Abstract
Mitochondrial function, including oxidative phosphorylation (OXPHOS), mitochondrial biogenesis, and mitochondria dynamics, are essential for the maintenance of renal health. Through modulation of mitochondrial function, the kidneys are able to sustain or recover acute kidney injury (AKI), chronic kidney disease (CKD), nephrotoxicity, nephropathy, and ischemia perfusion. Therapeutic improvement in mitochondrial function in the kidneys is related to the regulation of adenosine triphosphate (ATP) production, free radicals scavenging, decline in apoptosis, and inflammation. Dietary antioxidants, notably polyphenols present in fruits, vegetables, and plants, have attracted attention as effective dietary and pharmacological interventions. Considerable evidence shows that polyphenols protect against mitochondrial damage in different experimental models of kidney disease. Mechanistically, polyphenols regulate the mitochondrial redox status, apoptosis, and multiple intercellular signaling pathways. Therefore, this review attempts to focus on the role of polyphenols in the prevention or treatment of kidney disease and explore the molecular mechanisms associated with their pharmacological activity.
1. Introduction
Kidneys are one of the most energy-demanding organs and play a vital physiological role in the maintenance of salt and water homeostasis [1]. Kidneys receive approximately 25% of the cardiac output and are responsible for the regulation of blood pressure and continuous blood filtration [2]. Physiologically, kidneys consume about 7% of the total oxygen available for overall human function, indicating a significant role of mitochondria in their physiology [2]. Mitochondria are abundant in metabolically active organs, including kidneys, especially in the renal tubule cells [3,4]. Indeed, the kidney is a metabolically active organ containing more mitochondria per weight than any other human organ [5,6]. Acute and chronic kidney diseases, such as renal ischemia, toxicity, and acute injury, include underlying mitochondrial dysfunction [7,8,9]. Research has established links between both acute and chronic kidney diseases with impaired mitochondrial biogenesis, OXPHOS, and mitochondria mitophagy [10]. The mitochondrial dysfunction in kidneys is also linked to inflammation, apoptosis, and tissue injury, thus, contributing to mortality and morbidity rates [11]. Studies have shown that dietary patterns and dietetic components could modulate renal function and disease [12,13]. A diet rich in plants, vegetables, and fruits is related to a lower incidence of chronic diseases, such as cardiovascular disease, cancers, type 2 diabetes, and kidney disease(s) [14,15]. These biological functionalities are associated with the presence of active antioxidants, particularly polyphenols [15]. ‘Polyphenol’ is not a strict chemical term and is used to refer to flavonoids, tannins, and phenolic acids and their various chemically modified or polymerized derivatives [16]. Over the last two decades, multiple polyphenols have attracted attention as nephro-protective agents, particularly owing to their ability to maintain oxidative homeostasis and activate cytoprotective signaling in vivo (Figure 1) [17]. Recent studies have shown the therapeutic effects of bioactive compounds and their beneficial health effects; however, little effort has been put into summarizing the impact of polyphenol interventions on mitochondrial dysfunction in various renal diseases [12,18,19]. This literature review attempts to focus on the role of polyphenols in the prevention and/or treatment of kidney disease and explore the cellular mechanisms associated with their pharmacological activity. We mainly focus on preclinical studies, both cellular and animal, that displayed the ability of polyphenols to decrease physiological complications and enhance mitochondrial function.
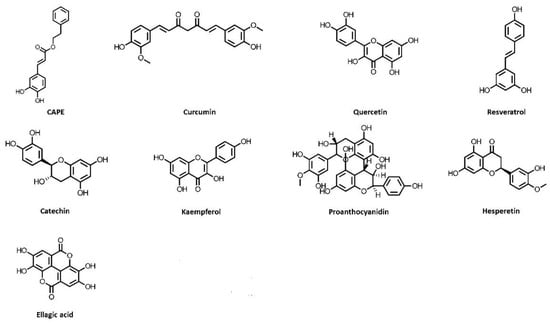
Figure 1.
Chemical structures of polyphenols, which exhibit kidney-protective activities. CAPE, caffeic acid phenethyl ester.
2. Bioavailability of Polyphenols
Recent studies have reinforced the health-promoting evidence of polyphenols based on diverse experimental models [20,21]. However, their chief problems are their low bioavailability and rapid metabolism [22]. Therefore, the bioavailability of polyphenols has been considered a significant limitation for their clinical evaluation and translations.
After polyphenol administration, oxidation, reduction, hydrolysis, and conjugation cause the production of different water-soluble conjugate metabolites, which can pass the enteric barrier for further distribution to organs [20,23]. These processes are mediated by lactase phlorizin hydrolase (LPH) and cytosolic β-glucosidase (CBG) [24]. Multidrug-resistance-associated proteins (MRP-1 and MRP-2) also play essential roles in polyphenol bioavailability and tissue accumulation [25]. During intestinal transit, MRP-2 on the apical surface of cells transports intracellular polyphenols to the lumen of the intestine. MRP-1, located in the vascular pole of enterocytes, promotes polyphenol passage from the enterocyte into the bloodstream [24]. MRP-3 and the glucose transporter 2 (GLUT2) efflux polyphenol metabolites from the enterocytes basolateral membrane to the portal circulation and reach the liver [24]. It is reported that small intestines can only absorb about 5–10% of the total polyphenol intake after deglycosylation [26]. About 90–95% of unmodified polyphenols and the conjugated forms pass through the intestinal tract to the large intestine for gut microbiota action. Gut microbiota can produce various metabolites to exert physiological impacts [27].
Despite the relatively few studies demonstrating lower mitochondrial uptake of polyphenols, their lipophilicity and pKa make them more suitable for mitochondrial enrichment [28]. A recent study showed that polyphenols were more bioavailable and could reach mitochondrial sites of action than previously assumed [29]. The pH value of cells affects the diffusion of polyphenols. Polyphenols are neutral phenols and form phenolate anions in the cytosol [29,30]. Their lipophilicity determines their ability to cross cell membranes and inner- and outer-mitochondrial membranes. Due to their pKa values close to the cytosol’s and mitochondria’s pH and distribution coefficients, many polyphenols can reach the mitochondrial matrix and release a proton in a relatively basic environment. [29]. At that time, phenolate anions move back down the electrochemical gradient to the relatively acidic intermembrane space. Protons are then transported from the inner-mitochondrial membrane to the matrix to regulate the electrochemical gradient (ΔΨm) [29,30]. In general, studies have shown that polyphenols are bioavailable and their metabolism via different mechanisms is responsible for their biological activities [31,32].
3. Mitochondria and Kidneys
3.1. Oxidative Phosphorylation (OXPHOS) System
Mitochondria are the central site for over 90% ATP production in cells [33,34]. ΔΨm in mitochondria is critical for mitochondrial function and is widely used as an indicator for mitochondrial function and oxidative stress [35]. The overproduction of reactive oxygen species (ROS), primarily superoxide anion (O2·−), during the transfer of electrons to oxygen, and a deficiency in antioxidant enzymes, such as superoxide dismutase (SOD) and glutathione (GSH) [36], leads to oxidative stress, mitochondrial dysfunction, and apoptosis [37]. Because mitochondrial ROS can inhibit multiple signaling pathways and prevent redox-dependent proteins’ proper function and activity, it is reported that mitochondrial ROS could be detrimental to cell survival and the health of a kidney cell [38]. ROS are produced in both the renal cortex and medulla, resulting in altering renal blood flow, inflammation, fibrotic changes, and proteinuria [39].
3.2. Mitochondrial Biogenesis
Mitochondrial biogenesis is an intricate and adaptive cellular response process [40]. It requires coordinated transcription and replication of mitochondrial DNA accompanied by the synthesis and import of proteins [5]. The mitochondrial biogenesis is regulated by the proliferator-activated receptor-gamma coactivator-1α (PGC-1α) family of transcriptional coactivators [12]. Mitochondrial biogenesis, respiration, fatty acid β-oxidation, and OXPHOS are all controlled by the interaction of PGC1-α with different transcription factors, such as nuclear respiratory factors 1 and 2 (Nrf1/2) and peroxisome proliferator-activated receptors (PPARα) [38]. The PGC-1α transcriptional coactivator is highly expressed in the proximal tubules of the kidney and plays a critical role in tubular homeostasis [11]. AMP-activated protein kinase (AMPK) and family of NAD+-dependent deacetylases known as Sirtuins (SIRT1–7), including SIRT1, are essential modulators of energy metabolism. AMPK with phosphorylation and SIRT1 through deacetylation can positively regulate PGC-1α [41,42,43]. Stimulation of PGC-1α through deacetylation or phosphorylation can stimulate the pathway followed by activation of nuclear transcription series factors, such as Nrf1, Nrf2, and transcription factor A mitochondria (TFAM) expression, consequently leading to mitochondria DNA (mtDNA) transcription and replication [44]. Moreover, PGC-1α activation improves the nicotinamide adenine dinucleotide (NAD+) biosynthesis, a key molecule critical for oxidative metabolism and cell protection [11]. It is reported that transgenic expression of PGC-1α leads to increased mitochondrial content and expression of mitochondrial genes. Conversely, loss of PGC-1α results in reducing the mitochondrial genes expression and causes mitochondrial dysfunction in mice [38]. There has been widespread evidence of reduced mitochondrial biogenesis as well as low PGC-1α levels in AKI and CKD [45]. Further, the Nrf2 antioxidant pathway was established to cope with CKD-induced oxidative stress in renal cells. Nrf2 is bound to its repressor under normal physiological conditions; under oxidative stress, Nrf2 is rapidly dissociated and translocated to the nucleus, encoding the antioxidant enzyme gene [46]. On the other hand, ROS, oxidative stress, and inflammation suppress the antioxidant potential of renal cells by suppressing the expression of Nrf2 [47]. Cellular homeostasis is integrated with function of mitochondria and biogenesis. It leads to metabolic syndrome, neurodegenerative diseases, and cancer if the intracellular pathway is malfunctioned [44]. According to the broad involvement of PGC-1α and Nrf1/2 as the important factors of mitochondria biogenesis, they can serve as a vital pharmacological target in metabolic diseases.
3.3. Mitochondrial Dynamics
To maintain cellular homeostasis and mitochondrial function, mitochondrial dynamics, such as division, fusion, and movement, are indispensable [48,49,50]. There are also fission proteins regulating mitochondrial dynamics, including mitochondrial fission 1 (Fis1), fusion proteins, and optical atrophy (OPA1) [7,51]. For the optimal function of mitochondria, there must be a balance between fission and fusion events, since imbalanced mitochondrial dynamics will eventually result in diseases, such as insulin resistance and type 2 diabetes, hypertension, cardiovascular diseases, and obesity [11,38,52]. Further, kidney disease and impairment are related to an increased mitochondrial fragmentation [53]. These findings suggest that a balanced mitochondrial fission and fusion is necessary for optimal mitochondrial function in kidney cells.
3.4. Mitophagy
Mitophagy is the autophagy of accumulated dysfunctional mitochondria modulated by PTEN-induced putative kinase 1 (PINK1)-parkin RBR E3 ubiquitin protein ligase (PARK2) pathways (ubiquitin-dependent mechanism) and B-cell lymphoma 2 (Bcl2) interacting protein 3 (ubiquitin-independent mechanism) [3,54,55,56]. There is an association between disturbed mitophagy and kidney diseases, such as acute kidney injury, diabetic nephropathy, and glomerulosclerosis [11]. In PINK1 and/or PARK2 knockout models, ROS production, inflammation, mitochondrial fragmentation, and cell apoptosis were enhanced in kidney cells, resulting in severe kidney injury. This suggests that PINK1 and PARK2 pathways act as a protective mechanism in AKI to maintain renal tubular integrity and kidney function [57].
4. Kidney and Mitochondria
Chronic and acute kidney injuries are linked with the production of ROS and reactive nitrogen species (RNS) [11]. Oxidative stress in AKI results from sepsis, ischemia-reperfusion injury, exposure to nephrotoxic reagents, and diabetic nephropathy. It was revealed that a balance between fission and fusion tended toward fission, contributing to mitochondrial fragmentation in AKI [58]. As a consequence, fragmentation could be related to the release of apoptotic factors, such as cytochrome C, activation of caspase, and apoptosis [53]. Additionally, AKI in cell and mouse models showed a decrease in mitophagy, ROS production, inflammation, and increase in mitochondrial damage [59]. Renal fibrosis and, consequently, CKD usually results from repeated or severe AKI [60,61,62]. Further, CKD may arise from environmental exposure to metal, pesticides, and infectious agents, decreased glomerular filtration rate, and higher urinary albumin excretion [63,64]. Enhanced fragmentation of mitochondria in kidney tubules, the reduced mitochondrial biogenesis, loss of mitochondria membrane potential (MMP), drop in ATP generation, and overproduction of mitochondrial ROS were reported in CKD [38,65]. Thus, CKD and AKI might perturb mitochondria biogenesis, dynamics, and mitophagy clearance. The conditions are all likely to lead to an accumulation of inflammatory cytokines, release of pro-apoptotic factors, and tissue damage [11].
An ischemic/reperfusion (I/R)-injury-induced AKI is a cellular injury that is triggered by a pathological condition that results in blood returning to tissues that have been ischemic [66]. I/R contributes to kidney dysfunction and AKI [67]. It is accompanied by inflammation, ROS and cytokine generation, lipid peroxidation, changes in mitochondrial function, and mitochondria injury [68,69]. I/R could increase the protein levels of pro-inflammatory factors, including tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β), and interleukin 6 (IL-6), and levels of the ROS and malondialdehyde (MDA), while decreasing SOD and GSH [70]. In mitochondria, cytochrome oxidase (complex IV) is able to catalyze electron transfer from cytochrome C to oxygen to produce a proton gradient for ATP synthesis [71]. ROS and lipid peroxidation products effectively inhibit mitochondrial complex IV activity [36,72], thus, influencing the electron flow across the electron transport chain and ATP production [73]. As a result of lipid peroxidation, different pathways lead to apoptosis and autophagy [74]. In another study, the Nrf2/heme oxygenase-1 (HO-1) signaling pathway decreased renal I/R injury by mediating oxidative stress [75]. Ca2+ at physiological concentrations is an essential regulator of mitochondrial energy metabolism [76]. Ca2+ influx into the mitochondria is a noteworthy factor in triggering mitochondrial ROS production [77]. Overproduction of ROS might result from increased mitochondrial Ca2+ accumulation, leading to inhibition of electron transport and/or increase in the enzymes responsible for ROS generation [78]. The mitochondrial Ca2+ load reduces the transmembrane potential and opens the mitochondrial permeability transition pore (MPT), damaging mitochondria and mitochondrial respiratory chains and subsequent ROS surge [79]. On the other hand, it was found that ischemic injury decreased the OXPHOS and Ca2+ uptake in kidney mitochondria, which could impact mitochondrial metabolism [69]. These studies demonstrated that I/R-induced inflammation, oxidative stress, and apoptosis might be related to kidney mitochondria. Acute kidney injury resulting from nephrotoxicity could damage mitochondria and, consequently, impair renal functions [80].
