

Activation of AMPK Entails the Protective Effect of Royal Jelly against High-Fat-Diet-Induced Hyperglycemia, Hyperlipidemia, and Non-Alcoholic Fatty Liver Disease in Rats
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Diets
2.3. Experimental Design
2.4. Oral Glucose Tolerance Test (OGTT)
2.5. Blood and Tissue Sampling
2.6. Hepatic and Stool Lipid Extraction and Preparation of Tissue Homogenates
2.7. Biochemical Analysis
2.8. Real-Time PCR
2.9. Western Blotting
2.10. Liver Histopathological Evaluation
2.11. Statistical Analysis
3. Results
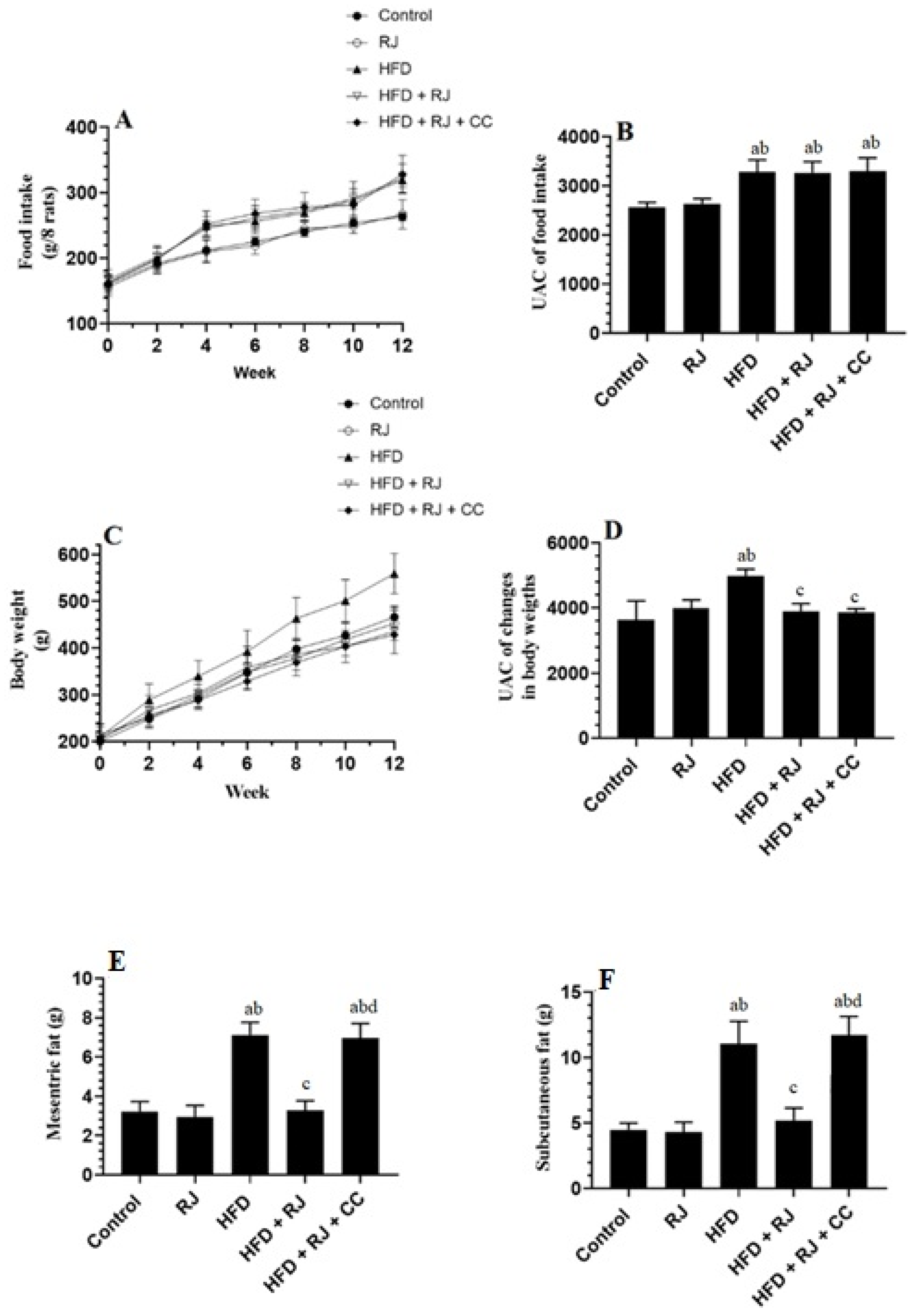
3.1. RJ Reduces Body and WAT Fat Weights with No Effect on Food Intake
3.2. RJ Attenuates Fasting Hyperglycemia, Insulinemia, and Insulin Resistance in HFD-Fed Rats
3.3. RJ Improves Adiponectin Levels and Ameliorates Systemic Inflammation and the Increase in Liver Marker Enzymes in HFD-Fed Rats
3.4. RJ Reverses Hyperlipidemia and Reduces Hepatic Lipid Levels in HFD-Fed Rats
3.5. RJ Attenuates Oxidative Stress and Inflammation and Improves Antioxidant Status in the Livers of HFD-Fed Rats
3.6. RJ Enhances the Activity (Phosphorylation) of AMPK and PPARα and Downregulates SREBP1c in the Livers of Control and HFD-Fed Rats
3.7. Histological Finding
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alswat, K.; Aljumah, A.A.; Sanai, F.M.; Abaalkhail, F.; Alghamdi, M.; Al Hamoudi, W.K.; Al Khathlan, A.; Al Quraishi, H.; Al Rifai, A.; Al Zaabi, M.; et al. Nonalcoholic fatty liver disease burden—Saudi Arabia and United Arab Emirates, 2017–2030. Saudi J. Gastroenterol. Off. J. Saudi Gastroenterol. Assoc. 2018, 24, 211–219. [Google Scholar]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, 47–64. [Google Scholar] [CrossRef] [Green Version]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Liu, W.; Baker, R.D.; Bhatia, T.; Zhu, L.; Baker, S.S. Pathogenesis of nonalcoholic steatohepatitis. Cell. Mol. Life Sci. 2016, 73, 1969–1987. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Kawada, N.; Japan Study Group of Nafld Jsg-Nafld. The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef]
- Zhang, C.H.; Zhou, B.G.; Sheng, J.Q.; Chen, Y.; Cao, Y.Q.; Chen, C. Molecular mechanisms of hepatic insulin resistance in nonalcoholic fatty liver disease and potential treatment strategies. Pharmacol. Res. 2020, 159, 104984. [Google Scholar] [CrossRef]
- Monserrat-Mesquida, M.; Quetglas-Llabrés, M.; Abbate, M.; Montemayor, S.; Mascaró, C.M.; Casares, M.; Tejada, S.; Abete, I.; Zulet, M.A.; Tur, J.A.; et al. Oxidative Stress and Pro-Inflammatory Status in Patients with Non-Alcoholic Fatty Liver Disease. Antioxidants 2020, 9, 759. [Google Scholar] [CrossRef]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 595371. [Google Scholar] [CrossRef] [PubMed]
