
A Brief Review of the Mechanisms of ?-Cell Dedifferentiation in Type 2 Diabetes


Abstract
:1. Introduction
2. Dedifferentiation of Pancreatic β-Cells
3. Trans-Differentiation of Pancreatic β-Cells
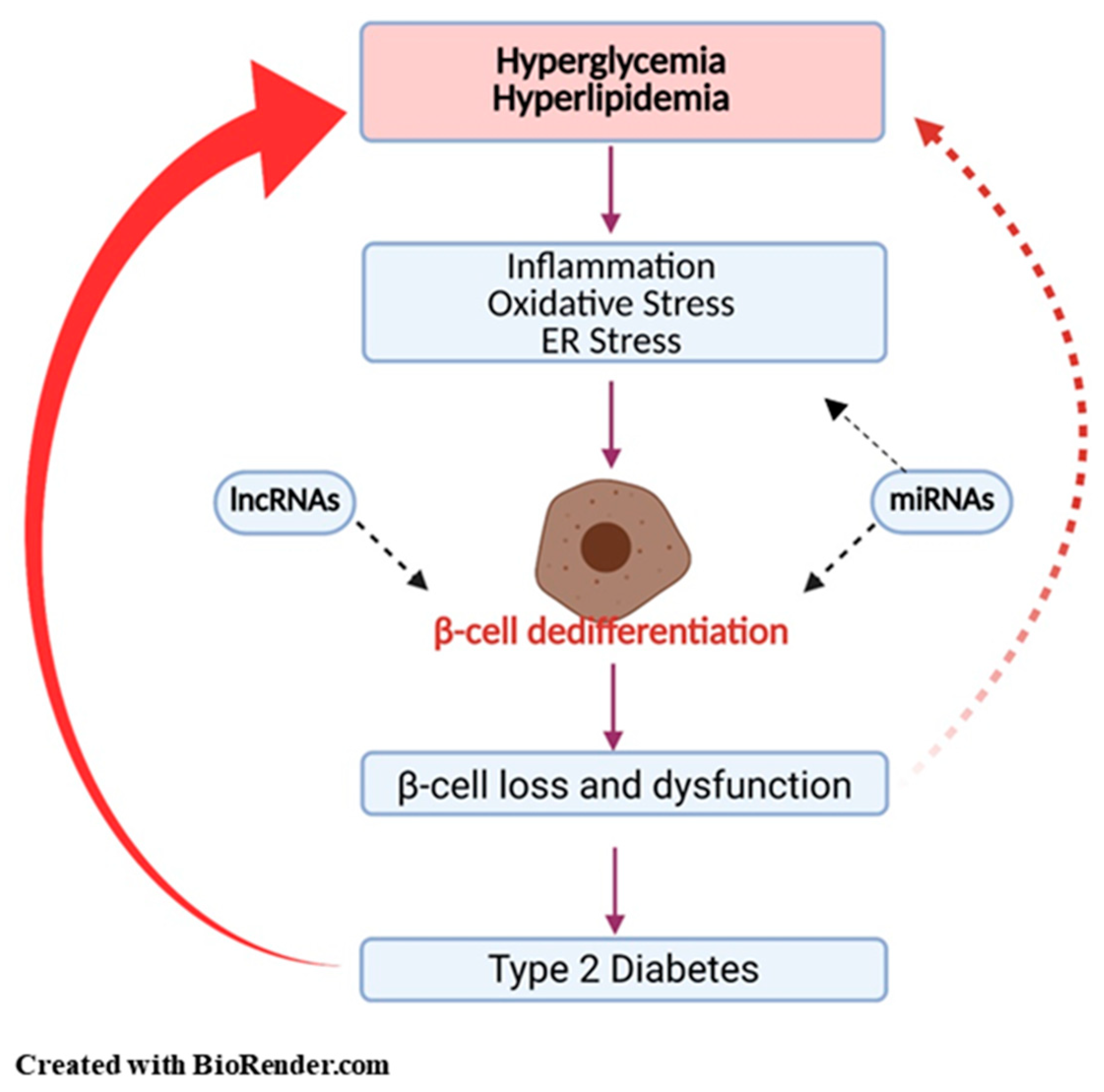
4. Potential Mechanisms Regulating β-Cell Dedifferentiation
4.1. Inflammation
4.2. Oxidative Stress
4.3. ER Stress
4.4. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ER | Endoplasmic reticulum |
| DM | Diabetes mellitus |
| T2DM | Type 2 diabetes |
| Glut 2 | Glucose transporter 2 |
| Pdx1 | Pancreatic and duodenal homeobox 1 |
| FoxO1 | Forkhead box protein O1 |
| MafA | V-maf musculoaponeurotic fibrosarcoma oncogene homolog A |
| HKI-III | Hexokinase |
| Ldha | Lactate dehydrogenase A |
| Ngn3 | Neurogenin 3 |
| Oct4 | Nanog Homoebox |
| Pou5f1 | POU domain class 5 transcription factor 1 |
| T1DM | Type 1 diabetes |
| SGLT2 | Sodium-glucose co-transporter type 2 |
| GLP-1 | Glucagon-like peptide-1 |
| IAPP | Islet amyloid polypeptide |
| IL-1β | Interleukin-1 beta |
| NLRP3 | NLR family pyrin domain containing 3 |
| IL7R | Interleukin 7 receptor |
| IL17R | Interleukin 17 receptor |
| CCL3 | CC chemokine Ligand 3 |
| CCL8 | CC chemokine ligand 8 |
| CXCL2 | CXC chemokine ligand 2 |
| CXCL11 | CXC chemokine ligand 11 |
| CXCL12 | CXC chemokine ligand 12 |
| COX-2 | Cyclooxygenase-2 |
| PGE2 | Prostaglandin E2 |
| Ucn3 | Urocortin 3 |
| H3K27 | 27th amino acid in Histone H3 |
| IL-1R2 | Interleukin 1 receptor 2 |
| MYD88 | Myeloid differentiation primary response 88 |
| IRAK | Interleukin-1 receptor associated kinase |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| IKKs | IκB kinases |
| NO | Nitric oxide |
| ROS | Reactive oxygen species |
| H2O2 | Hydrogen peroxide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| LOsG | Long-term oscillating glucose |
| UPR | Unfolded protein response |
| ERAD | ER-associated protein degradation |
| miRNAs | MicroRNAs |
| lncRNAs | Long non-coding RNAs |
| Aldh1a3 | Aldenyde dehydrogenase family 1, subfamily A3 |
References
- American Diabetes Association 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2021. Diabetes Care 2021, 44 (Suppl. S1), S15–S33. [Google Scholar] [CrossRef] [PubMed]
- Alejandro, E.U.; Gregg, B.; Blandino-Rosano, M.; Cras-Méneur, C.; Bernal-Mizrachi, E. Natural history of β-cell adaptation and failure in type 2 diabetes. Mol. Asp. Med. 2015, 42, 19–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.K.; Gao, N.; Gorski, R.K.; White, P.; Hardy, O.T.; Rafiq, K.; Brestelli, J.E.; Chen, G.; Stoeckert, J.C.J.; Kaestner, K.H. Expansion of adult beta-cell mass in response to increased metabolic demand is dependent on HNF-4. Genes Dev. 2007, 21, 756–769. [Google Scholar] [CrossRef] [Green Version]
- Khadra, A.; Schnell, S. Development, growth and maintenance of β-cell mass: Models are also part of the story. Mol. Asp. Med. 2015, 42, 78–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brereton, M.F.; Rohm, M.; Shimomura, K.; Holland, C.; Tornovsky-Babeay, S.; Dadon, D.; Iberl, M.; Chibalina, M.V.; Lee, S.; Glaser, B.; et al. Hyperglycaemia induces metabolic dysfunction and glycogen accumulation in pancreatic β-cells. Nat. Commun. 2016, 7, 13496. [Google Scholar] [CrossRef]