Cadmium is a toxic heavy metal, which has extensive nephrotoxic impact [81]. The expression of PGC-1α, Nrf1, SIRT1, and TFAM involved in mitochondrial biogenesis were impaired in cadmium-induced nephrotoxicity [82]. Nephrotoxicity caused mitochondrial fission by inhibiting mitochondrial membrane fusion and activating mitophagy mediated by the PINK/Parkin pathway [83]. Cadmium-induced renal impairment might alter tissue redox status by increasing lipid peroxidation products, such as MDA and nitrite oxide (NO), and decreasing SOD and catalase (CAT) enzymes in the kidneys [84]. This leads to disruption in mitochondria function, mitochondrial membrane potential, and eventually, renal hemostasis [82,85,86].
The antibiotic gentamycin is widely used to treat bacterial infections [87]. Nephrotoxicity caused by gentamycin also triggers ROS production in mitochondria, stimulating the opening of the MPT pore [88]. Thus, the MPT pore opening triggers the release of cytochrome C into cytosol which leads to swelling of mitochondria, activation of caspase cascade, and finally culminates to apoptosis [89]. In addition, the Bcl-2/Bcl-2-associated X (Bax) ratio, which is a vital factor to control cell apoptosis, decreased in the kidney following nephrotoxicity [90].
Anticancer drugs, such as cisplatin, cause DNA crosslinking and apoptosis [91]. Likewise, cisplatin-induced nephrotoxicity elevated protein oxidation and lipid peroxidation in the kidney mitochondria of rats, resulting from increasing ROS production or decreasing antioxidant status [92]. After cisplatin administration, the levels of the lipid peroxidation end-product MDA were significantly increased along with GSH and SOD depletion in rats [93]. The enhanced lipid peroxidation in mitochondria might cause decreased mitochondrial membrane fluidity, an increase in the distribution of negative surface charge, and an altered ionic membrane permeability [94]. Cisplatin triggers signaling cascades, such as p53, MAP kinase (MAPK), and nuclear factor kappa B (NF-κB), by ROS formation [95]. Further, cisplatin released pro-inflammatory cytokines, for instance, interleukin 12 (IL-12), TNF-α, and IL-1β to induce kidney damages [96]. Therefore, cisplatin was able to damage the kidney by generating oxidative stress, inflammation, DNA damage, apoptosis, and mitochondrial dysfunction [97].
Cyclosporine A is an immunosuppressive drug used to treat autoimmune diseases and to prevent organ rejection [98]. Studies indicated that cyclosporine A could cause acute and chronic nephrotoxicity by inhibiting mitochondrial respiration and decreasing ATP production in vivo and in vitro [99,100,101,102]. Cyclosporine A might suppress mitochondria biogenesis to induce nephrotoxicity [103]. Human kidney proximal tubule epithelial cells treated with cyclosporine A showed increased mitochondrial dysfunction and cellular death induced by H2O2. ROS production during H2O2 injury could activate the p53 pathway. In addition to binding DNA, activated p53 could accumulate in the mitochondrial matrix and trigger necrotic cell death by opening the MPT pore [104].
Doxorubicin, an anticancer agent, is widely used in the treatment of leukemia, breast cancer, and solid tumors [105]. Similar to other nephrotoxic drugs, there was an association between doxorubicin exposure and declining antioxidant parameters, such as glutathione peroxidase (GPx), SOD, and CAT, as well as SIRT1 activity [106,107]. Research showed that doxorubicin elevated thiobarbituric acid reactants (TBARS) and MDA, an indicator of oxidative damage [108]. NF-κB activation plays a critical role in the pathogenesis of doxorubicin-induced renal inflammation [109]. According to this, NF-κB was responsible for inflammatory reactions by mediating TNF-α, IL-1β, and IL-6 expressions in rats treated with doxorubicin [110]. The formation of superoxide radical(s) by doxorubicin exposure led to apoptosis [111,112]. Further, doxorubicin-treated animals showed cell death and apoptosis characterized by upregulation of Bax, down-regulation of Bcl2, increased mitochondrial permeability, and activation of caspase-3 in kidneys [106].
Diabetic nephropathy, a complication of microvascular in diabetes, could cause renal disease [113]. Redox changes are caused by persistent hyperglycemia and the accumulation of advanced glycation end products (AGEs) [114]. The resulting chronic inflammatory response leads to aberrant redox changes, albuminuria, proteinuria, glomerulosclerosis, and tubule-interstitial fibrosis [115]. Complications associated with diabetes are caused by ROS production, can damage mitochondrial DNA, and induce cell dysfunction [116,117]. These changes in renal cells, including glomerular endothelial cells, mesangial cells, and renal epithelial cells, disrupt ATP synthesis, cause intracellular calcium imbalances, and contribute to apoptosis and necrosis [118]. Diabetic rats’ kidney tissues showed higher levels of ROS, MDA, TNF-α, IL-6, and NF-κB p65 [119]. Apoptosis was also observed with higher Bax protein and cleaved caspase-3 levels, increased cytochrome c cytoplasmic levels, and Bcl2 down-regulation. In addition, the kidneys of diabetic rats revealed a significant decrease in the mRNA levels and nuclear levels of Nrf2, with a reduction in SOD mRNA levels and SOD and GSH protein levels. This disruption in cellular viability and oxidative homeostasis was possibly backed by hyperglycemia-induced ROS surge and depleted Nrf2 pool [120]. In diabetic nephropathy, the oxidative stress might increase GSH degradation or lower innate GSH synthesis. Moreover, ROS also lower the enzymatic activities of SOD and CAT [121]. Further, free radicals induced during diabetic nephropathy lowered the activity of AMPK and SIRT1, the critical regulators of PGC1α activity and energy metabolism of mitochondria [122]. The injury of the podocyte cells that cover the outer surfaces of glomerular capillaries, related to Nrf1 and mitochondrial dysfunction, contributed to diabetic kidney disease [123]. Studies have also shown that mitochondrial damage contributed to chronic and acute kidney injury as a result of a reduction in mitochondrial DNA, mitochondrial membrane potential, and ATP production along with increase in inflammation, and apoptosis [65].
5. Antioxidants and Kidney Diseases
5.1. Caffeic Acid Phenethyl Ester
Caffeic acid phenethyl ester (CAPE) is a natural phenolic compound possessing anti-inflammatory, antioxidant, and immunomodulatory effects [124]. CAPE exhibits a strong antioxidant potential by scavenging free radicals and facilitating oxidative homeostasis [125]. Further, CAPE improved OXPHOS of mitochondria through complex-I-dependent substrate(s) glutamate/malate [69]. It was later shown that CAPE pre-treatment protected complex II (SDH) activity and inhibited ROS formation at the Complex II F [68]. CAPE reduced Fe3+ (oxidized form of cytochrome C) into Fe2+, inhibiting the release of cytochrome C to cytosol and apoptosis. This protection decreased MDA and xanthine oxidase (XO), while increasing antioxidant enzyme GSH [68]. Therefore, CAPE inhibited lipid peroxidation in renal tissues [126]. Further, CAPE pre-treatment ameliorated mitochondrial swelling and dissipation of membrane potential following renal toxicity by cadmium [127]. Özeren et al. [128] showed that CAPE prevented kidney ischemia/reperfusion injury by inhibiting lipid peroxidation and improving mitochondrial Ca2+ uptake, resulting in improved mitochondrial energy metabolism [69]. Furthermore, CAPE treatment also boosted levels of NO from endothelial cells, thus preventing the pathological damage in ischemia [129]. Consequently, CAPE increased mitochondrial function to uptake calcium and boost OXPHOS [69,129]. Lastly, CAPE was able to lower oxidative stress, increase antioxidant enzyme activity and GSH content, and inhibit MPT pore opening, resulting in improved renal health [130]. Additionally, CAPE blocked ROS production and augmented the activity of antioxidant enzymes, such as SOD and CAT [126]. Since CAPE exhibits potent antioxidant, anti-inflammatory, and mitochondrial protective effects in kidney cells and tissues, this promotes CAPE as a promising new therapeutic agent that has the potential to protect the kidney from damage [126].
5.2. Curcumin
Curcumin is a natural polyphenol product derived from the rhizome of the Curcuma longa, exerting anti-inflammatory, antioxidative, anti-tumor, and anti-fibrotic effects [131]. The presence of conjugated double bonds in the curcumin structure allows it to donate an electron and scavenge ROS [132]. Curcumin has shown a protective effect in kidney damage models via its antioxidant activity, leading to the preservation of mitochondrial function [133]. Further, curcumin prevented mitochondrial dysfunction by protecting the mitochondrial respiratory complexes [134]. Some drugs, including gentamycin, reduce the activity of complexes I, II, and IV [134]. The complexes I and IV concentration and activities were recovered through curcumin treatment [134]. Consequently, the phosphorylation efficiency (Adenosine di-phosphate (ADP)/Oxygen) ratio in mitochondria oxidizing malate/glutamate and uncoupled respiration was recovered and redox homeostasis was maintained to prevent mitochondrial dysfunction. Curcumin suppresses TNF-α-mediated NF-κB activity in the development of chronic renal failure and inflammation [135,136]. Further, curcumin reduced interferon gamma (IFNγ) expression, but increased IL-10 levels in the renal ischemia/reperfusion model [137].
Curcumin also exhibited protective impacts against various nephrotoxic agents, such as cisplatin, gentamicin, and cadmium [138]. Particularly, curcumin treatment increased the PGC-1α levels and TFAM expression in nephrotoxcity-induced AKI [139,140]. Curcumin also protected the kidneys from oxidative stress in cisplatin-induced nephrotoxicity [141]. For example, curcumin attenuated oxidative stress and lipid peroxidation by scavenging ROS, restoring manganese superoxide dismutase (MnSOD) activity, enhancing glutathione s transferase (GST) activity, and modulating the GSH levels in kidney mitochondria [142]. Mechanistically, curcumin protected against cisplatin-induced oxidative damage by activating transcription factor EB (TFEB), leading to the regulation of autophagy and decreased levels of ROS after elimination of damaged mitochondria [143]. Moreover, curcumin was also able to restore the imbalance of mitochondrial dynamics in cisplatin nephrotoxicity through attenuation of Fis1 levels and restoring OPA1 levels [144]. Curcumin significantly regulated SIRT3, leading to mitochondrial integrity, a decrease in mitochondrial fission, and improved mitochondrial fusion. SIRT3 upregulation by curcumin also reduced dynamin-related protein 1 (DRP1) levels and prevented depolarization of the mitochondrial membrane in nephrotoxicity with cisplatin [142,145]. Further, curcumin treatment showed a higher number of normal-structure mitochondria and lower swollen mitochondria in gentamicin-induced kidney damage, owing to its ability to recover oxygen consumption of mitochondria [134]. Additionally, curcumin also ameliorated the MPT pore opening and protected them from the detrimental effects by preserving mitochondrial integrity [134]. Curcumin also showed protective effects in rats with a renal interstitial fibrosis model. In this study, curcumin inhibited the PI3K/Akt mammalian target of the rapamycin (mTOR) signaling pathway activation and upregulated essential proteins, mediating autophagosome formation. This led to suppressing the inflammatory response and mitochondrial dysfunction development [131]. Furthermore, the ability of curcumin to boost mitochondrial biogenesis warrants its exploration and use for renal disease [146].
5.3. Quercetin
Quercetin, a natural flavonoid abundant in fruits, vegetables, and leaves, is a potent antioxidant, which alleviates cell senescence by reducing oxidative stress [107,147]. Quercetin alleviates oxidative stress, prevents kidney damage, and inhibits renal inflammation in animal models of diabetic nephropathy [148]. Further, quercetin treatment prevented structural and functional damage of renal tissue and suppressed oxidative stress in the rats with tubulointerstitial necrosis and cadmium nephrotoxicity [149]. Recently, it was found that quercetin had chemo-protective and anti-apoptotic effects as a result of elevated expression of p53, p21, and p27 and lowered Bax expression in vitro [150]. Quercetin chelated metal ions, such as iron and copper, which were able to scavenge free radicals in in vitro experiments [151]. Quercetin also suppressed NF-κB, lipid peroxidation, and expression of pro-inflammatory matrix metalloproteases, whereas it might elevate nitric oxide levels and non-enzymatic antioxidant capacity of plasma [107]. Quercetin also ameliorated nephrectomy-induced oxidative stress by increasing GPx and decreasing MDA levels in rats [46,152]. In addition, quercetin restored mitochondrial function and protected against DNA double-strand breaks after doxorubicin treatment in H9c2 cells [153]. It was shown that quercetin could increase the expression of Nrf2 in the nucleus to enhance the encoding of antioxidant enzymes and gene expression of HO-1 in rats with CKD [46]. In renal interstitial fibrosis, quercetin significantly enhanced mitophagy by activating SIRT1 and inducing the PINK1-Parkin signaling pathway [153]. Moreover, a reduction in systolic blood pressure was associated with a reduction in epithelial Na+ channel (ENaCs) expression in the kidneys of hypertensive Dahl salt-sensitive rats treated with quercetin [154,155]. Based on the studies, quercetin can be considered a polyphenol with the ability to lower oxidative stress and apoptosis, while improving mitochondria mitophagy and biogenesis in the kidney.