- Golabi, P.; Bush, H.; Younossi, Z.M. Treatment Strategies for Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2017, 21, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.F. Nutrition Management Strategies for Nonalcoholic Fatty Liver Disease: Treatment and Prevention. Clin. Liver Dis. 2020, 15, 144–148. [Google Scholar] [CrossRef]
- Pydyn, N.; Miękus, K.; Jura, J.; Kotlinowski, J. New therapeutic strategies in nonalcoholic fatty liver disease: A focus on promising drugs for nonalcoholic steatohepatitis. Pharmacol. Rep. 2020, 72, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 2016, 311, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Hellberg, K.; Chaix, A.; Wallace, M.; Herzig, S.; Badur, M.G.; Lin, T.; Shokhirev, M.N.; Pinto, A.F.M.; Ross, D.S.; et al. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Rep. 2019, 26, 192–208.e6. [Google Scholar] [CrossRef] [Green Version]
- Strzyz, P. AMPK against NASH. Nat. Rev. Mol. Cell Biol. 2020, 21, 181. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; In Wong, K.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef] [PubMed]
- von Loeffelholz, C.; Coldewey, S.M.; Birkenfeld, A.L. A Narrative Review on the Role of AMPK on De Novo Lipogenesis in Non-Alcoholic Fatty Liver Disease: Evidence from Human Studies. Cells 2021, 10, 1822. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Pan, J.; Qu, N.; Lei, Y.; Han, J.; Zhang, J.; Han, D. The AMPK pathway in fatty liver disease. Front. Physiol. 2022, 13, 970292. [Google Scholar] [CrossRef]
- Maleki, V.; Jafari-Vayghan, H.; Saleh-Ghadimi, S.; Adibian, M.; Kheirouri, S.; Alizadeh, M. Effects of Royal jelly on metabolic variables in diabetes mellitus: A systematic review. Complement. Ther. Med. 2019, 43, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Balkanska, R.; Karadjova, I.; Ignatova, M. Comparative analyses of chemical composition of royal jelly and drone brood. Bulg. Chem. Commun. 2014, 46, 412–416. [Google Scholar]
- Tokunaga, K.H.; Yoshida, C.; Suzuki, K.M.; Maruyama, H.; Futamura, Y.; Araki, Y.; Mishima, S. Antihypertensive effect of peptides from royal jelly in spontaneously hypertensive rats. Biol. Pharm. Bull. 2004, 27, 189–192. [Google Scholar] [CrossRef] [Green Version]
- Viuda-Martos, M.; Pérez-Alvarez, J.A.; Fernández-López, J. Royal jelly: Health benefits and uses in medicine. In Bee Products-Chemical and Biological Properties; Springer: Cham, Switzerland, 2017; pp. 199–218. [Google Scholar]