- Stoffers, D.A. The Development of Beta-cell Mass: Recent Progress and Potential Role of GLP-1. Horm. Metab. Res. 2004, 36, 811–821. [Google Scholar] [CrossRef]
- Shanik, M.H.; Xu, Y.; Škrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin Resistance and Hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. S2), S262–S268. [Google Scholar] [CrossRef] [Green Version]
- Olokoba, A.B.; Obateru, O.A.; Olokoba, L.B. Type 2 Diabetes Mellitus: A Review of Current Trends. Oman Med. J. 2012, 27, 269–273. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Eldor, R.; Abdul-Ghani, M. Pathophysiologic Approach to Therapy in Patients with Newly Diagnosed Type 2 Diabetes. Diabetes Care 2013, 36, S127–S138. [Google Scholar] [CrossRef] [Green Version]
- John, A.N.; Morahan, G.; Jiang, F. Incomplete Re-Expression of Neuroendocrine Progenitor/Stem Cell Markers is a Key Feature of β-Cell Dedifferentiation. J. Neuroendocr. 2017, 29. [Google Scholar] [CrossRef] [Green Version]
- Amo-Shiinoki, K.; Tanabe, K.; Hoshii, Y.; Matsui, H.; Harano, R.; Fukuda, T.; Takeuchi, T.; Bouchi, R.; Takagi, T.; Hatanaka, M.; et al. Islet cell dedifferentiation is a pathologic mechanism of long-standing progression of type 2 diabetes. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β Cell Dedifferentiation as a Mechanism of Diabetic β Cell Failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Han, X. Death versus dedifferentiation: The molecular bases of beta cell mass reduction in type 2 diabetes. Semin. Cell Dev. Biol. 2020, 103, 76–82. [Google Scholar] [CrossRef]
- Weir, G.C.; Aguayo-Mazzucato, C.; Bonner-Weir, S. β-cell dedifferentiation in diabetes is important, but what is it? Islets 2013, 5, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moin, A.S.M.; Butler, A.E. Alterations in Beta Cell Identity in Type 1 and Type 2 Diabetes. Curr. Diabetes Rep. 2019, 19, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of β-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marselli, L.; Suleiman, M.; Masini, M.; Campani, D.; Bugliani, M.; Syed, F.; Martino, L.; Focosi, D.; Scatena, F.; Olimpico, F.; et al. Are we overestimating the loss of beta cells in type 2 diabetes? Diabetologia 2014, 57, 362–365. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Dai, C.; Guo, M.; Taylor, B.; Harmon, J.S.; Sander, M.; Robertson, R.P.; Powers, A.C.; Stein, R. Inactivation of specific β cell transcription factors in type 2 diabetes. J. Clin. Investig. 2013, 123, 3305–3316. [Google Scholar] [CrossRef] [Green Version]
- Efrat, S. Beta-Cell Dedifferentiation in Type 2 Diabetes: Concise Review. Stem Cells 2019, 37, 1267–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.E.; Dhawan, S.; Hoang, J.; Cory, M.; Zeng, K.; Fritsch, H.; Meier, J.J.; Rizza, R.A.; Butler, P.C. β-Cell Deficit in Obese Type 2 Diabetes, a Minor Role of β-Cell Dedifferentiation and Degranulation. J. Clin. Endocrinol. Metab. 2016, 101, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spijker, H.S.; Song, H.; Ellenbroek, J.H.; Roefs, M.M.; Engelse, M.A.; Bos, E.; Koster, A.J.; Rabelink, T.J.; Hansen, B.C.; Clark, A.; et al. Loss of β-Cell Identity Occurs in Type 2 Diabetes and Is Associated with Islet Amyloid Deposits. Diabetes 2015, 64, 2928–2938. [Google Scholar] [CrossRef] [Green Version]