5.4. Resveratrol
Resveratrol is a natural stilbenoid polyphenol found in grapes, blueberries, and peanuts [156]. It exhibits anti-inflammatory, anti-cancer, and anti-aging effects, both in cells and in animals [157]. Further, resveratrol has potential in the treatment of kidney diseases to improve overall health [34]. Studies observed that resveratrol enhanced the NADH entry into electron transport, thus, increasing the NAD+-NADH ratio, which might influence SIRT1 activity [72,158]. There is ample evidence indicating that resveratrol increased all SIRT1 target proteins, which were critical to mitochondrial function and oxidative stress reduction in kidneys [159]. Resveratrol-induced SIRT1 activity triggered a decrease in fibrosis, mesangial expansion, oxidative stress, and inflammatory cytokine levels, resulting in improved kidney function [160,161]. In the kidneys of SIRT1 KO db/db mice, the expression of pro-inflammatory factors mediated by NF-κB and signal transducer and activator of transcription 3 (STAT3) rose dramatically, supporting resveratrol-induced SIRT1’s crucial role in kidney inflammation [162]. Likewise, resveratrol protected against diabetic kidney disease in db/db mice with type 2 diabetes via an AMPK/SIRT1-independent mechanism [163]. The treatment of db/db mice with 20 mg resveratrol/kg/day for 12 weeks led to a reduction in kidney damage and modification of renal diabetes phenotypes [164]. A recent study revealed that resveratrol was essential in restoring mitochondrial function and biogenesis via SIRT1/PGC-1α activation in kidneys of diabetic mice [165]. It was shown that activation of SIRT1-dependent pathways by resveratrol attenuated kidney injury by upregulation of mitochondrial biogenesis factors [72]. Further, in chickens that were treated with resveratrol, Nrf2 signaling was activated to reverse renal oxidative damage caused by cadmium injury and activate downstream phase II detoxification factors, such as HO-1, NAD(P)H dehydrogenase quinone 1 (NQO1), and GSTs [82]. Likewise, Kim et al. proved that a reduction in oxidative stress through Nrf2 activation ameliorated renal function, proteinuria, and pathological changes in aging mice [157]. Alternatively, resveratrol treatment prevented a decrease in the activity of complex II and complex IV following hemorrhagic shock, which decreased ROS production and damage in a rat model of kidney disease [72]. Additionally, Hui et al. showed that resveratrol treatment raised MMP and activities of complex I and III; therefore, the production of ATP improved and reduced the generation of ROS in a rat model of CKD [34]. Further, Zhang et al. showed that resveratrol reversed mitochondrial injury, diminished the autophagic vacuole number, and ameliorated mitochondrial fission in chicken kidney [82]. In addition, by improving mitochondrial elongation, resveratrol facilitated autophagy, suppressed Parkin and PINK1 phosphorylation, and degraded mitochondria that were removed [82]. Generally, these studies suggested that treating renal injuries with resveratrol might attenuate nephrotoxicity, I/R, oxidative stress, and apoptosis, while increasing antioxidant enzyme activities. In addition, resveratrol treatment might affect mitochondrial biogenesis and dynamics in kidney diseases to ameliorate mitochondrial dysfunction and metabolic stress.
5.5. Catechin
Catechin, as a part of the flavonoids family, is present in plants, fruits, teas, red wine, and cacao [166]. In addition to having antioxidant properties, it also exhibits potent anti-inflammatory properties [167]. Catechin protects the kidneys by scavenging free radicals, inhibiting intracellular ROS, chelating redox-active metals, and enhancing antioxidant defense mechanisms [168,169]. In addition, catechin had the potential to prevent MMP loss and apoptosis by restoring the activity of mitochondrial complex I and ATP synthesis [170]. In SK-N-MC cells, catechin boosted the expression of anti-apoptotic protein Bcl-2 and inhibited the expression of apoptotic protein Bax [171,172].
Epigallocatechin gallate (EGCG) is a catechin esterified with gallic acid [173]. It is the major polyphenol in green tea with antioxidant activity in reducing mitochondrial oxidative stress [174,175]. It was found that EGCG restored mitochondrial electron transport chain function to normal in mouse kidney with cisplatin-induced damage [176]. Further, EGCG protected against renal injury caused by cisplatin through favoring mitochondrial antioxidant enzymes, such as MnSOD and GPx, and enhancing the anti-inflammatory effect [177]. Further, EGCG treatment significantly reduced DNA damage caused by p65 and P53 and modulated NF-κB nuclear accumulation in cisplatin nephrotoxicity [176]. In the rat model of obstructive nephropathy, treatment with EGCG inhibited NF-κB activation, while improving the phosphorylated IkappaB (IκB) protein and inducing Nrf2 nuclear translocation [177]. EGCG induced GST, GPx, and HO-1 expression, where they were able to eliminate or inactivate ROS and oxidative stress; thus, it could suppress oxidative stress and acute renal injury [178,179]. In a mouse model of nephrotoxicity, EGCG modulated the receptor Bax and Bcl-2 that attenuated cisplatin-induced apoptosis [180]. Thus, EGCG-induced modulation of NF-κB and Nrf2 is a critical element for oxidative stress and inflammation alleviation in acute kidney damage [177,181]. Furthermore, green tea polyphenols (polyphenol + catechin + EGCG) protected the rat kidneys from the oxidative damage caused by a high-fat diet via an SIRT3/MnSOD pathway mediated by PPARα [182]. It was suggested that green tea polyphenols increased PGC1-α and TFAM axis, mitochondria DNA, OXPHOS proteins, and SIRT1 activity related to a reduction in kidney injury and improvement in renal function after cyclosporine treatment of rats [103]. Eventually, EGCG and catechin had the ability to enhance mitochondria function by impacting biogenesis, dynamics, and OXPHOS to prevent or treat kidney diseases.
5.6. Kaempferol
Kaempferol, a natural flavonoid, is found in tea, vegetables, and fruits, such as broccoli, grapes, kale, tomatoes, and citrus fruits [183,184]. Kaempferol has antioxidant, anti-cancer and anti-inflammatory effects [97]. It was reported that kaempferol caused a significant decline in MDA levels, an indicator of oxidative stress, cytotoxicity, and renal damage in calcineurin inhibitor-induced renal injury and CKD [185]. In addition, kaempferol could lower lipid peroxidation and improve antioxidant defense activity [186]. Tumor necrosis-factor-receptor-associated factor 6 (TRAF6), a transcription factor upstream of NF-κB, is downregulated by kaempferol, reducing renal inflammation and fibrosis in renal tubular epithelial cells [187]. It was shown that pre-treatment of kaempferol reduced pro-inflammatory cytokine release, such as IL-12 and TNF-α, and regulated NF-κB levels by hindering the IkappaB kinase (IKK) phosphorylation and IκBα degradation; thus, it ameliorated the cisplatin-mediated inflammation in mouse kidney proximal tubule epithelial (TKPTS) cells [97]. Furthermore, kaempferol inhibited the p38, ERK, and c-Jun N-terminal kinase (JNK) activation, while augmenting Coenzyme Q (CoQ) biosynthesis and content [97]. Treatment with kaempferol increased GSH and SOD2, while reducing TNF-α and IL-6 in the kidneys of doxorubicin-treated rats [106]. Moreover, treatment and pre-treatment with kaempferol in rats increased nuclear accumulation of Nrf2, which was necessary for mitochondrial biogenesis, in contrast to the cisplatin- and doxorubicin-treated animals [106,180]. In addition, the protective effects of kaemepferol against streptozotocin-induced diabetic nephropathy could be attributed to its potent antioxidant effect, mediated by upregulation and activation of Nrf2 [188]. Overall, kaempferol can be a potential therapeutic used in treatment, preventing kidney mitochondria injury because it has anti-inflammatory and antioxidative properties.
5.7. Grape Seed Proanthocyanidin
Other plant polyphenols, such as grape seed proanthocyanidin extracts (GSPE), have strong therapeutic characteristics against oxidative stress and inflammatory damage [189,190]. The effects of GSPE on obese rats included the stimulation of energy expenditure, an increase in thermogenic capacity, and inhibiting mitochondrial dysfunction in brown adipose tissue [191]. Rats treated with GSPE had fewer mitochondrial degenerations, stabilized mitochondrial enzymes, and corrected mitochondrial dysfunction in myocardium and brown adipose tissue [191,192,193]. GSPE served to reduce proteinuria and podocyte injury as well as nephropathy progression in diabetic rats [194]. Further, the antioxidant capacity of GSPE enhanced the activity of SOD2 and CAT and decreased the levels of MDA and inflammatory cytokines, such as TNF-α and Monocyte chemoattractant protein (MCP1), in renal tissues of diabetic rats [195,196]. Additionally, GSPE was able to restore mitochondrial DNA and increase Nrf1 and TFAM RNA expression, which could suppress renal mitochondrial dysfunction [123]. In addition, GSPE protected diabetic podocytes from injury by restoring phospho-AMPK, SIRT1, and PGC-1α levels [123]. It was shown that protein SIRT1 was the therapeutic target of GSPE against H2O2 injury. GSPE upregulated the SIRT1 and re-established homeostasis of mitochondrial complexes I, II, III, and IV, enhanced antioxidant enzymes, such as SOD2, whereas it inhibited apoptosis factors, such as BAX and P53, in HEK-293 cells [197]. Further, GSPE increased GSH and TBARS and the protein levels of Nrf2, HO-1, and GST in diabetic kidney and nephrotoxicity [198,199]. By reducing ROS levels, GSPE protected kidneys from oxidative-stress-induced injury [195]. Further, GSPE inhibited NF-κB in I/R injuries in Rats; therefore, it reduced the markers of renal injury and oxidative damage and even inactivated the inflammatory pathway [200]. Thus, GSPE reduced renal damage in rats by activating the Nrf2 signaling pathway, which consequently improved the antioxidant capacity of the tissue [198]. These studies revealed that GSPE might be a safe therapeutic candidate to regulate mitochondrial dysfunction in kidney diseases.
5.8. Hesperetin
As a natural flavonoid found in citrus plants [201], hesperetin has antioxidant, cardiovascular regulation, and anti-cancer activities [93]. Oxidative stress and ROS generation are the significant factors in cisplatin-induced AKI [202]. Hesperetin reduces the renal MDA and NO levels and restores the antioxidant enzyme levels, such as GSH, CAT, GPx, and SOD, to the normal levels in rats with nephrotoxicity [93]. It was reported that the levels of MDA and NO in the kidneys reduced by hesperetin and the levels of antioxidant enzymes, such as GSH, CAT, GPx, and SOD, were restored to normal levels. Hesperetin significantly normalized the elevated level of inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, and, thus, protected the kidney from inflammatory insult in rats with nephrotoxicity [93,203]. Moreover, hesperetin inhibited phosphorylation of Akt in diabetic nephropathy, indicating that the PI3K/Akt pathway could be involved in the protective effects of hesperetin [204]. Hesperetin also inhibited JNK, ERK, and p38 phosphorylation, suggesting that it could inhibit cisplatin-induced inflammation [205]. Activating the Nrf2 signaling pathway by hesperetin significantly diminished the oxidative damage of ARPE-19 cells and promoted the SIRT6 expression to protect from I/R injury [206,207]. It was shown that hesperetin could inhibit the apoptosis induced by cisplatin, decrease Bax and caspase-3 expression, and increase Bcl-2 expression [208]. Overall, hesperetin protects against nephrotoxicity and diabetic kidney injury by inhibiting inflammation, oxidative stress, and apoptosis.
5.9. Ellagic Acid
Ellagic acid is a phenolic acid present in fruits and vegetables, such as raspberries, strawberries, walnuts, grapes, and blackcurrants [209]. The antioxidant effect of ellagic acid leads to scavenging O2·−, OH−, and lipid peroxide, therefore, inhibiting lipid peroxidation and improving the antioxidant status [210]. A study proved that ellagic acid reduced serum MDA levels and increased SOD levels, indicating that it alleviated diabetic nephropathy symptoms by reducing oxidative stress [211,212]. Ellagic acid was also reported to lower TNF-α and IL-1β levels in diabetic nephropathy and nephrotoxicity kidney injury mice, which might be mediated through NF-κB; therefore, ellagic acid could be a potent inhibitor of NF-κB activation [211,213]. Further, ellagic acid reduced the cellular membrane damage by scavenging the free radicals in rats with nephrotoxicity and nephropathy [90]. This protection was shown by covering depleted levels of SOD, GSH, CAT, and Bcl2 in the kidney, inhibiting caspase-3 activation and increasing the Bcl-2/Bax expression ratio. They found that ellagic acid significantly reduced the mitochondrial ROS content, reversed the swelling of mitochondrial kidney, and prevented loss of mitochondria membrane potential. Further, it was suggested that the anti-apoptotic effects of ellagic acid could be attributed to the upregulation of Nrf2 [90,120,214]. Additionally, Nrf2 could suppress inflammation by inhibiting TNF-α and NF-κB in diabetic nephropathy in cell lines, an animal model, or both [215]. It also activated different antioxidant enzymes, such as HO-1, NQO1, GST, and GSH [216,217]. The dysfunction of mesangial cells in diabetic nephropathy might be related to the PI3K/Akt signaling pathway activation inhibited by ellagic acid [218]. Ellagic acid treatment also triggered SIRT1 overexpression in renal tissues, which imparted renal tolerance to oxidative stress [214]. Moreover, ellagic acid-induced SIRT1 expression suppressed p53 and promoted cell survival via expression of antioxidant enzymes, such as CAT [214]. Overall, these results suggest that ellagic acid decreases renal inflammation and oxidative stress, leading to improved kidney function (Figure 2 and Table 1).
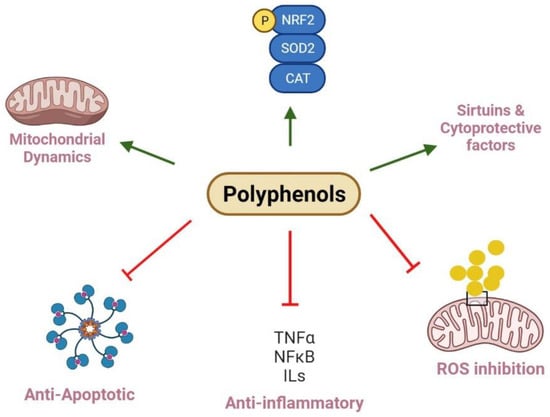
Figure 2.
Polyphenols and their roles in renal mitochondrial dysfunction. Polyphenols regulate mitochondria biogenesis and dynamics via increasing Nrf2 and PGC-1α expression and balancing fission and fusion events, while kidney diseases result from imbalanced mitochondria dynamics and reduce biogenesis. In this pathway, deacetylation of SIRT and phosphorylation of AMPK can positively regulate biogenesis. Polyphenols can decrease the protein levels of pro-inflammatory factors, including TNF-α, IL-1β, IL-6, and NF-κB to exert anti-inflammatory effects. They also improve mitochondria function and injury through inhibition of ROS generation. Polyphenols show protective effects through inhibiting MPT pore opening, which can trigger the release of cytochrome C into the cytosol, swelling of mitochondria, and activating of caspase cascade; finally, they reveal anti-apoptotic impacts. Further, polyphenols restore antioxidant enzyme levels such as SOD2 and CAT to normal levels, improve antioxidant status, and scavenge free radicals.

Table 1.
Preclinical evidence of polyphenols as a therapeutic approach for kidney disease.