- Kohno, K.; Okamoto, I.; Sano, O.; Arai, N.; Iwaki, K.; Ikeda, M.; Kurimoto, M. Royal jelly inhibits the production of proinflammatory cytokines by activated macrophages. Biosci. Biotechnol. Biochem. 2004, 68, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Saiga, A.; Sato, M.; Miyazawa, I.; Shibata, M.; Takahata, Y.; Morimatsu, F. Royal jelly supplementation improves lipoprotein metabolism in humans. J. Nutr. Sci. Vitaminol. 2007, 53, 345–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoshpey, B.; Djazayeri, S.; Amiri, F.; Malek, M.; Hosseini, A.F.; Hosseini, S.; Shidfar, S.; Shidfar, F. Effect of Royal Jelly Intake on Serum Glucose, Apolipoprotein A-I (ApoA-I), Apolipoprotein B (ApoB) and ApoB/ApoA-I Ratios in Patients with Type 2 Diabetes: A Randomized, Double-Blind Clinical Trial Study. Can. J. Diabetes 2016, 40, 324–328. [Google Scholar] [CrossRef]
- Yoshida, M.; Hayashi, K.; Watadani, R.; Okano, Y.; Tanimura, K.; Kotoh, J.; Sasaki, D.; Matsumoto, K.; Maeda, A. Royal jelly improves hyperglycemia in obese/diabetic KK-Ay mice. J. Vet. Med. Sci. 2017, 79, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, S.N.; Jazayeri, S.; Khoshpay, B.; Malek, M.; Hosseini, A.F.; Hosseini, S.; Shidfar, F. Royal jelly decreases blood pressure, serum glucose, and interleukin-6 in patients with type 2 diabetes on an iso-caloric diet. J. Nutr. Food Secur. 2017, 2, 300–307. [Google Scholar]
- Rezk, D.M. A comparative study on the effect of Royal jelly on blood glucose and serum lipids in streptozotocin induced diabetic rats. Eur. J. Pharm. Med. Res. 2018, 4, 39–44. [Google Scholar]
- National Research Council. Division on Earth and Life Studies; Institute for Laboratory Animal Research: Washington, DC, USA, 2011; p. 246. [Google Scholar]
- Van Herck, M.A.; Vonghia, L.; Francque, S.M. Animal Models of Nonalcoholic Fatty Liver Disease-A Starter’s Guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef] [Green Version]
- Yahya, M.A.; Alshammari, G.M.; Osman, M.A.; Al-Harbi, L.N.; Yagoub, A.E.A.; AlSedairy, S.A. Liquorice root extract and isoliquiritigenin attenuate high-fat diet-induced hepatic steatosis and damage in rats by regulating AMPK. Arch. Physiol. Biochem. 2022, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, E.; Nejati, V.; Azadbakht, M. Protective effect of royal jelly against renal damage in streptozotocin induced diabetic rats. Iran. J. Toxicol. 2015, 9, 1258–1263. [Google Scholar]
- Asgari, M.; Asle-Rousta, M.; Sofiabadi, M. Effect of royal jelly on blood glucose and lipids in streptozotocin induced type 1 diabetic rats. J. Arak Univ. Med. Sci. 2017, 20, 48–56. [Google Scholar]
