Sarcopoterium spinosum Inhibited the Development of Non-Alcoholic Steatosis and Steatohepatitis in Mice



Abstract
1. Introduction
2. Materials and Methods
2.1. S. spinosum Extract Preparation
2.2. Study Design
2.2.1. Steatosis Model
2.2.2. NASH Model
Prevention of NASH
Treatment of NASH
2.3. Glucose Tolerance Test (GTT)
2.4. Insulin Tolerance Test (ITT)
2.5. Analysis of mRNA Expression by Real-Time PCR
2.6. Measurement of Triglycerides (TG)
2.7. Serum Cholesterol
2.8. Hepatic Cholesterol Levels
2.9. Serum ALT (Alanine Transaminase) and AST (Aspartate Transaminase)
2.10. Histochemistry
2.11. Statistical Analysis
3. Results
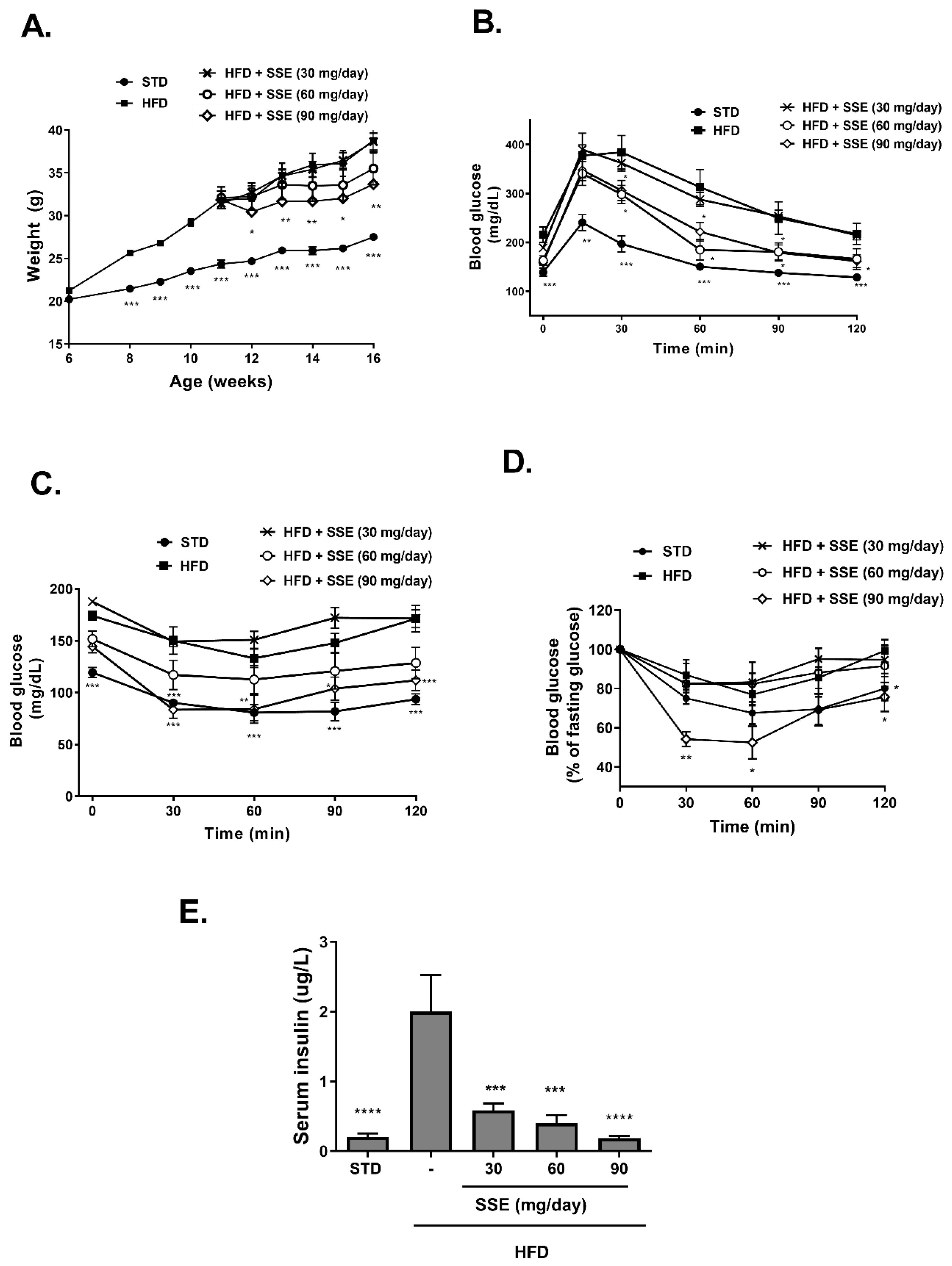
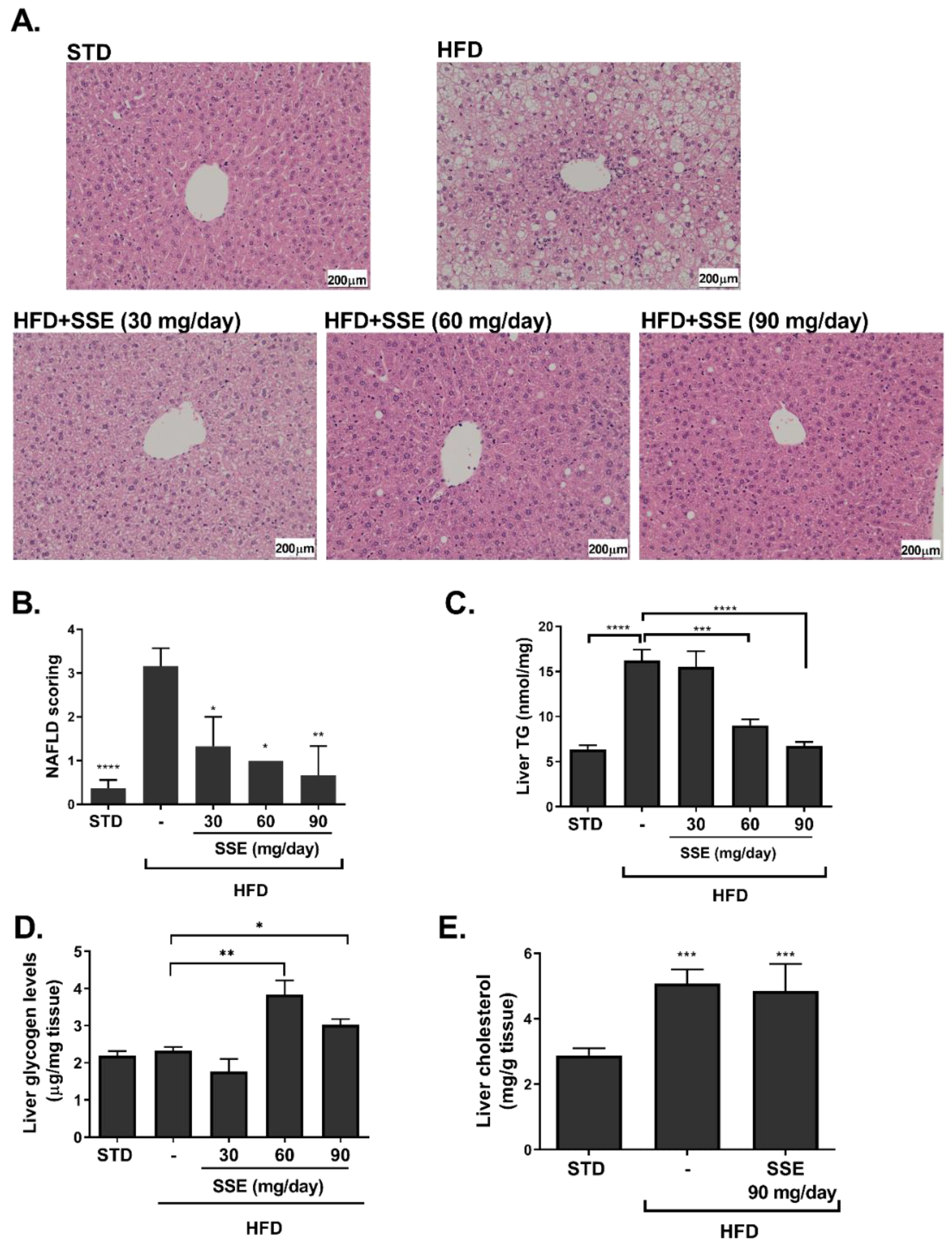
3.1. Effect of SSE on Hepatic Steatosis
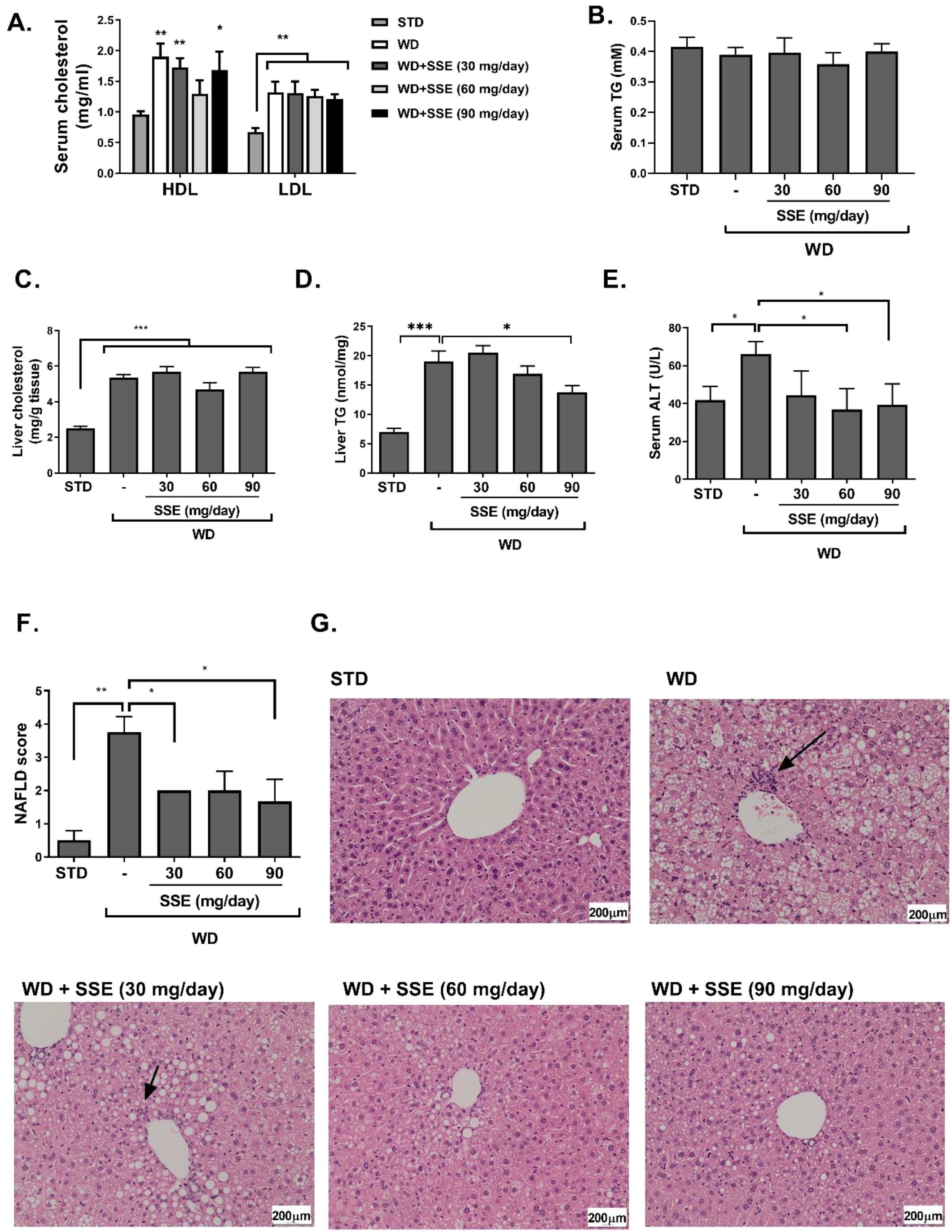
3.2. SSE for the Prevention of NASH
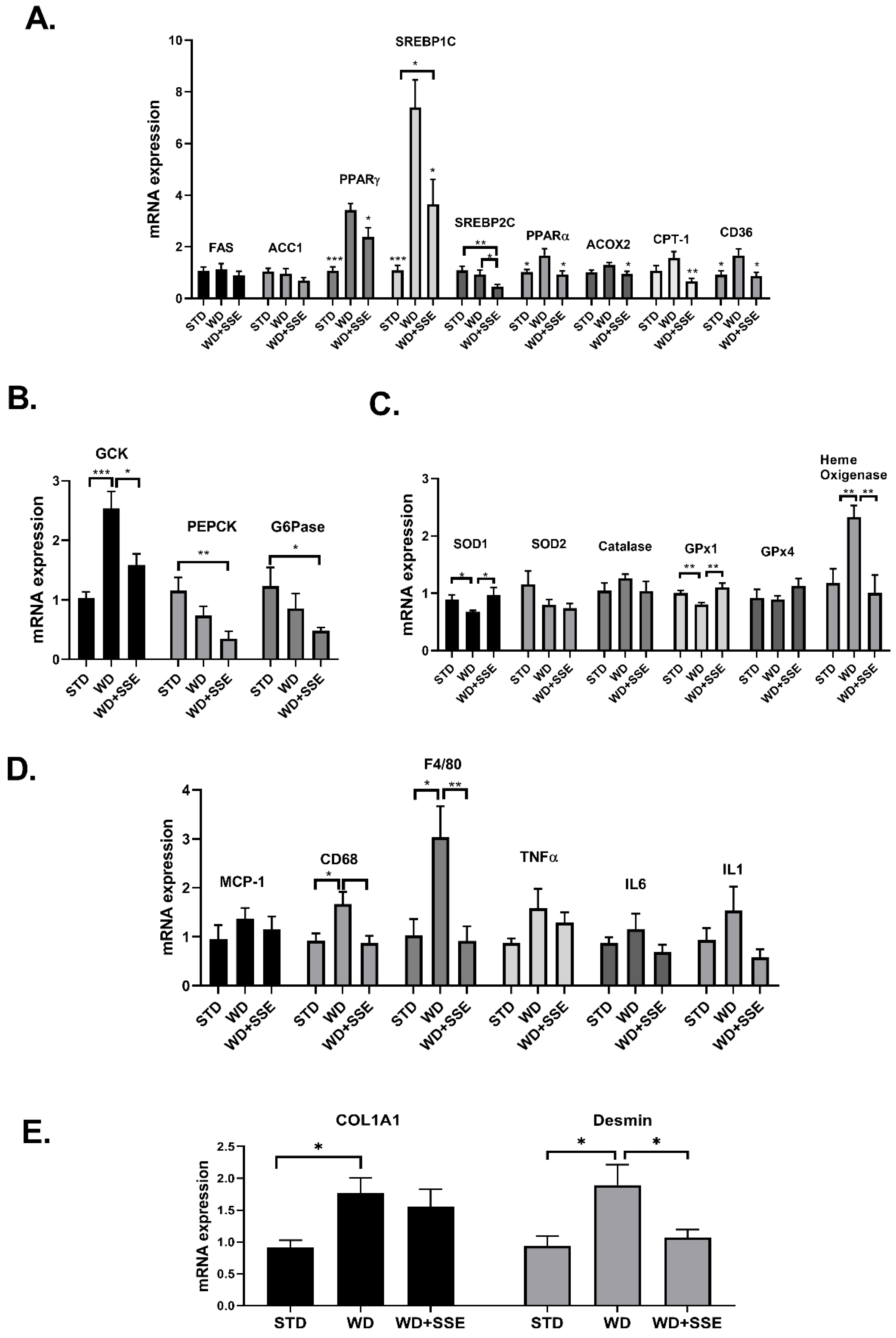
3.3. SSE for the Treatment of NASH
4. Discussion
5. Patents
Author Contributions
Funding
Conflicts of Interest
References
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.J. Nonalcoholic Fatty Liver Disease and Diabetes: An Epidemiological Perspective. Endocrinol. Metab. 2019, 34, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Yki-Jarvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Basaranoglu, M.; Basaranoglu, G.; Senturk, H. From fatty liver to fibrosis: A tale of “second hit”. World J. Gastroenterol. 2013, 19, 1158–1165. [Google Scholar] [CrossRef]