6. Discussion and Perspectives
As discussed above, dysfunctional mitochondrial biogenesis, dynamics, or OXPHOS is a vital underlying factor in renal mitochondrial damage [11]. Although the commonly used drugs, such as cisplatin, gentamycin, cyclosporine A, and doxorubicin, in clinical practice have anticancer, antibiotic, and anti-inflammatory effects, they have irreversible side effects on the kidney [225]. The current literature suggests that mitochondrial dysfunction adversely alters kidney function and worsens complications that may promote complex renal diseases [6]. Renal mitochondrial alterations are associated with cellular damage, oxidative stress, inflammation, and apoptosis [226]. Eventually, the disturbed renal mitochondrial homeostasis leads to CKD, AKI derived from nephrotoxicity and I/R, and nephropathy [11]. Overall, the available studies display the need to target mitochondrial dysfunctional to restore kidney function and stimulate renal repair or prevent further damage in renal tissues. Despite the fact that defective mitochondria are linked to kidney diseases, the pathogenic relationship and our knowledge of the impact of mitochondrial dysfunction in patients with kidney disease remain uncertain. In animal models of kidney damage, mitochondria-targeting therapeutics have been shown to preserve mitochondrial structures and functions [227]. Indeed, dietary antioxidants, such as vitamin C and E, polyunsaturated fatty acids (PUFA), probiotics, N-Acetylcysteine (NAC), and exercise, may be suitable therapeutics for mitochondrial oxidative damage [12,148]. Polyphenols have shown promising potential in specific kidney injuries and diseases in animal and cell studies [18,228,229,230]. These mitochondria-targeting antioxidants have been demonstrated to effectively decrease ROS accumulation, inhibit release of pro-inflammatory cytokines, and kidney injury, and favor mitochondrial biogenesis and kidney function in various renal disease models.
Primarily, the structure of polyphenols allows them to act as an antioxidant, as they are able to donate an electron and scavenge ROS to make them stable [68,133]. Moreover, recent research has revealed that polyphenols may have more specific cell signaling mechanisms than general antioxidant actions via complex mitochondria function regulation [231]. Emerging evidence indicates that polyphenols, such as resveratrol, quercetin, curcumin, EGCG, kaempferol, ellagic acid, hesperetin, and GSPE, restore mitochondrial biogenesis by stimulating PGC-1α, NRF1/2, and TFAM to improve kidney function [72,134,139,140,198,199]. On the other hand, the down-regulation of apoptotic proteins and release of cytochrome C by polyphenols, such as catechin, ellagic acid, hesperetin, quercetin, and EGCG, represent an anti-apoptotic mechanism and cyto-protective impacts to prevent kidney injury [90,150,182,208]. Notably, some polyphenols, including curcumin and caffeic acid, can ameliorate the MPT pore opening, consequently preserving mitochondrial integrity [126,134]. Another mitochondrial action restricted to catechin and resveratrol inhibits MMP loss and improves ATP production through mitochondria protein complexes [130,134]. Further, polyphenols, including caffeic acid, curcumin, resveratrol, catechin, EGCG, and GSPE, may directly prevent mitochondrial dysfunction in renal injuries by enhancing the activities of mitochondrial electron transport chain complexes [170,176,197]. In addition to acting as antioxidants, polyphenols’ action include direct up-regulation of antioxidant defense systems, such as SOD, CAT, GSH, and GPx, whereas they decrease MDA and the pro-inflammatory cytokines, such as IL-12 and TNF-α-modulated NF-κB [96,106,126,137,178,179]. Taken together, polyphenols can regulate the electron transport chain activity, improve oxygen consumption, maintain the mitochondrial membrane, and support ATP generation, probably by scavenging free radicals and inhibiting protein and lipid oxidation in nephrotoxicity, I/R, and nephropathy.
Although polyphenols are natural compounds and present themselves as therapeutic possibilities, more detailed studies on the dose of polyphenols for clinical intervention are recommended. Because most of the studies are based on animals and cells, thus, the safety and effectivity of polyphenols to restore kidney mitochondria should be examined in humans. Further, pre-treatment of some polyphenols, such as caffeic acid and kaempferol, diminished the treatment duration of kidney disease, particularly nephrotoxicity [68,97]. Therefore, further investigations are required to elucidate the exact effect of pre-treatment polyphenols as a prevention agent against kidney disease. It is necessary to analyze whether polyphenols alter mitochondrial dysfunction in kidney disease compared to standard medicine; hence, they can be used as an alternative treatment compared to chemical medicine with more minor side effects. Further, it is necessary to observe interactions between clinically used medicine and polyphenols to address the safety aspects of pharmacology. There is also a lack of data to present the impact of fruits, vegetables, cereals, nuts, and plant consumption on kidney health and its mitochondria function. Further, the production of foods rich in polyphenols, food fortification, and polyphenol supplementation plays a significant role in the pharmaceutical use of this strategy. Accordingly, extensive studies on the design of new dietary patterns should be carried out.
Author Contributions
Conceptualization, F.A., K.S.B. and J.W.; writing—original draft preparation and editing, F.A., K.S.B. and J.W; supervision and funding: J.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Natural Sciences and Engineering Research Council of Canada [grant numbers CRDPJ 532150-18].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vart, P.; Grams, M.E. Measuring and Assessing Kidney Function. Semin. Nephrol. 2016, 36, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Hartogh, D.J.D.; Tsiani, E. Health Benefits of Resveratrol in Kidney Disease: Evidence from In Vitro and In Vivo Studies. Nutrient 2019, 11, 1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, J.M. Mitochondria–Power Players in Kidney Function? Trends Endocrinol. Metab. 2016, 27, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Hoenig, M.P.; Zeidel, M.L. Homeostasis, the Milieu Intérieur, and the Wisdom of the Nephron. Clin. J. Am. Soc. Nephrol. 2014, 9, 1272–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Lianos, E.A.; Ma, J.; Lin, P.-H. Autophagy, Innate Immunity and Tissue Repair in Acute Kidney Injury. Int. J. Mol. Sci. 2016, 17, 662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D.R. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lameire, N.H.; Bagga, A.; Cruz, D.; De Maeseneer, J.; Endre, Z.; A Kellum, J.; Liu, K.D.; Mehta, R.L.; Pannu, N.; Van Biesen, W.; et al. Acute kidney injury: An increasing global concern. Lancet 2013, 382, 170–179. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.; Monsalve, M.; Ramos, A.; Sanchez-Niño, M.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef] [Green Version]
- Duann, P.; Lin, P.-H. Mitochondria Damage and Kidney Disease. Mitochondrial Dyn. Cardiovasc. Med. 2017, 982, 529–551. [Google Scholar] [CrossRef]
- Mafra, D.; Gidlund, E.-K.; Borges, N.A.; Magliano, D.C.; Lindholm, B.; Stenvinkel, P.; Von Walden, F. Bioactive food and exercise in chronic kidney disease: Targeting the mitochondria. Eur. J. Clin. Investig. 2018, 48, e13020. [Google Scholar] [CrossRef] [Green Version]
- Yi, W.; Xie, X.; Du, M.; Bu, Y.; Wu, N.; Yang, H.; Tian, C.; Xu, F.; Xiang, S.; Zhang, P.; et al. Green Tea Polyphenols Ameliorate the Early Renal Damage Induced by a High-Fat Diet via Ketogenesis/SIRT3 Pathway. Oxidative Med. Cell. Longev. 2017, 2017, 9032792. [Google Scholar] [CrossRef] [Green Version]
- Boeing, H.; Bechthold, A.; Bub, A.; Ellinger, S.; Haller, D.; Kroke, A.; Leschik-Bonnet, E.; Müller, M.J.; Oberritter, H.; Schulze, M.; et al. Critical review: Vegetables and fruit in the prevention of chronic diseases. Eur. J. Nutr. 2012, 51, 637–663. [Google Scholar] [CrossRef] [Green Version]
- Mehmood, A.; Zhao, L.; Wang, C.; Nadeem, M.; Raza, A.; Ali, N.; Shah, A.A. Management of hyperuricemia through dietary polyphenols as a natural medicament: A comprehensive review. Crit. Rev. Food Sci. Nutr. 2017, 59, 1433–1455. [Google Scholar] [CrossRef]
- Williamson, G. The role of polyphenols in modern nutrition. Nutr. Bull. 2017, 42, 226–235. [Google Scholar] [CrossRef]
- Adekunle, I.A.; Imafidon, C.E.; Oladele, A.A.; Ayoka, A.O. Ginger polyphenols attenuate cyclosporine-induced disturbances in kidney function: Potential application in adjuvant transplant therapy. Pathophysiology 2018, 25, 101–115. [Google Scholar] [CrossRef]
- Tovar-Palacio, C.; Noriega, L.G.; Mercado, A. Potential of Polyphenols to Restore SIRT1 and NAD+ Metabolism in Renal Disease. Nutrients 2022, 14, 653. [Google Scholar] [CrossRef]
- Bendokas, V.; Skemiene, K.; Trumbeckaite, S.; Stanys, V.; Passamonti, S.; Borutaite, V.; Liobikas, J. Anthocyanins: From Plant Pigments to Health Benefits at Mitochondrial Level: Reviews; Critical Reviews in Food Science and Nutrition; Taylor & Francis: Philadelphia, PA, USA, 2020; Volume 60. [Google Scholar]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (Poly) phenolics in Human Health: Structures, Bioavailability, and Evidence of Protective Effects Against Chronic Diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [Green Version]
- Teng, H.; Chen, L. Polyphenols and bioavailability: An update. Crit. Rev. Food Sci. Nutr. 2019, 59, 2040–2051. [Google Scholar] [CrossRef]
- Brglez Mojzer, E.; Knez Hrnčič, M.; Škerget, M.; Knez, Ž.; Bren, U. Polyphenols: Extraction Methods, Antioxidative Action, Bioavailability and Anticarcinogenic Effects. Molecules 2016, 21, 901. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.E.; Chowrimootoo, G.; Choudhury, R.; Debnam, E.S.; Srai, S.K.; Rice-Evans, C. The small intestine can both absorb and glucuronidate luminal flavonoids. FEBS Lett. 1999, 458, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Santhakumar, A.B.; Battino, M.; Alvarez-Suarez, J.M. Dietary polyphenols: Structures, bioavailability and protective effects against atherosclerosis. Food Chem. Toxicol. 2018, 113, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Leonarduzzi, G.; Testa, G.; Sottero, B.; Gamba, P.; Poli, G. Design and development of nanovehicle-based delivery systems for preventive or therapeutic supplementation with flavonoids. Curr. Med. Chem. 2010, 17, 74–95. [Google Scholar] [CrossRef]
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Bowey, E.; Adlercreutz, H.; Rowland, I. Metabolism of isoflavones and lignans by the gut microflora: A study in germ-free and human flora associated rats. Food Chem. Toxicol. 2003, 41, 631–636. [Google Scholar] [CrossRef]
- Naven, R.T.; Swiss, R.; Klug-McLeod, J.; Will, Y.; Greene, N. The Development of Structure-Activity Relationships for Mitochondrial Dysfunction: Uncoupling of Oxidative Phosphorylation. Toxicol. Sci. 2012, 131, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Stevens, J.F.; Revel, J.S.; Maier, C.S. Mitochondria-Centric Review of Polyphenol Bioactivity in Cancer Models. Antioxid. Redox Signal. 2018, 29, 1589–1611. [Google Scholar] [CrossRef]