- Nagy, C.; Einwallner, E. Study of in Vivo Glucose Metabolism in High-fat Diet-fed Mice Using Oral Glucose Tolerance Test (OGTT) and Insulin Tolerance Test (ITT). J. Vis. Exp. 2018, 131, 56672. [Google Scholar]
- Yoon, H.; Jeon, D.J.; Park, C.E.; You, H.S.; Moon, A.E. Relationship between homeostasis model assessment of insulin resistance and beta cell function and serum 25-hydroxyvitamin D in non-diabetic Korean adults. J. Clin. Biochem. Nutr. 2016, 59, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folch, J.; Lees, M.; Stanley, G.H.S. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Carnevali, L.C., Jr.; Eder, R.; Lira, F.S.; Lima, W.P.; Gonçalves, D.C.; Zanchi, N.E.; Nicastro, H.; Lavoie, J.M.; Seelaender, M.C. Effects of high-intensity intermittent training on carnitine palmitoyl transferase activity in the gastrocnemius muscle of rats. J. Med. Biol. Res. 2012, 45, 777–783. [Google Scholar] [CrossRef] [Green Version]
- Ghanbari, E.; Nejati, V.; Khazaei, M. Improvement in serum biochemical alterations and oxidative stress of liver and pancreas following use of royal jelly in streptozotocininduced diabetic rats. Cell J. (Yakhteh) 2016, 18, 362. [Google Scholar]
- Shidfar, F.; Jazayeri, S.; Mousavi, S.N.; Malek, M.; Hosseini, A.F.; Khoshpey, B. Does Supplementation with Royal Jelly Improve Oxidative Stress and Insulin Resistance in Type 2 Diabetic Patients? Iran. J. Public Health 2015, 44, 797–803. [Google Scholar]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Tangvarasittichai, S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J. Diabetes 2015, 6, 456–480. [Google Scholar] [CrossRef]
- Akash, M.S.; Rehman, K.; Chen, S. Role of inflammatory mechanisms in pathogenesis of type 2 diabetes mellitus. J. Cell. Biochem. 2013, 114, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lee, G.; Heo, S.Y.; Roh, Y.S. Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease. Antioxidants 2021, 11, 91. [Google Scholar] [CrossRef]
- Chiu, H.F.; Chen, B.K.; Lu, Y.Y.; Han, Y.C.; Shen, Y.C.; Venkatakrishnan, K.; Golovinskaia, O.; Wang, C.K. Hypocholesterolemic efficacy of royal jelly in healthy mild hypercholesterolemic adults. Pharm. Biol. 2016, 55, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Mobasseri, M.; Pourmoradian, S.; Mahdavi, R.; Faramarzi, E. Effects of royal jelly supplementation on lipid profile and high-sensitivity c-reactive protein levels in type-2 diabetic women: A pilot study. Curr. Top. Nutraceutical Res. 2014, 12, 101–105. [Google Scholar]