- Bensellam, M.; Jonas, J.-C.; Laybutt, D.R. Mechanisms of β-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143. [Google Scholar] [CrossRef] [Green Version]
- Lemaire, K.; Thorrez, L.; Schuit, F. Disallowed and Allowed Gene Expression: Two Faces of Mature Islet Beta Cells. Annu. Rev. Nutr. 2016, 36, 45–71. [Google Scholar] [CrossRef]
- Sun, J.; Ni, Q.; Xie, J.; Xu, M.; Zhang, J.; Kuang, J.; Wang, Y.; Ning, G.; Wang, Q. β-Cell Dedifferentiation in Patients with T2D With Adequate Glucose Control and Nondiabetic Chronic Pancreatitis. J. Clin. Endocrinol. Metab. 2018, 104, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Dunning, B.E.; Gerich, J.E. The Role of α-Cell Dysregulation in Fasting and Postprandial Hyperglycemia in Type 2 Diabetes and Therapeutic Implications. Endocr. Rev. 2007, 28, 253–283. [Google Scholar] [CrossRef] [Green Version]
- Jonas, J.-C.; Sharma, A.; Hasenkamp, W.; Ilkova, H.; Patanè, G.; Laybutt, R.; Bonner-Weir, S.; Weir, G.C. Chronic Hyperglycemia Triggers Loss of Pancreatic β Cell Differentiation in an Animal Model of Diabetes. J. Biol. Chem. 1999, 274, 14112–14121. [Google Scholar] [CrossRef] [Green Version]
- Tellez, N.; Vilaseca, M.; Martí, Y.; Pla, A.; Montanya, E. β-Cell dedifferentiation, reduced duct cell plasticity, and impaired β-cell mass regeneration in middle-aged rats. Am. J. Physiol. Metab. 2016, 311, E554–E563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.E.; Janson, J.; Soeller, W.C.; Butler, P.C. Increased -Cell Apoptosis Prevents Adaptive Increase in -Cell Mass in Mouse Model of Type 2 Diabetes: Evidence for Role of Islet Amyloid Formation Rather Than Direct Action of Amyloid. Diabetes 2003, 52, 2304–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosh, D.; Slack, J.M.W. How cells change their phenotype. Nat. Rev. Mol. Cell Biol. 2002, 3, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, F. The De-, Re-, and trans-differentiation of β-cells: Regulation and function. Semin. Cell Dev. Biol. 2020, 103, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Piran, R.; Lee, S.-H.; Li, C.-R.; Charbono, A.; Bradley, L.M.; Levine, F. Pharmacological induction of pancreatic islet cell transdifferentiation: Relevance to type I diabetes. Cell Death Dis. 2014, 5, e1357. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Cui, X.; Feng, J.; Gu, L.; Lang, S.; Wei, T.; Yang, J.; Liu, J.; Le, Y.; Wang, H.; et al. Dapagliflozin promotes beta cell regeneration by inducing pancreatic endocrine cell phenotype conversion in type 2 diabetic mice. Metabolism 2020, 111. [Google Scholar] [CrossRef]