- Spycher, S.; Smejtek, P.; Netzeva, T.I.; Escher, B.I. Toward a Class-Independent Quantitative Structure−Activity Relationship Model for Uncouplers of Oxidative Phosphorylation. Chem. Res. Toxicol. 2008, 21, 911–927. [Google Scholar] [CrossRef]
- Velderrain-Rodríguez, G.R.; Palafox-Carlos, H.; Wall-Medrano, A.; Ayala-Zavala, J.F.; Chen, C.-Y.O.; Robles-Sánchez, M.; Astiazaran-García, H.; Alvarez-Parrilla, E.; González-Aguilar, G.A. Phenolic compounds: Their journey after intake. Food Funct. 2014, 5, 189–197. [Google Scholar] [CrossRef]
- Hussain, M.B.; Hassan, S.; Waheed, M.; Javed, A.; Farooq, M.A.; Tahir, A. Bioavailability and Metabolic Pathway of Phenolic Compounds. 5. In Plant Physiological Aspects of Phenolic Compounds; Marcos, S.-H., Rosario, G.-M., Mariana, P.-T., Eds.; IntechOpen: Rijeka, Croatia, 2019. [Google Scholar]
- Barchiesi, A.; Bazzani, V.; Tolotto, V.; Elancheliyan, P.; Wasilewski, M.; Chacinska, A.; Vascotto, C. Mitochondrial Oxidative Stress Induces Rapid Intermembrane Space/Matrix Translocation of Apurinic/Apyrimidinic Endonuclease 1 Protein through TIM23 Complex. J. Mol. Biol. 2020, 432, 166713. [Google Scholar] [CrossRef]
- Hui, Y.; Lu, M.; Han, Y.; Zhou, H.; Liu, W.; Li, L.; Jin, R. Resveratrol improves mitochondrial function in the remnant kidney from 5/6 nephrectomized rats. Acta Histochem. 2017, 119, 392–399. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2018, 34, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Nistala, R.; Whaley-Connell, A.; Sowers, J.R. Redox Control of Renal Function and Hypertension. Antioxid. Redox Signal. 2008, 10, 2047–2089. [Google Scholar] [CrossRef] [Green Version]
- Rensvold, J.W.; Ong, S.-E.; Jeevananthan, A.; Carr, S.A.; Mootha, V.K.; Pagliarini, D.J. Complementary RNA and Protein Profiling Identifies Iron as a Key Regulator of Mitochondrial Biogenesis. Cell Rep. 2013, 3, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-L.; Kang, C.-H.; Wang, S.-G.; Lee, H.-M. α-Lipoic acid regulates lipid metabolism through induction of sirtuin 1 (SIRT1) and activation of AMP-activated protein kinase. Diabetologia 2012, 55, 1824–1835. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2013, 25, 138–145. [Google Scholar] [CrossRef]
- Chodari, L.; Aytemir, M.D.; Vahedi, P.; Alipour, M.; Vahed, S.Z.; Khatibi, S.M.H.; Ahmadian, E.; Ardalan, M.; Eftekhari, A. Targeting Mitochondrial Biogenesis with Polyphenol Compounds. Oxidative Med. Cell. Longev. 2021, 2021, 4946711. [Google Scholar] [CrossRef]
- Chambers, J.M.; Wingert, R.A. PGC-1α in Disease: Recent Renal Insights into a Versatile Metabolic Regulator. Cells 2020, 9, 2234. [Google Scholar] [CrossRef]
- Layal, K.; Perdhana, I.S.; Louisa, M.; Estuningtyas, A.; Soetikno, V. The effects of quercetin on oxidative stress and fibrosis markers in chronic kidney disease rat model. Med. J. Indones. 2017, 26, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Putti, R.; Sica, R.; Migliaccio, V.; Lionetti, L. Diet impact on mitochondrial bioenergetics and dynamics. Front. Physiol. 2015, 6, 109. [Google Scholar] [CrossRef] [Green Version]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Lahera, V.; Heras, N.D.L.; Farre, A.L.; Manucha, W.; Ferder, L. Role of Mitochondrial Dysfunction in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.-G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M.; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2014, 25, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2012, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.J.; Parikh, S.M. Mitochondrial Metabolism in Acute Kidney Injury. Semin. Nephrol. 2020, 40, 101–113. [Google Scholar] [CrossRef]
- Tang, C.; Han, H.; Yan, M.; Zhuohua, Z.; Liu, J.; Liu, Z.; Chengyuan, T.; Tan, J.; Liu, Y.; Liu, H.; et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018, 14, 880–897. [Google Scholar] [CrossRef] [Green Version]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Bravi, C.A.; Vertosick, E.; Benfante, N.; Tin, A.; Sjoberg, D.; Hakimi, A.A.; Touijer, K.; Montorsi, F.; Eastham, J.; Russo, P.; et al. Impact of Acute Kidney Injury and Its Duration on Long-term Renal Function After Partial Nephrectomy. Eur. Urol. 2019, 76, 398–403. [Google Scholar] [CrossRef]
- Forni, L.G.; Darmon, M.; Ostermann, M.; Straaten, H.M.O.-V.; Pettilä, V.; Prowle, J.; Schetz, M.; Joannidis, M. Renal recovery after acute kidney injury. Intensiv. Care Med. 2017, 43, 855–866. [Google Scholar] [CrossRef]
- Scammell, M.K.; Sennett, C.M.; Petropoulos, Z.; Kamal, J.; Kaufman, J.S. Environmental and Occupational Exposures in Kidney Disease. Semin. Nephrol. 2019, 39, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.Y.; Sanders, A.P.; Saland, J.M.; Wright, R.O.; Arora, M. Environmental exposures and pediatric kidney function and disease: A systematic review. Environ. Res. 2017, 158, 625–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granata, S.; Gassa, A.D.; Tomei, P.; Lupo, A.; Zaza, G. Mitochondria: A new therapeutic target in chronic kidney disease. Nutr. Metab. 2015, 12, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amani, H.; Habibey, R.; Shokri, F.; Hajmiresmail, S.J.; Akhavan, O.; Mashaghi, A.; Pazoki-Toroudi, H. Selenium nanoparticles for targeted stroke therapy through modulation of inflammatory and metabolic signaling. Sci. Rep. 2019, 9, 6044. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Chen, Z.; Weng, X.; Chen, H.; Du, Y.; Diao, C.; Liu, X.; Wang, L. Enhancer of zeste homolog 2 modulates oxidative stress-mediated pyroptosis in vitro and in a mouse kidney ischemia-reperfusion injury model. FASEB J. 2019, 34, 835–852. [Google Scholar] [CrossRef] [Green Version]
- Kamarauskaite, J.; Baniene, R.; Trumbeckas, D.; Strazdauskas, A.; Trumbeckaite, S. Caffeic Acid Phenethyl Ester Protects Kidney Mitochondria against Ischemia/Reperfusion Induced Injury in an In Vivo Rat Model. Antioxidants 2021, 10, 747. [Google Scholar] [CrossRef]
- Trumbeckaite, S.; Pauziene, N.; Trumbeckas, D.; Jievaltas, M.; Baniene, R. Caffeic Acid Phenethyl Ester Reduces Ischemia-Induced Kidney Mitochondrial Injury in Rats. Oxidative Med. Cell. Longev. 2017, 2017, 1697018. [Google Scholar] [CrossRef]
- Liu, Q.; Liang, X.; Liang, M.; Qin, R.; Qin, F.; Wang, X. Ellagic Acid Ameliorates Renal Ischemic-Reperfusion Injury Through NOX4/JAK/STAT Signaling Pathway. Inflammation 2019, 43, 298–309. [Google Scholar] [CrossRef]
- Roede, J.R.; Jones, D.P. Reactive species and mitochondrial dysfunction: Mechanistic significance of 4-hydroxynonenal. Environ. Mol. Mutagen. 2010, 51, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Guan, Y.; Karamercan, M.A.; Ye, L.; Bhatti, T.; Becker, L.B.; Baur, J.A.; Sims, C.A. Resveratrol Rescues Kidney Mitochondrial Function Following Hemorrhagic Shock. Shock 2015, 44, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Chong, S.J.F.; Low, I.C.C.; Pervaiz, S. Mitochondrial ROS and involvement of Bcl-2 as a mitochondrial ROS regulator. Mitochondrion 2014, 19, 39–48. [Google Scholar] [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Liu, X.; Wang, M.; Chen, H.; Chen, Z.; Qiu, T. Oxymatrine ameliorates renal ischemia-reperfusion injury from oxidative stress through Nrf2/HO-1 pathway. Acta Cir. Bras. 2015, 30, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Kumaran, K.S.; Prince, P.S.M. Caffeic acid protects rat heart mitochondria against isoproterenol-induced oxidative damage. Cell Stress Chaperones 2010, 15, 791–806. [Google Scholar] [CrossRef] [Green Version]
- Camello-Almaraz, C.; Gomez-Pinilla, P.J.; Pozo, M.J.; Camello, P.J. Mitochondrial reactive oxygen species and Ca2+ signaling. Am. J. Physiol. Cell Physiol. 2006, 291, C1082–C1088. [Google Scholar] [CrossRef] [Green Version]
- Robert, F.F.; Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.-S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Chen, W.; Hu, R.; Feng, H. The Injury and Therapy of Reactive Oxygen Species in Intracerebral Hemorrhage Looking at Mitochondria. Oxidative Med. Cell. Longev. 2016, 2016, 2592935. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of Acute Kidney Injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef] [Green Version]
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The Effects of Cadmium Toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, C.; Ge, J.; Lv, M.-W.; Talukder, M.; Guo, K.; Li, Y.-H.; Li, J.-L. Ameliorative effects of resveratrol against cadmium-induced nephrotoxicity via modulating nuclear xenobiotic receptor response and PINK1/Parkin-mediated Mitophagy. Food Funct. 2020, 11, 1856–1868. [Google Scholar] [CrossRef]
- Liu, L.; Tao, R.; Huang, J.; He, X.; Qu, L.; Jin, Y.; Zhang, S.; Fu, Z. Hepatic oxidative stress and inflammatory responses with cadmium exposure in male mice. Environ. Toxicol. Pharmacol. 2015, 39, 229–236. [Google Scholar] [CrossRef]
- Karaca, S.; Eraslan, G. The Effects of Flaxseed Oil on Cadmium-Induced Oxidative Stress in Rats. Biol. Trace Elem. Res. 2013, 155, 423–430. [Google Scholar] [CrossRef]
- Cannino, G.; Ferruggia, E.; Rinaldi, A.M. Proteins participating to the post-transcriptional regulation of the mitochondrial cytochrome c oxidase subunit IV via elements located in the 3′UTR. Mitochondrion 2009, 9, 471–480. [Google Scholar] [CrossRef]
- Xu, S.; Pi, H.; Zhang, L.; Zhang, N.; Li, Y.; Zhang, H.; Tang, J.; Li, H.; Feng, M.; Deng, P.; et al. Melatonin prevents abnormal mitochondrial dynamics resulting from the neurotoxicity of cadmium by blocking calcium-dependent translocation of Drp1 to the mitochondria. J. Pineal Res. 2016, 60, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Pham, D.T.N.; Kim, Y.-M. Alternative strategies for the application of aminoglycoside antibiotics against the biofilm-forming human pathogenic bacteria. Appl. Microbiol. Biotechnol. 2020, 104, 1955–1976. [Google Scholar] [CrossRef]
- Walker, P.D.; Shah, S.V. Gentamicin enhanced production of hydrogen peroxide by renal cortical mitochondria. Am. J. Physiol. Physiol. 1987, 253, C495–C499. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.-Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepand, M.R.; Ghahremani, M.H.; Razavi-Azarkhiavi, K.; Aghsami, M.; Rajabi, J.; Keshavarz-Bahaghighat, H.; Soodi, M. Ellagic acid confers protection against gentamicin-induced oxidative damage, mitochondrial dysfunction and apoptosis-related nephrotoxicity. J. Pharm. Pharmacol. 2016, 68, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhong, X.; Yuan, H.; Guo, Y.; Song, D.; Qi, F.; Zhu, Z.; Wang, X.; Guo, Z. Interfering in apoptosis and DNA repair of cancer cells to conquer cisplatin resistance by platinum (iv) prodrugs. Chem. Sci. 2020, 11, 3829–3835. [Google Scholar] [CrossRef]
- Waseem, M.; Kaushik, P.; Parvez, S. Mitochondria-Mediated mitigatory role of curcumin in cisplatin-induced nephrotoxicity. Cell Biochem. Funct. 2013, 31, 678–684. [Google Scholar] [CrossRef]
- Kumar, M.; Dahiya, V.; Kasala, E.R.; Bodduluru, L.N.; Lahkar, M. The renoprotective activity of hesperetin in cisplatin induced nephrotoxicity in rats: Molecular and biochemical evidence. Biomed. Pharmacother. 2017, 89, 1207–1215. [Google Scholar] [CrossRef]
