controlled-released mechanisms. J. Agric. Food Chem. 2018, 66, 11265–11272. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, M.J.; Beach, E.S.; Zimmerman, J.B.; Anastas, P.T. Green chemistry and green engineering: A framework for sustainable technology development. Annu. Rev. Environ. Resour. 2011, 36, 271–293. [Google Scholar] [CrossRef]
- Mazzei, L.; Broll, V.; Casali, L.; Silva, M.; Braga, D.; Grepioni, F.; Baltrusaitis, J.; Ciurli, S. Multifunctional urea cocrystal with combined ureolysis and nitrification inhibiting capabilities for enhanced nitrogen management. ACS Sustain. Chem. Eng. 2019, 7, 13369–13378. [Google Scholar] [CrossRef]
- Casali, L.; Mazzei, L.; Shemchuk, O.; Sharma, L.; Honer, K.; Grepioni, F.; Ciurli, S.; Braga, D.; Baltrusaitis, J. Novel dual-action plant fertilizer and urease inhibitor: Urea·catechol cocrystal. Characterization and environmental reactivity. ACS Sustain. Chem. Eng. 2019, 7, 2852–2859. [Google Scholar] [CrossRef]
- Honer, K.; Kalfaoglu, E.; Pico, C.; McCann, J.; Baltrusaitis, J. Mechanosynthesis of magnesium and calcium salt–urea ionic cocrystal fertilizer materials for improved nitrogen management. ACS Sustain. Chem. Eng. 2017, 5, 8546–8550. [Google Scholar] [CrossRef]
- Barčauskaitė, K.; Brazienė, Z.; Avižienytė, D.; Silva, M.; Drapanauskaite, D.; Honer, K.; Gvildienė, K.; Slinksiene, R.; Jancaitiene, K.; Mazeika, R.; et al. Mechanochemically synthesized gypsum and gypsum drywall waste cocrystals with urea for enhanced environmental sustainability fertilizers. J. Environ. Chem. Eng. 2020, 8, 103965. [Google Scholar] [CrossRef]
- Pereira, E.I.; Minussi, F.B.; Da Cruz, C.C.T.; Bernardi, A.C.C.; Ribeiro, C. Urea–montmorillonite-extruded nanocomposites: A novel slow-release material. J. Agric. Food Chem. 2012, 60, 5267–5272. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Liu, Y.; Yang, G.; Zhang, A. Water- and fertilizer-integrated hydrogel derived from the polymerization of acrylic acid and urea as a slow-release N fertilizer and water retention in agriculture. J. Agric. Food Chem. 2018, 66, 5762–5769. [Google Scholar] [CrossRef] [PubMed]
- Honer, K.; Pico, C.; Baltrusaitis, J. Reactive mechanosynthesis of urea ionic cocrystal fertilizer materials from abundant low solubility magnesium- and calcium-containing minerals. ACS Sustain. Chem. Eng. 2018, 6, 4680–4687. [Google Scholar] [CrossRef]
- Casali, L.; Mazzei, L.; Shemchuk, O.; Honer, K.; Grepioni, F.; Ciurli, S.; Braga, D.; Baltrusaitis, J. Smart urea ionic co-crystals with enhanced urease inhibition activity for improved nitrogen cycle management. Chem. Commun. 2018, 54, 7637–7640. [Google Scholar] [CrossRef] [PubMed]
- Julien, P.A.; Germann, L.S.; Titi, H.M.; Etter, M.; Dinnebier, R.E.; Sharma, L.; Baltrusaitis, J. In situ monitoring of mechanochemical synthesis of calcium urea phosphate fertilizer cocrystal reveals highly effective water-based autocatalysis. Chem. Sci. 2020, 11, 2350–2355. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, B.; Sinha, A.S.; Despera, J.; Aakeröy, C.B. Modulating the physical properties of solid forms of urea using co-crystallization technology. Chem. Commun. 2018, 54, 4657–4660. [Google Scholar] [CrossRef] [PubMed]
- Adassooriya, N.M.; Mahanta, S.P.; Thakuria, R. Mechanochemistry as an emerging tool for the preparation of sustained release urea cocrystals as a nitrogen source. CrystEngComm 2022, 24, 1679–1689. [Google Scholar] [CrossRef]
- Zhao, J.; Jia, C.; Fang, X.; Xiao, A.; Zhang, H. Utilization of industrial by-product gypsum to prepare urea gypsum cocrystals as a sustained release fertilizer: A review. Asia-Pac. J. Chem. Eng. 2024, e3066. [Google Scholar] [CrossRef]
- Malinowski, P.; Biskupski, A.; Głowiński, J. Preparation methods of calcium sulphate and urea adduct. Pol. J. Chem. Technol. 2007, 9, 111–114. [Google Scholar] [CrossRef]
- Borowik, M.; Malinowski, P.; Biskupski, A.; Dawidowicz, M.; Schab, S.; Rusek, P.; Igras, J.; Kesik, K. Production technology of nitrogen-sulphur-calcium fertilizers on the base of urea and phosphogypsum. Chemik 2012, 66, 525–534. [Google Scholar]