- Spijker, H.S.; Ravelli, R.B.; Mommaas-Kienhuis, A.M.; Van Apeldoorn, A.A.; Engelse, M.A.; Zaldumbide, A.; Bonner-Weir, S.; Rabelink, T.J.; Hoeben, R.C.; Clevers, H.; et al. Conversion of Mature Human β-Cells into Glucagon-Producing α-Cells. Diabetes 2013, 62, 2471–2480. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 Maintains β Cell Identity and Function by Repressing an α Cell Program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, C.S.; Stein, R.W. Evidence for Loss in Identity, De-Differentiation, and Trans-Differentiation of Islet β-Cells in Type 2 Diabetes. Front. Genet. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moin, A.S.M.; Dhawan, S.; Cory, M.; Butler, P.C.; Rizza, R.A.; Butler, A.E. Increased Frequency of Hormone Negative and Polyhormonal Endocrine Cells in Lean Individuals with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 3628–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedel, M.J.; Asadi, A.; Wang, R.; Ao, Z.; Warnock, G.L.; Kieffer, T.J. Immunohistochemical characterisation of cells co-producing insulin and glucagon in the developing human pancreas. Diabetologia 2011, 55, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Ehses, J.A.; Böni-Schnetzler, M.; Faulenbach, M.; Donath, M.Y. Macrophages, cytokines and β-cell death in Type 2 diabetes. Biochem. Soc. Trans. 2008, 36 Pt 3, 340–342. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Fu, W.; Lee, Y.S.; Olefsky, J.M. The role of macrophages in obesity-associated islet inflammation and β-cell abnormalities. Nat. Rev. Endocrinol. 2020, 16, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, P.; Suleiman, M.; De Luca, C.; Baronti, W.; Bosi, E.; Tesi, M.; Marselli, L. A direct look at the dysfunction and pathology of the β cells in human type 2 diabetes. Semin. Cell Dev. Biol. 2020, 103, 83–93. [Google Scholar] [CrossRef] [PubMed]
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Gong, Q.; Mima, A. Inflammatory Regulation in Diabetes and Metabolic Dysfunction. J. Diabetes Res. 2017, 2017, 5165268. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Liang, R.; Liu, T.; Wang, L.; Zou, J.; Liu, N.; Liu, Y.; Cai, X.; Liu, Y.; Ding, X.; et al. Opposing effects of IL-1β/COX-2/PGE2 pathway loop on islets in type 2 diabetes mellitus. Endocr. J. 2019, 66, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ni, Q.; Sun, J.; Xu, M.; Xie, J.; Zhang, J.; Fang, Y.; Ning, G.; Wang, Q. Paraneoplastic β Cell Dedifferentiation in Nondiabetic Patients with Pancreatic Cancer. J. Clin. Endocrinol. Metab. 2019, 105, e1489–e1503. [Google Scholar] [CrossRef] [PubMed]
- Urizar, A.I.; Prause, M.; Wortham, M.; Sui, Y.; Thams, P.; Sander, M.; Christensen, G.L.; Billestrup, N. Beta-cell dysfunction induced by non-cytotoxic concentrations of Interleukin-1β is associated with changes in expression of beta-cell maturity genes and associated histone modifications. Mol. Cell. Endocrinol. 2019, 496, 110524. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Lamacchia, C.; Palmer-Lourenco, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef]
- Karin, M. How NF-κB is activated: The role of the IκB kinase (IKK) complex. Oncogene 1999, 18, 6867–6874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, M.; Knoch, K.-P.; Diedisheim, M.; Petzold, A.; Cattan, P.; Bugliani, M.; Marchetti, P.; Choudhary, P.; Huang, G.-C.; Bornstein, S.R.; et al. Virus-like infection induces human β cell dedifferentiation. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- King, A.J.F.; Guo, Y.; Cai, N.; Hollister-Lock, J.; Morris, B.; Salvatori, A.; Corbett, J.A.; Bonner-Weir, S.; Shoelson, S.E.; Weir, G.C. Sustained NF-κB Activation and Inhibition in β-Cells Have Minimal Effects on Function and Islet Transplant Outcomes. PLoS ONE 2013, 8, e77452. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Böni-Schnetzler, M.; Donath, M.Y. The Role of Inflammation in β-cell Dedifferentiation. Sci. Rep. 2017, 7, 6285. [Google Scholar] [CrossRef] [PubMed]