- Engin, A. Non-Alcoholic Fatty Liver Disease. Adv. Exp. Med. Biol. 2017, 960, 443–467. [Google Scholar] [CrossRef]
- Khan, R.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in NAFLD. Hepatology 2018. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V., Sr.; et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007, 6, 69–78. [Google Scholar] [CrossRef]
- Gastaldelli, A. Insulin resistance and reduced metabolic flexibility: Cause or consequence of NAFLD? Clin. Sci. 2017, 131, 2701–2704. [Google Scholar] [CrossRef] [PubMed]
- Ota, T.; Takamura, T.; Kurita, S.; Matsuzawa, N.; Kita, Y.; Uno, M.; Akahori, H.; Misu, H.; Sakurai, M.; Zen, Y.; et al. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology 2007, 132, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Hage Hassan, R.; Bourron, O.; Hajduch, E. Defect of insulin signal in peripheral tissues: Important role of ceramide. World J. Diabetes 2014, 5, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Sugimoto, K.; Inui, H.; Fukusato, T. Current pharmacological therapies for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2015, 21, 3777–3785. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, Z.; Dafni, A.; Friedman, J.; Palevitch, D. Plants used for the treatment of diabetes in Israel. J. Ethnopharmacol. 1987, 19, 145–151. [Google Scholar] [CrossRef]