- Brekalo, I.; Martinez, V.; Karadeniz, B.; Orešković, P.; Drapanauskaite, D.; Vriesema, H.; Stenekes, R.; Etter, M.; Dejanović, I.; Baltrusaitis, J.; et al. Scale-up of agrochemical urea-gypsum cocrystal synthesis using thermally controlled mechanochemistry. ACS Sustain. Chem. Eng. 2022, 10, 6743–6754. [Google Scholar] [CrossRef]
- Whittaker, C.W.; Lundstrom, F.O.; Hendricks, S.B. Reaction between urea and gypsum. Ind. Eng. Chem. 1933, 25, 1280–1282. [Google Scholar] [CrossRef]
- Xiao, A.; Jia, C.; Fang, X.; Zhao, J.; Zhang, H. Simultaneous effect of Na2EDTA on the phase transformation and morphology evolution during the transformation of gypsum into α-calcium sulfate hemihydrate. New J. Chem. 2024, 48, 4473–4481. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, A.; Ma, F.; Liu, J.; Xiao, G.; Xu, X. Amendment of saline–alkaline soil with flue-gas desulfurization gypsum in the Yinchuan plain, northwest China. Sustainability 2023, 15, 8658. [Google Scholar] [CrossRef]
- Wang, J.; Yang, P. Potential flue gas desulfurization gypsum utilization in agriculture: A comprehensive review. Renew. Sustain. Energy Rev. 2018, 82, 1969–1978. [Google Scholar] [CrossRef]
- Watts, D.B.; Dick, W.A. Sustainable uses of FGD gypsum in agricultural systems: Introduction. J. Environ. Qual. 2014, 43, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ma, Y.; Liu, Y.; Li, J.; Deng, J.; Rao, X.; Zhang, Y. The combined effects of nitrogen fertilizer, humic acid, and gypsum on yield-scaled greenhouse gas emissions from a coastal saline rice field. Environ. Sci. Pollut. Res. 2019, 26, 19502–19511. [Google Scholar] [CrossRef] [PubMed]
- Acuña-Guzman, S.F.; Norton, L.D. Upcycling of FGD gypsum into a product to reduce interrill erosion: A study assessing methods of soil surface application. Sustainability 2023, 15, 1977. [Google Scholar] [CrossRef]
- Villiers, J.P.R.; Boeyens, J.C.A. Crystal structure of a calcium sulfate-urea complex. J. Cryst. Mol. Struct. 1975, 5, 215–226. [Google Scholar] [CrossRef]
- Frazier, A.W.; Lehr, J.R.; Smith, J.P. Urea-monocalcium phosphate, a component of mixed fertilizers. J. Agric. Food Chem. 1967, 15, 345–347. [Google Scholar] [CrossRef]
- Frazier, A.W. The phase system CaO-CO(NH2)2-N2O5-H2O at 25 °C. Fertil. Res. 1992, 32, 157–160. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CaO | SO3 | Fe2O3 | Al2O3 | SiO2 | MgO | K2O | Na2O | F | Cl | TiO2 | CuO | ZnO | SrO | BaO | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 40.66 | 46.33 | 0.29 | 5.51 | 3.64 | 2.48 | 0.19 | 0.18 | 0.14 | 0.32 | 0.05 | 0.02 | 0.01 | 0.05 | 0.13 | 100.00 |
| Urea | Gypsum–Urea | |||
|---|---|---|---|---|
| 25 °C | 30 °C | 35 °C | ||
| Day 1 |  |  |  |  |
| Day 6 |  |  |  |  |
| Day 10 |  |  |  |  |
| Day 15 |  |  |  |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, C.; Zhao, J.; Fang, X.; Wang, P.; Xiao, A.; Zhang, H. Preparation of Gypsum–Urea with Enhanced Sustainability from Flue Gas Desulfurization Gypsum in Saturated Urea Solution. Sustainability 2024, 16, 6208. https://doi.org/10.3390/su16146208
Jia C, Zhao J, Fang X, Wang P, Xiao A, Zhang H. Preparation of Gypsum–Urea with Enhanced Sustainability from Flue Gas Desulfurization Gypsum in Saturated Urea Solution. Sustainability. 2024; 16(14):6208. https://doi.org/10.3390/su16146208
Chicago/Turabian StyleJia, Caiyun, Jiang Zhao, Xiaoxia Fang, Pujun Wang, Anni Xiao, and Haijun Zhang. 2024. "Preparation of Gypsum–Urea with Enhanced Sustainability from Flue Gas Desulfurization Gypsum in Saturated Urea Solution" Sustainability 16, no. 14: 6208. https://doi.org/10.3390/su16146208
APA StyleJia, C., Zhao, J., Fang, X., Wang, P., Xiao, A., & Zhang, H. (2024). Preparation of Gypsum–Urea with Enhanced Sustainability from Flue Gas Desulfurization Gypsum in Saturated Urea Solution. Sustainability, 16(14), 6208. https://doi.org/10.3390/su16146208