- Boni-Schnetzler, M.; Boller, S.; Debray, S.; Bouzakri, K.; Meier, D.T.; Prazak, R.; Kerr-Conte, J.; Pattou, F.; Ehses, J.A.; Schuit, F.C.; et al. Free Fatty Acids Induce a Proinflammatory Response in Islets via the Abundantly Expressed Interleukin-1 Receptor I. Endocrinology 2009, 150, 5218–5229. [Google Scholar] [CrossRef]
- Wu, L.; Nicholson, W.; Knobel, S.M.; Steffner, R.J.; May, J.M.; Piston, D.W.; Powers, A.C. Oxidative Stress Is a Mediator of Glucose Toxicity in Insulin-secreting Pancreatic Islet Cell Lines. J. Biol. Chem. 2004, 279, 12126–12134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Tanaka, Y.; Takahashi, H. Glucose Toxicity in -Cells: Type 2 Diabetes, Good Radicals Gone Bad, and the Glutathione Connection. Diabetes 2003, 52, 581–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Cui, Q.; Yang, B.; Hou, Y.; Wang, H.; Xu, Y.; Wang, D.; Zhang, Q.; Pi, J. The impairment of glucose-stimulated insulin secretion in pancreatic β-cells caused by prolonged glucotoxicity and lipotoxicity is associated with elevated adaptive antioxidant response. Food Chem. Toxicol. 2017, 100, 161–167. [Google Scholar] [CrossRef]
- Harmon, J.S.; Bogdani, M.; Parazzoli, S.D.; Mak, S.S.M.; Oseid, E.A.; Berghmans, M.; Leboeuf, R.C.; Robertson, R.P. β-Cell-Specific Overexpression of Glutathione Peroxidase Preserves Intranuclear MafA and Reverses Diabetes in db/db Mice. Endocrinology 2009, 150, 4855–4862. [Google Scholar] [CrossRef] [Green Version]
- Stańczyk, M.; Gromadzińska, J.; Wasowicz, W. Roles of reactive oxygen species and selected antioxidants in regulation of cellular metabolism. Int. J. Occup. Med. Environ. Health 2005, 18, 15–26. [Google Scholar]
- Elsner, M.; Gehrmann, W.; Lenzen, S. Peroxisome-Generated Hydrogen Peroxide as Important Mediator of Lipotoxicity in Insulin-Producing Cells. Diabetes 2010, 60, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Morgan, D.; Oliveira-Emilio, H.R.; Keane, D.; Hirata, A.E.; Da Rocha, M.S.; Bordin, S.; Curi, R.; Newsholme, P.; Carpinelli, A.R. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 2007, 50, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Ly, L.D.; Xu, S.; Choi, S.-K.; Ha, C.-M.; Thoudam, T.; Cha, S.-K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.-K.; Park, K.-S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel Excess and β-Cell Dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Supale, S.; Li, N.; Brun, T.; Maechler, P. Mitochondrial dysfunction in pancreatic β cells. Trends Endocrinol. Metab. 2012, 23, 477–487. [Google Scholar] [CrossRef]