- Rozenberg, K.; Smirin, P.; Sampson, S.R.; Rosenzweig, T. Insulin-sensitizing and insulin-mimetic activities of Sarcopoterium spinosum extract. J. Ethnopharmacol. 2014, 155, 362–372. [Google Scholar] [CrossRef]
- Smirin, P.; Taler, D.; Abitbol, G.; Brutman-Barazani, T.; Kerem, Z.; Sampson, S.R.; Rosenzweig, T. Sarcopoterium spinosum extract as an antidiabetic agent: In vitro and in vivo study. J. Ethnopharmacol. 2010, 129, 10–17. [Google Scholar] [CrossRef]
- Rozenberg, K.; Rosenzweig, T. Sarcopoterium spinosum extract improved insulin sensitivity in mice models of glucose intolerance and diabetes. PLoS ONE 2018, 13, e0196736. [Google Scholar] [CrossRef]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef]
- Kanuri, G.; Bergheim, I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). Int. J. Mol. Sci. 2013, 14, 11963–11980. [Google Scholar] [CrossRef]
- Bril, F.; Lomonaco, R.; Orsak, B.; Ortiz-Lopez, C.; Webb, A.; Tio, F.; Hecht, J.; Cusi, K. Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology 2014, 59, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Livero, F.A.; Acco, A. Molecular basis of alcoholic fatty liver disease: From incidence to treatment. Hepatol. Res. 2016, 46, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; de Almeida, T.P.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Correction: Mouse Models of Diet-Induced Nonalcoholic Steatohepatitis Reproduce the Heterogeneity of the Human Disease. PLoS ONE 2015, 10, e0132315. [Google Scholar] [CrossRef] [PubMed]