- Szewczyk, A. Mitochondria as a Pharmacological Target. Pharmacol. Rev. 2002, 54, 101–127. [Google Scholar] [CrossRef] [Green Version]
- Sung, M.J.; Kim, D.H.; Jung, Y.J.; Kang, K.P.; Lee, A.S.; Lee, S.; Kim, W.; Davaatseren, M.; Hwang, J.-T.; Kim, H.-J.; et al. Genistein protects the kidney from cisplatin-induced injury. Kidney Int. 2008, 74, 1538–1547. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Beltrán, C.E.; Mukhopadhyay, P.; Horváth, B.; Rajesh, M.; Tapia, E.; García-Torres, I.; Pedraza-Chaverri, J.; Pacher, P. Sulforaphane, a natural constituent of broccoli, prevents cell death and inflammation in nephropathy. J. Nutr. Biochem. 2012, 23, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, W.; Sun, X.; Wang, Y.; Zhou, M. Kaempferol ameliorates Cisplatin induced nephrotoxicity by modulating oxidative stress, inflammation and apoptosis via ERK and NF-κB pathways. AMB Express 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Ciarcia, R.; Damiano, S.; Florio, A.; Spagnuolo, M.; Zacchia, E.; Squillacioti, C.; Mirabella, N.; Florio, S.; Pagnini, U.; Garofano, T.; et al. The Protective Effect of Apocynin on Cyclosporine A-Induced Hypertension and Nephrotoxicity in Rats. J. Cell. Biochem. 2015, 116, 1848–1856. [Google Scholar] [CrossRef]
- Tedesco, D.; Haragsim, L. Cyclosporine: A Review. J. Transplant. 2012, 2012, 230386. [Google Scholar] [CrossRef] [Green Version]
- Niemann, C.U.; Saeed, M.; Akbari, H.; Jacobsen, W.; Benet, L.Z.; Christians, U.; Serkova, N.; Saeed, M. Close Association Between the Reduction in Myocardial Energy Metabolism and Infarct Size: Dose-Response Assessment of Cyclosporine. J. Pharmacol. Exp. Ther. 2002, 302, 1123–1128. [Google Scholar] [CrossRef] [Green Version]
- Serkova, N.; Jacobsen, W.; Niemann, C.U.; Litt, L.; Benet, L.Z.; Leibfritz, D.; Christians, U. Sirolimus, but not the structurally related RAD (everolimus), enhances the negative effects of cyclosporine on mitochondrial metabolism in the rat brain. J. Cereb. Blood Flow Metab. 2001, 133, 875–885. [Google Scholar] [CrossRef]
- Serkova, N.; Klawitter, J.; Niemann, C.U. Organ-Specific response to inhibition of mitochondrial metabolism by cyclosporine in the rat. Transpl. Int. 2003, 16, 748–755. [Google Scholar] [CrossRef]
- Rehman, H.; Krishnasamy, Y.; Haque, K.; Thurman, R.G.; Lemasters, J.J.; Schnellmann, R.G.; Zhong, Z. Green Tea Polyphenols Stimulate Mitochondrial Biogenesis and Improve Renal Function after Chronic Cyclosporin A Treatment in Rats. PLoS ONE 2013, 8, e65029. [Google Scholar] [CrossRef]
- Moon, D.; Kim, J. Cyclosporin A aggravates hydrogen peroxide-induced cell death in kidney proximal tubule epithelial cells. Anat. Cell Biol. 2019, 52, 312–323. [Google Scholar] [CrossRef]
- Wu, Q.; Li, W.; Zhao, J.; Sun, W.; Yang, Q.; Chen, C.; Xia, P.; Zhu, J.; Zhou, Y.; Huang, G.; et al. Apigenin ameliorates doxorubicin-induced renal injury via inhibition of oxidative stress and inflammation. Biomed. Pharmacother. 2021, 137, 111308. [Google Scholar] [CrossRef]
- Alagal, R.I.; AlFaris, N.A.; Alshammari, G.M.; Altamimi, J.Z.; AlMousa, L.A.; Yahya, M.A. Kaempferol attenuates doxorubicin-mediated nephropathy in rats by activating SIRT1 signaling. J. Funct. Foods 2021, 89, 104918. [Google Scholar] [CrossRef]
- Kocahan, S.; Dogan, Z.; Erdemli, E.; Taskin, E. Protective Effect of Quercetin against Oxidative Stress-Induced Toxicity Associated with Doxorubicin and Cyclophosphamide in Rat Kidney and Liver Tissue. Iran. J. Kidney Dis. 2017, 11, 124–131. [Google Scholar] [PubMed]
- Chénais, B.; Andriollo, M.; Guiraud, P.; Belhoussine, R.; Jeannesson, P. Oxidative stress involvement in chemically induced differentiation of K562 cells. Free. Radic. Biol. Med. 2000, 28, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Sutariya, B.; Saraf, M. α-asarone reduce proteinuria by restoring antioxidant enzymes activities and regulating necrosis factor κB signaling pathway in doxorubicin-induced nephrotic syndrome. Biomed. Pharmacother. 2017, 98, 318–324. [Google Scholar] [CrossRef]
- Hekmat, A.S.; Chenari, A.; Alipanah, H.; Javanmardi, K. Protective effect of alamandine on doxorubicin-induced nephrotoxicity in rats. BMC Pharmacol. Toxicol. 2021, 22, 31. [Google Scholar] [CrossRef]
- Pedrycz, A.; Czerny, K. Immunohistochemical study of proteins linked to apoptosis in rat fetal kidney cells following prepregnancy adriamycin administration in the mother. Acta Histochem. 2008, 110, 519–523. [Google Scholar] [CrossRef]
- Pedrycz, A.; Wieczorski, M.; Czerny, K. Late effects of adriamycin single dose on fetal rat kidney—Ultrastructural assessment. Environ. Toxicol. Pharmacol. 2005, 20, 157–160. [Google Scholar] [CrossRef]
- de Zeeuw, D.; Remuzzi, G.; Parving, H.-H.; Keane, W.F.; Zhang, Z.; Shahinfar, S.; Snapinn, S.; Cooper, M.E.; Mitch, W.E.; Brenner, B.M. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: Lessons from RENAAL. Kidney Int. 2004, 65, 2309–2320. [Google Scholar] [CrossRef] [Green Version]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, B.K.; Pandey, K.B.; Abidi, A.B.; Rizvi, S.I. Markers of Oxidative Stress during Diabetes Mellitus. J. Biomark. 2013, 2013, 378790. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative Stress as a Major Culprit in Kidney Disease in Diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef] [Green Version]
- A Nath, K.; Norby, S.M. Reactive oxygen species and acute renal failure. Am. J. Med. 2000, 109, 665–678. [Google Scholar] [CrossRef]
- Fernandes, S.M.; Cordeiro, P.M.; Watanabe, M.; da Fonseca, C.D.; Vattimo, M.D.F.F. The role of oxidative stress in streptozotocin-induced diabetic nephropathy in rats. Arch. Endocrinol. Metab. 2016, 60, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.; Suchal, K.; Khan, S.I.; Bhatia, J.; Kishore, K.; Dinda, A.K.; Arya, D.S. Apigenin ameliorates streptozotocin-induced diabetic nephropathy in rats via MAPK-NF-κB-TNF-α and TGF-β1-MAPK-fibronectin pathways. Am. J. Physiol. Physiol. 2017, 313, F414–F422. [Google Scholar] [CrossRef] [Green Version]
- Altamimi, J.Z.; AlFaris, N.A.; Alshammari, G.M.; Alagal, R.I.; Aljabryn, D.H.; Aldera, H.; Alrfaei, B.M.; Alkhateeb, M.A.; Yahya, M.A. Ellagic acid protects against diabetic nephropathy in rats by regulating the transcription and activity of Nrf2. J. Funct. Foods 2021, 79, 104397. [Google Scholar] [CrossRef]
- Abdou, H.M.; Elkader, H.-T.A.E.A. The potential therapeutic effects of Trifolium alexandrinum extract, hesperetin and quercetin against diabetic nephropathy via attenuation of oxidative stress, inflammation, GSK-3β and apoptosis in male rats. Chem. Interact. 2021, 352, 109781. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Cai, X.; Dai, X.; Ding, Y.; Jiang, Y.; Li, Y.; Zhang, Z.; Li, Y. Grape seed proanthocyanidin extracts ameliorate podocyte injury by activating peroxisome proliferator-activated receptor-γ coactivator 1α in low-dose streptozotocin-and high-carbohydrate/high-fat diet-induced diabetic rats. Food Funct. 2014, 5, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Bankova, V.; Trusheva, B.; Popova, M. Caffeic Acid Phenethyl Ester (CAPE)—Natural Sources, Analytical Procedures and Synthetic Approaches. Comptes Rendus Lacademie Bulg. Sci. 2018, 71, 1157–1169. [Google Scholar] [CrossRef]
- Zhang, P.; Tang, Y.; Li, N.-G.; Zhu, Y.; Duan, J.-A. Bioactivity and Chemical Synthesis of Caffeic Acid Phenethyl Ester and Its Derivatives. Molecules 2014, 19, 16458–16476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akyol, S.; Ugurcu, V.; Altuntas, A.; Hasgul, R.; Cakmak, O.; Akyol, O. Caffeic Acid Phenethyl Ester as a Protective Agent against Nephrotoxicity and/or Oxidative Kidney Damage: A Detailed Systematic Review. Sci. World J. 2014, 2014, 561971. [Google Scholar] [CrossRef] [Green Version]
- Erdemli, H.K.; Akyol, S.; Armutcu, F.; Gulec, M.A.; Canbal, M.; Akyol, O. Melatonin and caffeic acid phenethyl ester in the regulation of mitochondrial function and apoptosis: The basis for future medical approaches. Life Sci. 2016, 148, 305–312. [Google Scholar] [CrossRef]
- Ozeren, M.; Sucu, N.; Tamer, L.; Aytacoglu, B.; Bayri, O.; Dondas, A.; Ayaz, L.; Dikmengil, M. Caffeic acid phenethyl ester (CAPE) supplemented St. Thomas’ hospital cardioplegic solution improves the antioxidant defense system of rat myocardium during ischemia-reperfusion injury. Pharmacol. Res. 2005, 52, 258–263. [Google Scholar] [CrossRef]
- Migliori, M.; Cantaluppi, V.; Mannari, C.; Bertelli, A.A.E.; Medica, D.; Quercia, A.D.; Navarro, V.; Scatena, A.; Giovannini, L.; Biancone, L.; et al. Caffeic Acid, a Phenol Found in White Wine, Modulates Endothelial Nitric Oxide Production and Protects from Oxidative Stress-Associated Endothelial Cell Injury. PLoS ONE 2015, 10, e0117530. [Google Scholar] [CrossRef]
- Teixeira, J.; Deus, C.M.; Borges, F.; Oliveira, P.J. Mitochondria: Targeting mitochondrial reactive oxygen species with mitochondriotropic polyphenolic-based antioxidants. Int. J. Biochem. Cell Biol. 2018, 97, 98–103. [Google Scholar] [CrossRef]
- Lu, M.; Li, H.; Liu, W.; Zhang, X.; Li, L.; Zhou, H. Curcumin attenuates renal interstitial fibrosis by regulating autophagy and retaining mitochondrial function in unilateral ureteral obstruction rats. Basic Clin. Pharmacol. Toxicol. 2020, 128, 594–604. [Google Scholar] [CrossRef]
- Shakeri, A.; Cicero, A.F.G.; Panahi, Y.; Mohajeri, M.; Sahebkar, A. Curcumin: A naturally occurring autophagy modulator. J. Cell. Physiol. 2019, 234, 5643–5654. [Google Scholar] [CrossRef]
- Avila-Rojas, S.H.; Lira-León, A.; Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Pedraza-Chaverri, J. Role of Autophagy on Heavy Metal-Induced Renal Damage and the Protective Effects of Curcumin in Autophagy and Kidney Preservation. Medicina 2019, 55, 360. [Google Scholar] [CrossRef] [Green Version]
- Negrette-Guzmán, M.; García-Niño, W.R.; Tapia, E.; Zazueta, C.; Huerta-Yepez, S.; León-Contreras, J.C.; Hernández-Pando, R.; Aparicio-Trejo, O.E.; Madero, M.; Pedraza-Chaverri, J. Curcumin Attenuates Gentamicin-Induced Kidney Mitochondrial Alterations: Possible Role of a Mitochondrial Biogenesis Mechanism. Evid. Based Complement. Altern. Med. 2015, 2015, 917435. [Google Scholar] [CrossRef]
- Iglesias, D.E.; Cremonini, E.; Oteiza, P.I.; Fraga, C.G. Curcumin Mitigates TNFα-Induced Caco-2 Cell Monolayer Permeabilization Through Modulation of NF-κB, ERK1/2, and JNK Pathways. Mol. Nutr. Food Res. 2022, 66, 2101033. [Google Scholar] [CrossRef]
- Ghosh, S.; Banerjee, S.; Sil, P.C. The beneficial role of curcumin on inflammation, diabetes and neurodegenerative disease: A recent update. Food Chem. Toxicol. 2015, 83, 111–124. [Google Scholar] [CrossRef]
- Liu, F.-H.; Ni, W.-J.; Wang, G.-K.; Zhang, J.-J. Protective role of curcumin on renal ischemia reperfusion injury via attenuating the inflammatory mediators and Caspase-3. Cell. Mol. Biol. 2016, 62, 95–99. [Google Scholar] [PubMed]
- Avila-Rojas, S.H.; Aparicio-Trejo, O.E.; Briones-Herrera, A.; Medina-Campos, O.N.; Reyes-Fermín, L.M.; Martínez-Klimova, E.; León-Contreras, J.C.; Hernández-Pando, R.; Tapia, E.; Pedraza-Chaverri, J. Alterations in mitochondrial homeostasis in a potassium dichromate model of acute kidney injury and their mitigation by curcumin. Food Chem. Toxicol. 2020, 145, 111774. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. Punctum on two different transcription factors regulated by PGC-1α: Nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 4137–4146. [Google Scholar] [CrossRef]