- Petelin, A.; Kenig, S.; Kopinč, R.; Deželak, M.; Černelič Bizjak, M.; Pražnikar, Z.J. Effects of Royal Jelly Administration on Lipid Profile, Satiety, Inflammation, and Antioxidant Capacity in Asymptomatic Overweight Adults. Evid. Based Complement. Altern. Med. 2019, 2019, 4969720. [Google Scholar] [CrossRef] [Green Version]
- You, M.M.; Chen, Y.F.; Pan, Y.M.; Liu, Y.C.; Tu, J.; Wang, K.; Hu, F.L. Royal Jelly Attenuates LPS-Induced Inflammation in BV-2 Microglial Cells through Modulating NF-κB and p38/JNK Signaling Pathways. Mediat. Inflamm. 2018, 2018, 7834381. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Sakai, H. Anti-Cancer and Protective Effects of Royal Jelly for Therapy-Induced Toxicities in Malignancies. Int. J. Mol. Sci. 2018, 19, 3270. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Hafez, S.M.N.; Rifaai, R.A.; Abdelzaher, W.Y. Possible protective effect of royal jelly against cyclophosphamide induced prostatic damage in male albino rats; a biochemical, histological and immuno-histo-chemical study. Biomed. Pharmacother. 2017, 90, 15–23. [Google Scholar] [CrossRef]
- Aslan, Z.; Aksoy, L. Anti-inflammatory effects of royal jelly on ethylene glycol induced renal inflammation in rats. Int. Braz. J. Urol. 2015, 41, 1008–1013. [Google Scholar] [CrossRef] [Green Version]
- Karaca, T.; Şimşek, N.; Uslu, S.; Kalkan, Y.; Can, I.; Kara, A.; Yörük, M. The effect of royal jelly on CD3(+), CD5(+), CD45(+) T-cell and CD68(+) cell distribution in the colon of rats with acetic acid-induced colitis. Allergol. Immunopathol. 2012, 40, 357–361. [Google Scholar] [CrossRef]
- Kocot, J.; Kiełczykowska, M.; Luchowska-Kocot, D.; Kurzepa, J.; Musik, I. Antioxidant Potential of Propolis, Bee Pollen, and Royal Jelly: Possible Medical Application. Oxid. Med. Cell. Longev. 2018, 2018, 7074209. [Google Scholar] [CrossRef]
- Gu, H.; Song, I.B.; Han, H.J.; Lee, N.Y.; Cha, J.Y.; Son, Y.K.; Kwon, J. Antioxidant Activity of Royal Jelly Hydrolysates Obtained by Enzymatic Treatment. Korean J. Food Sci. Anim. Resour. 2018, 38, 135–142. [Google Scholar]
- Guo, H.; Ekusa, A.; Iwai, K.; Yonekura, M.; Takahata, Y.; Morimatsu, F. Royal jelly peptides inhibit lipid peroxidation in vitro and in vivo. J. Nutr. Sci. Vitaminol. 2008, 54, 191–195. [Google Scholar] [CrossRef] [Green Version]
- Silici, S.; Ekmekcioglu, O.; Eraslan, G.; Demirtas, A. Antioxidative effect of royal jelly in cisplatin-induced testes damage. Urology 2009, 74, 545–551. [Google Scholar] [CrossRef]
- Cemek, M.; Aymelek, F.; Büyükokuroğlu, M.E.; Karaca, T.; Büyükben, A.; Yilmaz, F. Protective potential of Royal Jelly against carbon tetrachloride induced-toxicity and changes in the serum sialic acid levels. Food Chem. Toxicol. 2010, 48, 2827–2832. [Google Scholar] [CrossRef]