- Zhang, J.; An, H.; Ni, K.; Chen, B.; Li, H.; Li, Y.; Sheng, G.; Zhou, C.; Xie, M.; Chen, S.; et al. Glutathione prevents chronic oscillating glucose intake-induced β-cell dedifferentiation and failure. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.-P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. FoxO1 protects against pancreatic β cell failure through NeuroD and MafA induction. Cell Metab. 2005, 2, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Sakano, D.; Uefune, F.; Tokuma, H.; Sonoda, Y.; Matsuura, K.; Takeda, N.; Nakagata, N.; Kume, K.; Shiraki, N.; Kume, S. VMAT2 Safeguards β-Cells Against Dopamine Cytotoxicity Under High-Fat Diet–Induced Stress. Diabetes 2020, 69, 2377–2391. [Google Scholar] [CrossRef]
- Lai, E.; Teodoro, T.; Volchuk, A. Endoplasmic Reticulum Stress: Signaling the Unfolded Protein Response. Physiology 2007, 22, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab. 2011, 22, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, U.; Kim, H.-J.; Kim, J.-Y.; Lee, I.-K. Guards and Culprits in the Endoplasmic Reticulum: Glucolipotoxicity andβ-Cell Failure in Type II Diabetes. Exp. Diabetes Res. 2011, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Scheuner, D.; Kaufman, R.J. The Unfolded Protein Response: A Pathway That Links Insulin Demand with β-Cell Failure and Diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef] [Green Version]
- Herbert, T.P.; Laybutt, D.R. A Reevaluation of the Role of the Unfolded Protein Response in Islet Dysfunction: Maladaptation or a Failure to Adapt? Diabetes 2016, 65, 1472–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.-J.; Lin, C.-Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High Expression Rates of Human Islet Amyloid Polypeptide Induce Endoplasmic Reticulum Stress–Mediated β-Cell Apoptosis, a Characteristic of Humans with Type 2 but Not Type 1 Diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.-Y.; Jun, H.-S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.-H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes Res. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Jaikaran, E.T.; Clark, A. Islet amyloid and type 2 diabetes: From molecular misfolding to islet pathophysiology. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2001, 1537, 179–203. [Google Scholar] [CrossRef] [Green Version]
- Cao, P.; Marek, P.; Noor, H.; Patsalo, V.; Tu, L.-H.; Wang, H.; Abedini, A.; Raleigh, D.P. Islet amyloid: From fundamental biophysics to mechanisms of cytotoxicity. FEBS Lett. 2013, 587, 1106–1118. [Google Scholar] [CrossRef] [Green Version]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet Amyloid in Type 2 Diabetes, and the Toxic Oligomer Hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.Y.; Luzuriaga, J.; Bensellam, M.; Biden, T.J.; Laybutt, D.R. Failure of the Adaptive Unfolded Protein Response in Islets of Obese Mice Is Linked with Abnormalities in β-Cell Gene Expression and Progression to Diabetes. Diabetes 2012, 62, 1557–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negi, S.; Jetha, A.; Aikin, R.; Hasilo, C.; Sladek, R.; Paraskevas, S. Analysis of Beta-Cell Gene Expression Reveals Inflammatory Signaling and Evidence of Dedifferentiation following Human Islet Isolation and Culture. PLoS ONE 2012, 7, e30415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupsco, A.; Schlenk, D. Oxidative