- Winzell, M.S.; Magnusson, C.; Ahren, B. Temporal and dietary fat content-dependent islet adaptation to high-fat feeding-induced glucose intolerance in mice. Metabolism 2007, 56, 122–128. [Google Scholar] [CrossRef]
- Turner, N.; Kowalski, G.M.; Leslie, S.J.; Risis, S.; Yang, C.; Lee-Young, R.S.; Babb, J.R.; Meikle, P.J.; Lancaster, G.I.; Henstridge, D.C.; et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 2013, 56, 1638–1648. [Google Scholar] [CrossRef]
- Farese, R.V., Jr.; Zechner, R.; Newgard, C.B.; Walther, T.C. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab. 2012, 15, 570–573. [Google Scholar] [CrossRef]
- Jelenik, T.; Kaul, K.; Sequaris, G.; Flogel, U.; Phielix, E.; Kotzka, J.; Knebel, B.; Fahlbusch, P.; Horbelt, T.; Lehr, S.; et al. Mechanisms of Insulin Resistance in Primary and Secondary Nonalcoholic Fatty Liver. Diabetes 2017, 66, 2241–2253. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef]
- Santoleri, D.; Titchenell, P.M. Resolving the Paradox of Hepatic Insulin Resistance. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 447–456. [Google Scholar] [CrossRef]
- Ben-Shachar, M.; Rozenberg, K.; Skalka, N.; Wollman, A.; Michlin, M.; Rosenzweig, T. Activation of Insulin Signaling in Adipocytes and Myotubes by Sarcopoterium Spinosum Extract. Nutrients 2019, 11, 1396. [Google Scholar] [CrossRef]