- Almeer, R.S.; AlBasher, G.I.; Alarifi, S.; Alkahtani, S.; Ali, D.; Abdel Moneim, A.E. Royal jelly attenuates cadmium-induced nephrotoxicity in male mice. Sci. Rep. 2019, 9, 5825. [Google Scholar] [CrossRef] [Green Version]
- You, M.M.; Liu, Y.C.; Chen, Y.F.; Pan, Y.M.; Miao, Z.N.; Shi, Y.Z.; Si, J.J.; Chen, M.L.; Hu, F.L. Royal jelly attenuates nonalcoholic fatty liver disease by inhibiting oxidative stress and regulating the expression of circadian genes in ovariectomized rats. J. Food Biochem. 2020, 44, e13138. [Google Scholar] [CrossRef]
- Zhu, Y.Y.; Meng, X.C.; Zhou, Y.J.; Zhu, J.X.; Chang, Y.N. Major royal jelly proteins alleviate non-alcoholic fatty liver disease in mice model by regulating disordered metabolic pathways. J. Food Biochem. 2022, 46, e14214. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Tsiaousi, E. The role of adiponectin in the pathogenesis and treatment of non-alcoholic fatty liver disease. Diabetes Obes. Metab. 2010, 12, 365–383. [Google Scholar] [CrossRef]
- Habegger, K.M.; Hoffman, N.J.; Ridenour, C.M.; Brozinick, J.T.; Elmendorf, J.S. AMPK enhances insulin-stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology 2012, 153, 2130–2141. [Google Scholar] [CrossRef]
- Jansen, T.; Kvandová, M.; Daiber, A.; Stamm, P.; Frenis, K.; Schulz, E.; Münzel, T.; Kröller-Schön, S. The AMP-Activated Protein Kinase Plays a Role in Antioxidant Defense and Regulation of Vascular Inflammation. Antioxidants 2020, 9, 525. [Google Scholar] [CrossRef]
- Ding, X.; Jian, T.; Li, J.; Lv, H.; Tong, B.; Li, J.; Meng, X.; Ren, B.; Chen, J. Chicoric Acid Ameliorates Nonalcoholic Fatty Liver Disease via the AMPK/Nrf2/NFκB Signaling Pathway and Restores Gut Microbiota in High-Fat-Diet-Fed Mice. Oxid. Med. Cell. Longev. 2020, 2020, 9734560. [Google Scholar] [CrossRef]
- Li, S.; Qian, Q.; Ying, N.; Lai, J.; Feng, L.; Zheng, S.; Jiang, F.; Song, Q.; Chai, H.; Dou, X. Activation of the AMPK-SIRT1 pathway contributes to protective effects of Salvianolic acid A against lipotoxicity in hepatocytes and NAFLD in mice. Front. Pharmacol. 2020, 11, 560905. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Control | RJ | HFD | HFD + RJ | HFD + RJ + CC |
|---|---|---|---|---|---|
| Final liver weight (g) | 4.6 ± 0.5 | 4.3 ± 0.42 | 6.7± 0.61 ab | 4.8 ± 0.38 c | 6.3 ± 0.82 abd |
| Adiponectin (µg/mL) | 39.4 ± 4.2 | 55.5 ± 5.7 a | 24.7 ± 3.8 ab | 39.8 ± 4.3 bc | 38.9 ± 4.7 abd |
| Leptin (ng/mL) | 22.4 ± 3.4 | 25.4 ± 4.1 | 56.3 ± 4.9 ab | 29.4 ± 3.7 abc | 49.3± 5.4 abd |