- Liu, H.; Li, S.; Liu, X.; Chen, Y.; Deng, H. SIRT3 Overexpression Inhibits Growth of Kidney Tumor Cells and Enhances Mitochondrial Biogenesis. J. Proteome Res. 2018, 17, 3143–3152. [Google Scholar] [CrossRef]
- Ridzuan, N.R.A.; Rashid, N.A.; Othman, F.; Budin, S.B.; Hussan, F.; Teoh, S.L. Protective Role of Natural Products in Cisplatin-Induced Nephrotoxicity. Mini-Rev. Med. Chem. 2019, 19, 1134–1143. [Google Scholar] [CrossRef]
- Ortega-Domínguez, B.; Aparicio-Trejo, O.E.; García-Arroyo, F.E.; León-Contreras, J.C.; Tapia, E.; Molina-Jijón, E.; Hernández-Pando, R.; Sanchez-Lozada, L.-G.; Barrera-Oviedo, D.; Pedraza-Chaverri, J. Curcumin prevents cisplatin-induced renal alterations in mitochondrial bioenergetics and dynamic. Food Chem. Toxicol. 2017, 107, 373–385. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Xu, J.; Lu, Y.; Jiang, J.; Wang, L.; Shen, H.-M.; Xia, D. Curcumin targets the TFEB-lysosome pathway for induction of autophagy. Oncotarget 2016, 7, 75659–75671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Jijón, E.; Aparicio-Trejo, O.E.; Rodriguez-Munoz, R.; León-Contreras, J.C.; Cárdenas-Aguayo, M.D.C.; Medina-Campos, O.N.; Tapia, E.; Sanchez-Lozada, L.-G.; Hernández-Pando, R.; Reyes, J.L.; et al. The nephroprotection exerted by curcumin in maleate-induced renal damage is associated with decreased mitochondrial fission and autophagy. BioFactors 2016, 42, 686–702. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3–dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarenga, L.D.A.; Leal, V.D.O.; Borges, N.A.; de Aguiar, A.S.; Faxén-Irving, G.; Stenvinkel, P.; Lindholm, B.; Mafra, D. Curcumin—A promising nutritional strategy for chronic kidney disease patients. J. Funct. Foods 2017, 40, 715–721. [Google Scholar] [CrossRef]
- Malavolta, M.; Pierpaoli, E.; Giacconi, R.; Costarelli, L.; Piacenza, F.; Basso, A.; Cardelli, M.; Provinciali, M. Pleiotropic Effects of Tocotrienols and Quercetin on Cellular Senescence: Introducing the Perspective of Senolytic Effects of Phytochemicals. Curr. Drug Targets 2016, 17, 447–459. [Google Scholar] [CrossRef]
- Roumeliotis, S.; Roumeliotis, A.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Dietary Antioxidant Supplements and Uric Acid in Chronic Kidney Disease: A Review. Nutrients 2019, 11, 1911. [Google Scholar] [CrossRef] [Green Version]
- Renugadevi, J.; Prabu, S.M. Quercetin protects against oxidative stress-related renal dysfunction by cadmium in rats. Exp. Toxicol. Pathol. 2010, 62, 471–481. [Google Scholar] [CrossRef]
- Ko, C.-C.; Chen, Y.-J.; Chen, C.-T.; Liu, Y.-C.; Cheng, F.-C.; Hsu, K.-C.; Chow, L.-P. Chemical Proteomics Identifies Heterogeneous Nuclear Ribonucleoprotein (hnRNP) A1 as the Molecular Target of Quercetin in Its Anti-cancer Effects in PC-3 Cells. J. Biol. Chem. 2014, 289, 22078–22089. [Google Scholar] [CrossRef] [Green Version]
- Symonowicz, M.; Kolanek, M. Flavonoids and Their Properties to form Chelate Complexes; Lodz University of Technology Repository: Łódź, Poland, 2012. [Google Scholar]
- Padma, V.V.; Baskaran, R.; Roopesh, R.S.; Poornima, P. Quercetin attenuates lindane induced oxidative stress in wistar rats. Mol. Biol. Rep. 2012, 39, 6895–6905. [Google Scholar] [CrossRef]
- Liu, T.; Yang, Q.; Zhang, X.; Qin, R.; Shan, W.; Zhang, H.; Chen, X. Quercetin alleviates kidney fibrosis by reducing renal tubular epithelial cell senescence through the SIRT1/PINK1/mitophagy axis. Life Sci. 2020, 257, 118116. [Google Scholar] [CrossRef]
- Aoi, W.; Niisato, N.; Miyazaki, H.; Marunaka, Y. Flavonoid-Induced reduction of ENaC expression in the kidney of Dahl salt-sensitive hypertensive rat. Biochem. Biophys. Res. Commun. 2004, 315, 892–896. [Google Scholar] [CrossRef]
- Zhang, D.; Li, S.; Cruz, P.; Kone, B.C. Sirtuin 1 Functionally and Physically Interacts with Disruptor of Telomeric Silencing-1 to Regulate α-ENaC Transcription in Collecting Duct. J. Biol. Chem. 2009, 284, 20917–20926. [Google Scholar] [CrossRef] [Green Version]
- Ashkar, F.; Eftekhari, M.H.; Tanideh, N.; Koohpeyma, F.; Mokhtari, M.; Irajie, C.; Iraji, A. Effect of hydroalcoholic extract of Berberis integerrima and resveratrol on ovarian morphology and biochemical parameters in Letrozole-induced polycystic ovary syndrome rat model: An experimental study. Int. J. Reprod. Biomed. (IJRM) 2020, 18, 637. [Google Scholar] [CrossRef]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Jang, I.A.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Resveratrol, an Nrf2 activator, ameliorates aging-related progressive renal injury. Aging 2018, 10, 83–99. [Google Scholar] [CrossRef] [Green Version]
- Sack, M.N.; Finkel, T. Mitochondrial Metabolism, Sirtuins, and Aging. Cold Spring Harb. Perspect. Biol. 2012, 4, a013102. [Google Scholar] [CrossRef] [Green Version]
- Danz, E.D.B.; Skramsted, J.; Henry, N.; Bennett, J.A.; Keller, R.S. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free. Radic. Biol. Med. 2009, 46, 1589–1597. [Google Scholar] [CrossRef]
- Jang, I.-A.; Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Effects of resveratrol on the renin-angiotensin system in the aging kidney. Nutrients 2018, 10, 1741. [Google Scholar] [CrossRef] [Green Version]
- Albertoni, G.; Schor, N. Resveratrol plays important role in protective mechanisms in renal disease—Mini-Review. J. Bras. Nefrol. 2015, 37, 106–114. [Google Scholar] [CrossRef]
- Saldanha, J.F.; Leal, V.D.O.; Stenvinkel, P.; Carraro-Eduardo, J.C.; Mafra, D. Resveratrol: Why Is It a Promising Therapy for Chronic Kidney Disease Patients? Oxid. Med. Cell. Longev. 2013, 2013, 963217. [Google Scholar] [CrossRef]
- Kitada, M.; Kume, S.; Imaizumi, N.; Koya, D. Resveratrol Improves Oxidative Stress and Protects Against Diabetic Nephropathy Through Normalization of Mn-SOD Dysfunction in AMPK/SIRT1-Independent Pathway. Diabetes 2011, 60, 634–643. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Lim, J.H.; Youn, H.H.; Hong, Y.A.; Yang, K.S.; Park, H.S.; Chung, S.; Koh, S.H.; Shin, S.J.; Choi, B.S.; et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK–SIRT1–PGC1α axis in db/db mice. Diabetologia 2012, 56, 204–217. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Chi, Y.; Kang, Y.; Lu, H.; Niu, H.; Liu, W.; Li, Y. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1α mediated attenuation of mitochondrial oxidative stress. J. Cell. Physiol. 2018, 234, 5033–5043. [Google Scholar] [CrossRef]
- Grzesik, M.; Naparło, K.; Bartosz, G.; Sadowska-Bartosz, I. Antioxidant properties of catechins: Comparison with other antioxidants. Food Chem. 2018, 241, 480–492. [Google Scholar] [CrossRef]
- Crespy, V.; Williamson, G. A Review of the Health Effects of Green Tea Catechins in In Vivo Animal Models. J. Nutr. 2004, 134, 3431S–3440S. [Google Scholar] [CrossRef]
- Li, X.; Jiang, X.; Sun, J.; Zhu, C.; Li, X.; Tian, L.; Liu, L.; Bai, W. Cytoprotective effects of dietary flavonoids against cadmium-induced toxicity. Ann. N. Y. Acad. Sci. 2017, 1398, 5–19. [Google Scholar] [CrossRef]
- Zhang, T.; Mu, Y.; Yang, M.; Al Maruf, A.; Li, P.; Li, C.; Dai, S.; Lu, J.; Dong, Q. (+)-Catechin prevents methylglyoxal-induced mitochondrial dysfunction and apoptosis in EA. hy926 cells. Arch. Physiol. Biochem. 2016, 123, 121–127. [Google Scholar] [CrossRef]
- Silva Santos, L.F.; Stolfo, A.; Calloni, C.; Salvador, M. Catechin and epicatechin reduce mitochondrial dysfunction and oxidative stress induced by amiodarone in human lung fibroblasts. J. Arrhythmia 2016, 33, 220–225. [Google Scholar] [CrossRef]
- Gheysarzadeh, A.; Yazdanparast, R. STAT5 Reactivation by Catechin Modulates H2O2-Induced Apoptosis Through miR-182/FOXO1 Pathway in SK-N-MC Cells. Cell Biophys. 2014, 71, 649–656. [Google Scholar] [CrossRef]
- Shahid, A.; Ali, R.; Ali, N.; Hasan, S.K.; Bernwal, P.; Afzal, S.M.; Vafa, A.; Sultana, S. Modulatory effects of catechin hydrate against genotoxicity, oxidative stress, inflammation and apoptosis induced by benzo (a) pyrene in mice. Food Chem. Toxicol. 2016, 92, 64–74. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.R.; Nabavi, S.F.; Daglia, M.; Rastrelli, L. Epigallocatechin gallate and mitochondria—A story of life and death. Pharmacol. Res. 2016, 104, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, E.K.; Kelsey, N.A.; Doyle, J.; Breed, E.; Bouchard, R.J.; Loucks, F.A.; Harbison, R.A.; Linseman, D.A. Green Tea Epigallocatechin 3-Gallate Accumulates in Mitochondria and Displays a Selective Antiapoptotic Effect Against Inducers of Mitochondrial Oxidative Stress in Neurons. Antioxid. Redox Signal. 2009, 11, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Shankar, S.; Srivastava, R.K. Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, B.; Du, F.; Su, X.; Sun, G.; Zhou, G.; Bian, X.; Liu, N. Epigallocatechin-3-Gallate Attenuates Oxidative Stress and Inflammation in Obstructive Nephropathy via NF-κB and Nrf2/HO-1 Signalling Pathway Regulation. Basic Clin. Pharmacol. Toxicol. 2015, 117, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Peng, A. The Green Tea Polyphenol (−)-epigallocatechin-3-gallate and its beneficial roles in chronic kidney disease. J. Transl. Intern. Med. 2016, 4, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yu, J.F.; Zhao, C.G.; Sui, F.X.; Teng, X.; BIN Wu, Y. Therapeutic potential of EGCG on acute renal damage in a rat model of obstructive nephropathy. Mol. Med. Rep. 2013, 7, 1096–1102. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [Green Version]
- Sahin, K.; Tuzcu, M.; Gencoglu, H.; Dogukan, A.; Timurkan, M.; Sahin, N.; Aslan, A.; Kucuk, O. Epigallocatechin-3-gallate activates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Life Sci. 2010, 87, 240–245. [Google Scholar] [CrossRef]
- Pan, H.; Chen, J.; Shen, K.; Wang, X.; Wang, P.; Fu, G.; Meng, H.; Wang, Y.; Jin, B. Mitochondrial Modulation by Epigallocatechin 3-Gallate Ameliorates Cisplatin Induced Renal Injury through Decreasing Oxidative/Nitrative Stress, Inflammation and NF-kB in Mice. PLoS ONE 2015, 10, e0124775. [Google Scholar] [CrossRef] [Green Version]
- Hui, Y.; Zuo, X.Z.; Tian, C.; Liang, D.; Yi, W.J.; Chen, Z.; Zhang, P.W.; Ding, S.B.; Ying, C.J. Green tea polyphenols attenuate high-fat diet-induced renal oxidative stress through SIRT3-dependent deacetylation. Biomed. Environ. Sci. 2015, 28, 455–459. [Google Scholar]
- Devi, K.P.; Malar, D.S.; Nabavi, S.F.; Sureda, A.; Xiao, J.; Nabavi, S.M.; Daglia, M. Kaempferol and inflammation: From chemistry to medicine. Pharmacol. Res. 2015, 99, 1–10. [Google Scholar] [CrossRef]
- Calderon-Montaño, J.M.; Burgos-Morón, E.; Perez-Guerrero, C.; Lopez-Lazaro, M. A Review on the Dietary Flavonoid Kaempferol. Mini-Rev. Med. Chem. 2011, 11, 298–344. [Google Scholar] [CrossRef]
- Ali, A.S.; Almalki, A.S.; Alharthy, B.T. Effect of Kaempferol on Tacrolimus-Induced Nephrotoxicity and Calcineurin B1 Expression Level in Animal Model. J. Exp. Pharmacol. 2020, 12, 397–407. [Google Scholar] [CrossRef]
- Imran, M.; Rauf, A.; Shah, Z.A.; Saeed, F.; Imran, A.; Arshad, M.U.; Ahmad, B.; Bawazeer, S.; Atif, M.; Peters, D.G.; et al. Chemo-preventive and therapeutic effect of the dietary flavonoid kaempferol: A comprehensive review. Phytother. Res. 2018, 33, 263–275. [Google Scholar] [CrossRef]