- Benard, O.; Lim, J.; Apontes, P.; Jing, X.; Angeletti, R.H.; Chi, Y. Impact of high-fat diet on the proteome of mouse liver. J. Nutr. Biochem. 2016, 31, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Air, E.L.; Strowski, M.Z.; Benoit, S.C.; Conarello, S.L.; Salituro, G.M.; Guan, X.M.; Liu, K.; Woods, S.C.; Zhang, B.B. Small molecule insulin mimetics reduce food intake and body weight and prevent development of obesity. Nat. Med. 2002, 8, 179–183. [Google Scholar] [CrossRef]
- Plum, L.; Belgardt, B.F.; Bruning, J.C. Central insulin action in energy and glucose homeostasis. J. Clin. Investig. 2006, 116, 1761–1766. [Google Scholar] [CrossRef]
- Loh, K.; Zhang, L.; Brandon, A.; Wang, Q.; Begg, D.; Qi, Y.; Fu, M.; Kulkarni, R.; Teo, J.; Baldock, P.; et al. Insulin controls food intake and energy balance via NPY neurons. Mol. Metab. 2017, 6, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Farrell, G.C.; Haczeyni, F.; Chitturi, S. Pathogenesis of NASH: How Metabolic Complications of Overnutrition Favour Lipotoxicity and Pro-Inflammatory Fatty Liver Disease. Adv. Exp. Med. Biol. 2018, 1061, 19–44. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, R.; Xiong, Y.; Du, F.; Zhu, S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. 2017, 16, 203. [Google Scholar] [CrossRef]
- Finck, B.N. Targeting Metabolism, Insulin Resistance, and Diabetes to Treat Nonalcoholic Steatohepatitis. Diabetes 2018, 67, 2485–2493. [Google Scholar] [CrossRef]