Stress, Unfolded Protein Response, and Apoptosis in Developmental Toxicity. Int. Rev. Cell Mol. Biol. 2015, 317, 1–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Koulajian, K.; Schuiki, I.; Zhang, L.; Desai, T.; Ivovic, A.; Wang, P.; Robson-Doucette, C.; Wheeler, M.B.; Minassian, B.; et al. Glucose-induced beta cell dysfunction in vivo in rats: Link between oxidative stress and endoplasmic reticulum stress. Diabetologia 2012, 55, 1366–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPierre, M.P.; Stoffel, M. MicroRNAs as stress regulators in pancreatic beta cells and diabetes. Mol. Metab. 2017, 6, 1010–1023. [Google Scholar] [CrossRef]
- Grieco, G.; Brusco, N.; Licata, G.; Fignani, D.; Formichi, C.; Nigi, L.; Sebastiani, G.; Dotta, F. The Landscape of microRNAs in βCell: Between Phenotype Maintenance and Protection. Int. J. Mol. Sci. 2021, 22, 803. [Google Scholar] [CrossRef]
- Melkman-Zehavi, T.; Oren, R.; Kredo-Russo, S.; Shapira, T.; Mandelbaum, A.D.; Rivkin, N.; Nir, T.; Lennox, K.A.; Behlke, M.A.; Dor, Y.; et al. miRNAs control insulin content in pancreatic β-cells via downregulation of transcriptional repressors. EMBO J. 2011, 30, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Lynn, F.C.; Skewes-Cox, P.; Kosaka, Y.; McManus, M.T.; Harfe, B.D.; German, M.S. MicroRNA Expression Is Required for Pancreatic Islet Cell Genesis in the Mouse. Diabetes 2007, 56, 2938–2945. [Google Scholar] [CrossRef] [Green Version]
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; Macdonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nat. Cell Biol. 2004, 432, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; You, W.; Wang, H.; Li, Y.; Qiao, N.; Shi, Y.; Zhang, C.; Bleich, D.; Han, X. MicroRNA-24/MODY Gene Regulatory Pathway Mediates Pancreatic β-Cell Dysfunction. Diabetes 2013, 62, 3194–3206. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; You, Y.-H.; Jung, S.; Suh-Kim, H.; Lee, I.-K.; Cho, J.-H.; Yoon, K.-H. miRNA-30a-5p-mediated silencing of Beta2/NeuroD expression is an important initial event of glucotoxicity-induced beta cell dysfunction in rodent models. Diabetologia 2013, 56, 847–855. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Mohan, R.; Özcan, S.; Tang, X. MicroRNA-30d Induces Insulin Transcription Factor MafA and Insulin Production by Targeting Mitogen-activated Protein 4 Kinase 4 (MAP4K4) in Pancreatic β-Cells. J. Biol. Chem. 2012, 287, 31155–31164. [Google Scholar] [CrossRef] [Green Version]
- Baroukh, N.; Ravier, M.A.; Loder, M.K.; Hill, E.V.; Bounacer, A.; Scharfmann, R.; Rutter, G.A.; Van Obberghen, E. MicroRNA-124a Regulates Foxa2 Expression and Intracellular Signaling in Pancreatic β-Cell Lines. J. Biol. Chem. 2007, 282, 19575–19588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tattikota, S.G.; Rathjen, T.; Hausser, J.; Khedkar, A.; Kabra, U.D.; Pandey, V.; Sury, M.; Wessels, H.-H.; Mollet, I.G.; Eliasson, L.; et al. miR-184 Regulates Pancreatic β-Cell Function According to Glucose Metabolism. J. Biol. Chem. 2015, 290, 20284–20294. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Chen, J.; Xu, G.; Grayson, T.B.; Thielen, L.A.; Shalev, A. miR-204 Controls Glucagon-Like Peptide 1 Receptor Expression and Agonist Function. Diabetes 2017, 67, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 2013, 19, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Latreille, M.; Hausser, J.; Stützer, I.; Zhang, Q.; Hastoy, B.; Gargani, S.; Kerr-Conte, J.; Pattou, F.; Zavolan, M.; Esguerra, J.L.; et al. MicroRNA-7a regulates pancreatic β cell function. J. Clin. Investig. 2014, 124, 2722–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Mohan, R.; Chen, X.; Matson, K.; Waugh, J.; Mao, Y.; Zhang, S.; Li, W.; Tang, X.; Satin, L.S.; et al. microRNA-483 Protects Pancreatic β-Cells by Targeting ALDH1A3. Endocrinology 2021, 162. [Google Scholar] [CrossRef]
- Goyal, N.; Kesharwani, D.; Datta, M. Lnc-ing non-coding RNAs with metabolism and diabetes: Roles of lncRNAs. Cell. Mol. Life Sci. 2018, 75, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Akerman, I.; Tu, Z.; Beucher, A.; Rolando, D.M.; Sauty-Colace, C.; Benazra, M.; Nakic, N.; Yang, J.; Wang, H.; Pasquali, L.; et al. Human Pancreatic β Cell lncRNAs Control Cell-Specific Regulatory Networks. Cell Metab. 2017, 25, 400–411. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| microRNA (miR) | Mechanism of Action | Models | References |
|---|---|---|---|
| miR-7 | Regulation of genes associated with β-cell identity | Transgenic mice overexpressing miR-7a in β-cells. | [90] |
| miR-24 | Regulation of MODY gene regulatory pathway Regulation of genes associated with β-cell identity | Overexpression of miR-24 in islets | [82] |
| miR-30 | -Targeting in the UTRs of β2/Neuro D -Targeting mitogen-activated protein 4 kinase 4 | -Glucotoxicity-exposed primary rat islets and INS-1 cells. -or miR-30 knock-down diabetic mice. -Overexpression of miR-30 in β-cells | [83,84] |
| miR-124 | Regulation Foxa2-Pdx gene expression | -Overexpressed or down-regulated MIN6 β-cells. -Human pancreatic islets | [85,86] |
| miR-184 | Inhibition of miR375 | MIN6 cells overexpressing miR-184 | [87] |
| miR-204 | Inhibition of MafA or Regulation of genes associated with β-cell identity | β-cells and islets | [88,89] |
| miR-375 | Combination with other β-cell enriched miRNAs | β-cells and islets | [81] |
| miR-483 | Targeting in the UTRs of aldehyde dehydrogenase family 1, subfamily A3 (Aldh1a3) | miR-483 deletion mice | [91] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khin, P.-P.; Lee, J.-H.; Jun, H.-S. A Brief Review of the Mechanisms of ?-Cell Dedifferentiation in Type 2 Diabetes. Nutrients 2021, 13, 1593. https://doi.org/10.3390/nu13051593
Khin P-P, Lee J-H, Jun H-S. A Brief Review of the Mechanisms of ?-Cell Dedifferentiation in Type 2 Diabetes. Nutrients. 2021; 13(5):1593. https://doi.org/10.3390/nu13051593
Chicago/Turabian StyleKhin, Phyu-Phyu, Jong-Han Lee, and Hee-Sook Jun. 2021. "A Brief Review of the Mechanisms of ?-Cell Dedifferentiation in Type 2 Diabetes" Nutrients 13, no. 5: 1593. https://doi.org/10.3390/nu13051593
APA StyleKhin, P.-P., Lee, J.-H., & Jun, H.-S. (2021). A Brief Review of the Mechanisms of ?-Cell Dedifferentiation in Type 2 Diabetes. Nutrients, 13(5), 1593. https://doi.org/10.3390/nu13051593