| Serum TNF-α (pg/mL) | 83.2 ± 7.8 | 91.2 ± 8.5 | 322 ± 19 ab | 125 ± 11.2 abc | 311± 28.2 abd |
| Serum IL-6 (pg/mL) | 46.3 ± 5.1 | 49.2 ± 5.3 | 123 ± 13.1 ab | 66.2 ± 6.2 abc | 118± 10.2 abd |
| Serum ALT (U/L) | 24.5 ± 3.8 | 27.3 ± 5.7 | 88.2 ± 5.5 ab | 32.3 ± 4.7 abc | 82.4 ± 7.5 abd |
| AST (U/L) | 35.4 ± 4.3 | 31.3 ± 4.9 | 102 ± 9.2 ab | 54.3 ± 5.4 abc | 110 ± 11.2 abd |
| γ-GTT (U/L) | 21.4 ± 4.5 | 25.6 ± 3.8 a | 69.3 ± 3.2 ab | 37.3 ± 4.1 abc | 73.2 ± 6.9 abd |
| Parameter | Control | RJ | HFD | HFD + RJ | HFD + RJ + CC | |
|---|---|---|---|---|---|---|
| Serum | TGs (mg/dL) | 43.3 ± 5.1 | 34.3 ± 3.2 a | 89.3 ± 7.8 ab | 47.6 ± 5.4 bc | 83.4 ± 6.5 abd |
| CHOL (mg/dL) | 84.3 ± 7.5 | 68.5 ± 5.3 a | 176 ± 13.4 ab | 81.1 ± 7.6 bc | 183 ± 15.6 abd | |
| LDL-c (mg/dL) | 43.2 ± 4.6 | 35.4 ± 3.5 a | 98.2 ± 6.5 ab | 54.5 ± 5.3 abc | 89.3 ± 7.3 abd | |
| HDL-c (mg/dL) | 22.4 ± 4.2 | 19.8± 3.7 | 12.2 ± 2.1 ab | 24.5 ± 0.67 abc | 14.3 ± 1.4 abd | |
| FFAs (µmol/L) | 882 ± 55.8 | 632± 43.7 a | 1632 ± 150.5 ab | 829 ± 77.2 bc | 1722 ± 125.2 abd | |
| Liver | TGs (mg/g) | 3.6 ± 0.4 | 2.43 ± 0.2 a | 7.6 ± 0.6 ab | 4.3 ± 0.5 abc | 7.9 ± 0.6 abd |
| CHOL (mg/dL) | 4.7 ± 0.5 | 3.5 ± 0.3 a | 8.6 ± 0.7 ab | 4.3 ± 0.5 abc | 7.9 ± 0.8 abd | |
| FFAs (µmol/L) | 245± 22.2 | 143± 13.2 a | 565 ± 43.4 ab | 251 ± 19.4 bc | 632 ± 46.7 abd | |
| Stool | TGs (mg/g dry) | 2.4 ± 0.35 | 2.6 ± 0.27 | 6.34 ± 0.73 ab | 5.9 ± 0.53 ab | 6.7 ± 0.74 ab |
| CHOL (mg/g dry) | 4.32 ± 0.53 | 4.02 ± 0.38 | 8.34 ± 0.82 ab | 7.7 ± 0.93 ab | 8.3 ± 0.7 ab |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Felemban, A.H.; Alshammari, G.M.; Yagoub, A.E.A.; Al-Harbi, L.N.; Alhussain, M.H.; Yahya, M.A. Activation of AMPK Entails the Protective Effect of Royal Jelly against High-Fat-Diet-Induced Hyperglycemia, Hyperlipidemia, and Non-Alcoholic Fatty Liver Disease in Rats. Nutrients 2023, 15, 1471. https://doi.org/10.3390/nu15061471
Felemban AH, Alshammari GM, Yagoub AEA, Al-Harbi LN, Alhussain MH, Yahya MA. Activation of AMPK Entails the Protective Effect of Royal Jelly against High-Fat-Diet-Induced Hyperglycemia, Hyperlipidemia, and Non-Alcoholic Fatty Liver Disease in Rats. Nutrients. 2023; 15(6):1471. https://doi.org/10.3390/nu15061471
Chicago/Turabian StyleFelemban, Alaa Hasanain, Ghedeir M. Alshammari, Abu ElGasim Ahmed Yagoub, Laila Naif Al-Harbi, Maha H. Alhussain, and Mohammed Abdo Yahya. 2023. "Activation of AMPK Entails the Protective Effect of Royal Jelly against High-Fat-Diet-Induced Hyperglycemia, Hyperlipidemia, and Non-Alcoholic Fatty Liver Disease in Rats" Nutrients 15, no. 6: 1471. https://doi.org/10.3390/nu15061471