- Rozenberg, K.; Wollman, A.; Ben-Shachar, M.; Argaev-Frenkel, L.; Rosenzweig, T. Anti-inflammatory effects of Sarcopoterium spinosum extract. J. Ethnopharmacol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxidative Med. Cell Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef] [PubMed]
- Bachrach, Z.Y. Ethnobotanical studies of Sarcopoterium spinosum in Israel. ISR J. Plant Sci. 2007, 55, 111–114. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| ACC1 | CATGAACACCCAGAGCATTG | ATTTGTCGTAGTGGCCGTTC |
| ACOX2 | AAGTGGCCAGGTTTCTGATG | TCTTGGTGTGGCGAGATACA |
| CAT | TGACAAAATGCTTCAGGGCC | CTGGTTGTCATGCATGCACA |
| CD36 | AGCAGCTGCACCACATATCTAC | GGAACCAAACTGAGGAATGG |
| CD68 | CCAACAAAACCAAGGTCCAG | TGTATTCCACCGCCATGTAG |
| COL1A1 | GTGTTCCCTACTCAGCCGTC | TCCGTACTCGAACGGGAATC |
| CPT-1 | GATGTGGACCTGCATTCCTT | TCCTTGTAATGTGCGAGCTG |
| Desmin | CAGAGGCTCAAGGCCAAACTA | AGGGATTCGATTCTGCGCTC |
| FAS | TTGCTGGCACTACAGAATGC | AACAGCCTCAGAGCGACAAT |
| F4/80 | CTGTAACCGGATGGCAAACT | ATGGCCAAGGCAAGACATAC |
| Gck | GCAGAAGGGAACAACATCGT | TGGCGGTCTTCATAGTAGCA |
| G6Pase | GATTCCGGTGTTTGAACGTC | GTAGAATCCAAGCGCGAAAC |
| GPx1 | TTGGTGATTACTGGCTGCAC | CCATCTGAGGGGATTTTCCT |
| GPx4 | GAGCCCATTCCTGAACCTTT | CGATGTCCTTGGCTGAGAAT |
| HMOX1 | CAGAGCCGTCTCGAGCATAG | AAATCCTGGGGCATGCTGTC |
| IL1β | GCCCATCCTCTGTGACTCAT | AGGCCACAGGTATTTTGTCG |
| IL6 | AAGCCAGAGTCCTTCAGAGAGA | GGAAATTGGGGTAGGAAGGA |
| MCP-1 | CACTCACCTGCTGCTACTCATT | TCTGGACCCATTCCTTCTTG |