- Luo, W.; Chen, X.; Ye, L.; Chen, X.; Jia, W.; Zhao, Y.; Samorodov, A.V.; Zhang, Y.; Hu, X.; Zhuang, F.; et al. Kaempferol attenuates streptozotocin-induced diabetic nephropathy by downregulating TRAF6 expression: The role of TRAF6 in diabetic nephropathy. J. Ethnopharmacol. 2020, 268, 113553. [Google Scholar] [CrossRef]
- Alshehri, A.S. Kaempferol attenuates diabetic nephropathy in streptozotocin-induced diabetic rats by a hypoglycaemic effect and concomitant activation of the Nrf-2/Ho-1/antioxidants axis. Arch. Physiol. Biochem. 2021, 127, 1–14. [Google Scholar] [CrossRef]
- Devi, S.A.; Chandrasekar, B.S.; Manjula, K.; Ishii, N. Grape seed proanthocyanidin lowers brain oxidative stress in adult and middle-aged rats. Exp. Gerontol. 2011, 46, 958–964. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, H.; Ramachandran, S.; Waypa, G.B.; Yin, J.-J.; Li, C.-Q.; Han, M.; Huang, H.-H.; Sharp, W.W.; Hoek, T.L.V.; et al. Grape Seed Proanthocyanidins Ameliorate Doxorubicin-Induced Cardiotoxicity. Am. J. Chin. Med. 2010, 38, 569–584. [Google Scholar] [CrossRef]
- Pajuelo, D.; Quesada, H.; Díaz, S.; Fernández-Iglesias, A.; Arola-Arnal, A.; Bladé, C.; Salvado, J.; Arola, L. Chronic dietary supplementation of proanthocyanidins corrects the mitochondrial dysfunction of brown adipose tissue caused by diet-induced obesity in Wistar rats. Br. J. Nutr. 2011, 107, 170–178. [Google Scholar] [CrossRef]
- Cheng, M.; Gao, H.-Q.; Xu, L.; Li, B.-Y.; Zhang, H.; Li, X.-H. Cardioprotective Effects of Grape Seed Proanthocyanidins Extracts in Streptozocin Induced Diabetic Rats. J. Cardiovasc. Pharmacol. 2007, 50, 503–509. [Google Scholar] [CrossRef]
- Karthikeyan, K.; Bai, B.S.; Devaraj, S.N. Grape seed proanthocyanidins ameliorates isoproterenol-induced myocardial injury in rats by stabilizing mitochondrial and lysosomal enzymes: An in vivo study. Life Sci. 2007, 81, 1615–1621. [Google Scholar] [CrossRef]
- Li, X.; Xu, L.; Gao, H.; Li, B.; Cheng, M. Effects of grape seed proanthocyanidins extracts on AGEs and expression of bone morphogenetic protein-7 in diabetic rats. J. Nephrol. 2008, 21, 722–733. [Google Scholar]
- Bao, L.; Zhang, Z.; Dai, X.; Ding, Y.; Jiang, Y.; Li, Y.; Li, Y. Effects of grape seed proanthocyanidin extract on renal injury in type 2 diabetic rats. Mol. Med. Rep. 2014, 11, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Kadye, R.; Kramer, A.H.; Joos-Vandewalle, J.; Parsons, M.; Njengele, Z.; Hoppe, H.; Prinsloo, E. Guardian of the Furnace: Mitochondria, TRAP1, ROS and stem cell maintenance. IUBMB Life 2013, 66, 42–45. [Google Scholar] [CrossRef]
- Rigotti, M.; Cerbaro, A.F.; Silva, I.D.R.D.; Agostini, F.; Branco, C.S.; Moura, S.; Salvador, M. Grape seed proanthocyanidins prevent H2O2-induced mitochondrial dysfunction and apoptosis via SIRT 1 activation in embryonic kidney cells. J. Food Biochem. 2020, 44, e13147. [Google Scholar] [CrossRef]
- Ding, Y.; Li, H.; Li, Y.; Liu, D.; Zhang, L.; Wang, T.; Liu, T.; Ma, L.; De La Puerta, R. Protective Effects of Grape Seed Proanthocyanidins on the Kidneys of Diabetic Rats through the Nrf2 Signalling Pathway. Evid. Based Complement. Altern. Med. 2020, 2020, 5205903. [Google Scholar] [CrossRef]
- Yousef, M.; Saad, A.; El-Shennawy, L. Protective effect of grape seed proanthocyanidin extract against oxidative stress induced by cisplatin in rats. Food Chem. Toxicol. 2009, 47, 1176–1183. [Google Scholar] [CrossRef]
- Wei, R.; Ding, R.; Tang, L.; Wang, Y. Grape Seed Proanthocyanidin Extract Reduces Renal Ischemia/Reperfusion Injuries in Rats. Am. J. Med. Sci. 2012, 343, 452–457. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, S.; Cheng, H.; Lv, H.; Cheng, G.; Ci, X. Nrf2-Mediated liver protection by esculentoside A against acetaminophen toxicity through the AMPK/Akt/GSK3β pathway. Free Radic. Biol. Med. 2016, 101, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Xu, Y.; Tang, L.; Yang, X.; Chen, Z.; Wei, Y.; Shao, X.; Shao, X.; Xin, Z.; Cai, B.; et al. Astragalus Polysaccharide Attenuates Cisplatin-Induced Acute Kidney Injury by Suppressing Oxidative Damage and Mitochondrial Dysfunction. BioMed Res. Int. 2020, 2020, 2851349. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, Z.; Wang, J.; Zhu, H. Antioxidative effects of hesperetin against lead acetate-induced oxidative stress in rats. Indian J. Pharmacol. 2013, 45, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, B.; Guo, F.; Li, Z.; Qin, G. Involvement of the TGFβ1- ILK-Akt signaling pathway in the effects of hesperidin in type 2 diabetic nephropathy. Biomed. Pharmacother. 2018, 105, 766–772. [Google Scholar] [CrossRef]
- Chen, X.; Wei, W.; Li, Y.; Huang, J.; Ci, X. Hesperetin relieves cisplatin-induced acute kidney injury by mitigating oxidative stress, inflammation and apoptosis. Chem. Interact. 2019, 308, 269–278. [Google Scholar] [CrossRef]
- Zhu, C.; Dong, Y.; Liu, H.; Ren, H.; Cui, Z. Hesperetin protects against H2O2-triggered oxidative damage via upregulation of the Keap1-Nrf2/HO-1 signal pathway in ARPE-19 cells. Biomed. Pharmacother. 2017, 88, 124–133. [Google Scholar] [CrossRef]
- Zhang, W.; Wei, R.; Zhang, L.; Tan, Y.; Qian, C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience 2017, 366, 95–104. [Google Scholar] [CrossRef]
- Rabb, H.; Griffin, M.D.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2015, 27, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Mutanen, M.; Pajari, A.-M.; Päivärinta, E.; Misikangas, M.; Rajakangas, J.; Marttinen, M.; Oikarinen, S. Berries as chemopreventive dietary constituents—A mechanistic approach with the ApcMin/+ mouse. Asia Pac. J. Clin. Nutr. 2008, 17, 123–125. [Google Scholar]
- Iino, T.; Ogawa, Y.; Tashima, K.; Kato, S.; Takeuchi, K. Less damaging effect of whisky in rat stomachs in comparison with pure ethanol: Role of ellagic acid, the nonalcoholic ingredient. Gastroenterology 2001, 120, A150. [Google Scholar] [CrossRef]
- Zhou, B.; Li, Q.; Wang, J.; Chen, P.; Jiang, S. Ellagic acid attenuates streptozocin induced diabetic nephropathy via the regulation of oxidative stress and inflammatory signaling. Food Chem. Toxicol. 2018, 123, 16–27. [Google Scholar] [CrossRef]
- Polce, S.A.; Burke, C.; França, L.M.; Kramer, B.; Paes, A.M.D.A.; Carrillo-Sepulveda, M.A. Ellagic Acid Alleviates Hepatic Oxidative Stress and Insulin Resistance in Diabetic Female Rats. Nutrients 2018, 10, 531. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, A.; Kulkarni, V.H.; Chakraborty, M.; Habbu, P.V.; Ray, A. Ellagic acid restored lead-induced nephrotoxicity by anti-inflammatory, anti-apoptotic and free radical scavenging activities. Heliyon 2021, 7, e05921. [Google Scholar] [CrossRef]
- Mohammed, E.T.; Hashem, K.S.; Abdelazem, A.Z.; Foda, F.A.M.A. Prospective Protective Effect of Ellagic Acid as a SIRT1 Activator in Iron Oxide Nanoparticle-Induced Renal Damage in Rats. Biol. Trace Elem. Res. 2020, 198, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Adelusi, T.I.; Du, L.; Hao, M.; Zhou, X.; Xuan, Q.; Apu, C.; Sun, Y.; Lu, Q.; Yin, X. Keap1/Nrf2/ARE signaling unfolds therapeutic targets for redox imbalanced-mediated diseases and diabetic nephropathy. Biomed. Pharmacother. 2020, 123, 109732. [Google Scholar] [CrossRef] [PubMed]
- Aslan, A.; Gok, O.; Beyaz, S.; Ağca, C.A.; Erman, O.; Zerek, A. Ellagic acid prevents kidney injury and oxidative damage via regulation of Nrf-2/NF-κB signaling in carbon tetrachloride induced rats. Mol. Biol. Rep. 2020, 47, 7959–7970. [Google Scholar] [CrossRef] [PubMed]
- Dizakar, S.A.; Saribas, G.S.; Tekcan, A. Effects of ellagic acid in the testes of streptozotocin induced diabetic rats. Drug Chem. Toxicol. 2021, 44, 1–8. [Google Scholar] [CrossRef]
- Lin, W.; Liu, G.; Kang, X.; Guo, P.; Shang, Y.; Du, R.; Wang, X.; Chen, L.; Yue, R.; Kong, F.; et al. Ellagic acid inhibits high glucose-induced injury in rat mesangial cells via the PI3K/Akt/FOXO3a signaling pathway. Exp. Ther. Med. 2021, 22, 1017. [Google Scholar] [CrossRef]
- Soto-Urquieta, M.G.; López-Briones, S.; Pérez-Vázquez, V.; Saavedra-Molina, A.; A González-Hernández, G.; Ramírez-Emiliano, J. Curcumin restores mitochondrial functions and decreases lipid peroxidation in liver and kidneys of diabetic db/db mice. Biol. Res. 2014, 47, 74. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yang, Y.; Zou, X.; Zheng, Z.; Zhang, J. Curcumin ameliorates CKD-induced mitochondrial dysfunction and oxidative stress through inhibiting GSK-3β activity. J. Nutr. Biochem. 2020, 83, 108404. [Google Scholar] [CrossRef]
- Fu, B.; Zhao, J.; Peng, W.; Wu, H.; Zhang, Y. Resveratrol rescues cadmium-induced mitochondrial injury by enhancing transcriptional regulation of PGC-1α and SOD2 via the Sirt3/FoxO3a pathway in TCMK-1 cells. Biochem. Biophys. Res. Commun. 2017, 486, 198–204. [Google Scholar] [CrossRef]
- Wang, H.; Guan, Y.; Widlund, A.L.; Becker, L.B.; Baur, J.A.; Reilly, P.M.; Sims, C.A. Resveratrol ameliorates mitochondrial dysfunction but increases the risk of hypoglycemia following hemorrhagic shock. J. Trauma Acute Care Surg. 2014, 77, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Gao, Y.; Zhang, Q.; Wei, S.; Chen, Z.; Dai, X.; Zeng, Z.; Zhao, K. SIRT1/3 activation by resveratrol attenuates acute kidney injury in a septic rat model. Oxidative Med. Cell. Longev. 2016, 2016, 7296092. [Google Scholar] [CrossRef]
- Wongmekiat, O.; Peerapanyasut, W.; Kobroob, A. Catechin supplementation prevents kidney damage in rats repeatedly exposed to cadmium through mitochondrial protection. Naunyn-Schmiedebergs Arch. Pharmacol. 2018, 391, 385–394. [Google Scholar] [CrossRef]
- Barnett, L.M.A.; Cummings, B.S. Cellular and Molecular Mechanisms of Kidney Toxicity. Semin. Nephrol. 2019, 39, 141–151. [Google Scholar] [CrossRef]
- Small, D.M.; Coombes, J.S.; Bennett, N.; Johnson, D.W.; Gobe, G.C. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology 2012, 17, 311–321. [Google Scholar] [CrossRef]
- Tábara, L.C.; Poveda, J.; Martin-Cleary, C.; Selgas, R.; Arduan, A.O.; Sanchez-Niño, M.D. Mitochondria-Targeted therapies for acute kidney injury. Expert Rev. Mol. Med. 2014, 16, e13. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, R.; Bosco, C. Oxidative stress and protective effects of polyphenols: Comparative studies in human and rodent kidney. A review. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2006, 142, 317–327. [Google Scholar] [CrossRef]
- Yeh, W.-J.; Hsia, S.-M.; Lee, W.-H.; Wu, C.-H. Polyphenols with antiglycation activity and mechanisms of action: A review of recent findings. J. Food Drug Anal. 2016, 25, 84–92. [Google Scholar] [CrossRef]
- Vargas, F.; Romecín, P.; Guillen, A.I.G.; Wangesteen, R.; Vargas-Tendero, P.; Paredes, M.D.; Atucha, N.M.; García-Estañ, J. Flavonoids in Kidney Health and Disease. Front. Physiol. 2018, 9, 394. [Google Scholar] [CrossRef] [Green Version]
- Virgili, F.; Marino, M. Regulation of cellular signals from nutritional molecules: A specific role for phytochemicals, beyond antioxidant activity. Free Radic. Biol. Med. 2008, 45, 1205–1216. [Google Scholar] [CrossRef]
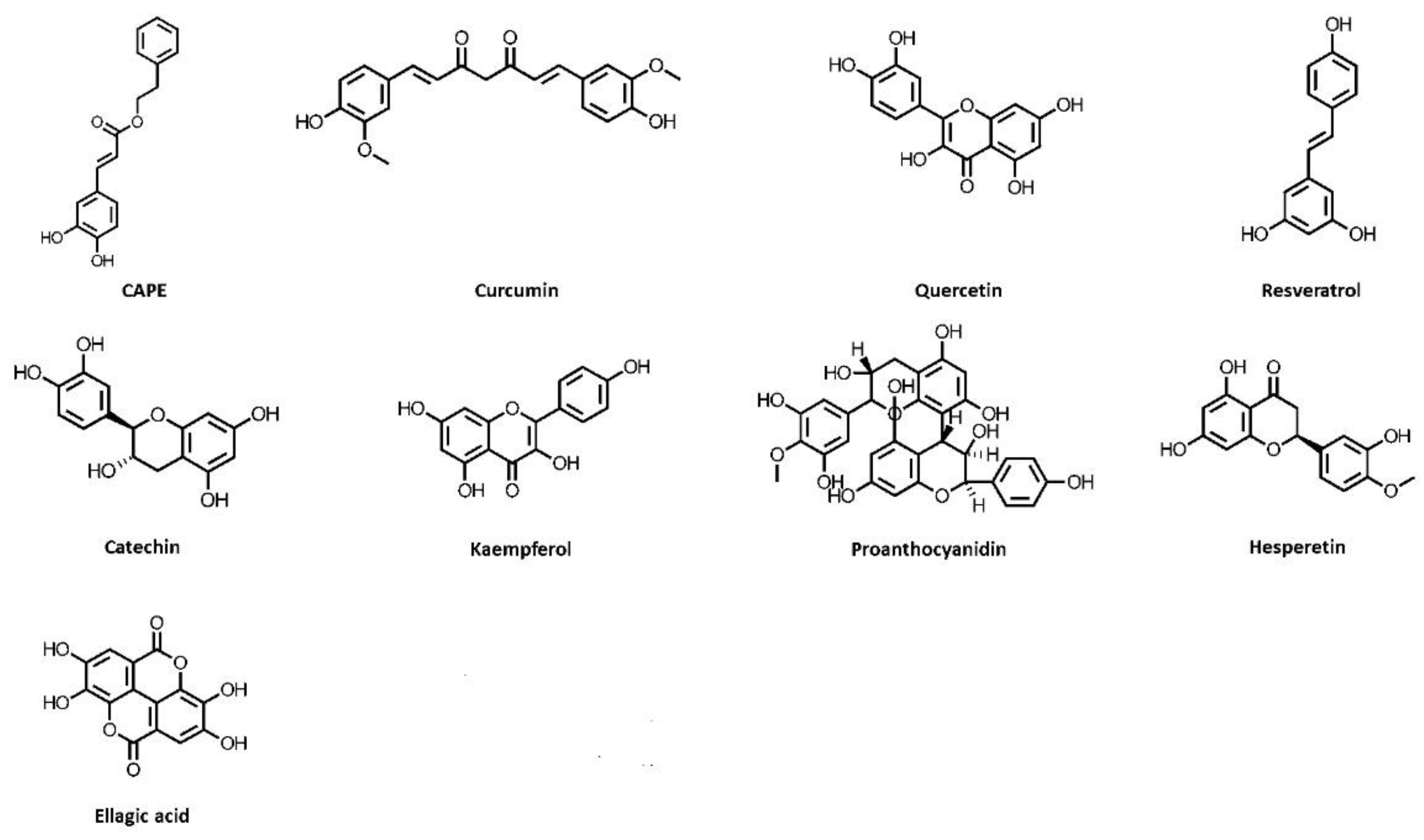
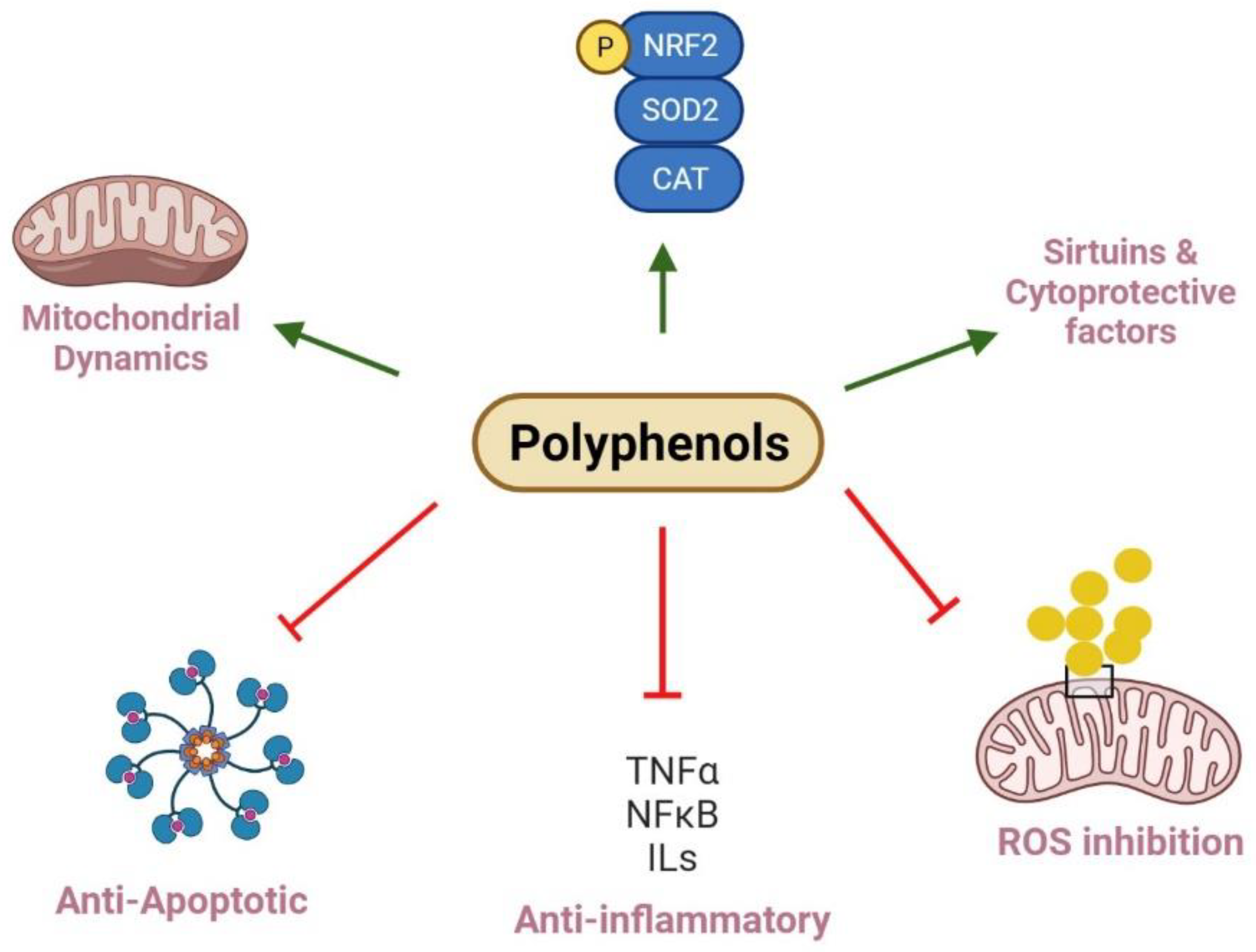
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).