| PEPCK | AGCCTTTGGTCAACAACTGG | GTTATGCCCAGGATCAGCAT |
| PPARα | ATGCCAGTACTGCCGTTTTC | CCGAATCTTTCAGGTCGTGT |
| PPARγ | CAGGCCTCATGAAGAACCTT | ACCCTTGCATCCTTCACAAG |
| SOD1 | CGGATGAAGAGAGGCATGTT | CACCTTTGCCCAAGTCATCT |
| SOD2 | GCGGTCGTGTAAACCTCAAT | GATCTGCGCGTTAATGTGTG |
| SREBP2 | AGAGGCGGACAACACACAAT | ACGCCAGACTTGTGCATCTT |
| SREBP1c | AAGAGCCCTGCACTTCTTGA | CCACAAAGAAACGGTGACCT |
| TNFα | TCTACTGAACTTCGGGGTGA | CACTTGGTGGTTTGCTACGA |
| RPS29 | TCGTTGGGCGTCTGAAGGCAA | CGGAAGCACTGGCGGCACAT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wollman, A.; Daniel, T.; Rosenzweig, T. Sarcopoterium spinosum Inhibited the Development of Non-Alcoholic Steatosis and Steatohepatitis in Mice. Nutrients 2019, 11, 3044. https://doi.org/10.3390/nu11123044
Wollman A, Daniel T, Rosenzweig T. Sarcopoterium spinosum Inhibited the Development of Non-Alcoholic Steatosis and Steatohepatitis in Mice. Nutrients. 2019; 11(12):3044. https://doi.org/10.3390/nu11123044
Chicago/Turabian StyleWollman, Ayala, Tehila Daniel, and Tovit Rosenzweig. 2019. "Sarcopoterium spinosum Inhibited the Development of Non-Alcoholic Steatosis and Steatohepatitis in Mice" Nutrients 11, no. 12: 3044. https://doi.org/10.3390/nu11123044
APA StyleWollman, A., Daniel, T., & Rosenzweig, T. (2019). Sarcopoterium spinosum Inhibited the Development of Non-Alcoholic Steatosis and Steatohepatitis in Mice. Nutrients, 11(12), 3044. https://doi.org/10.3390/nu11123044