
The Influence of Non-Pharmacological and Pharmacological Interventions on the Course of Autosomal Dominant Polycystic Kidney Disease

 , ,
, , 
Abstract
1. Introduction
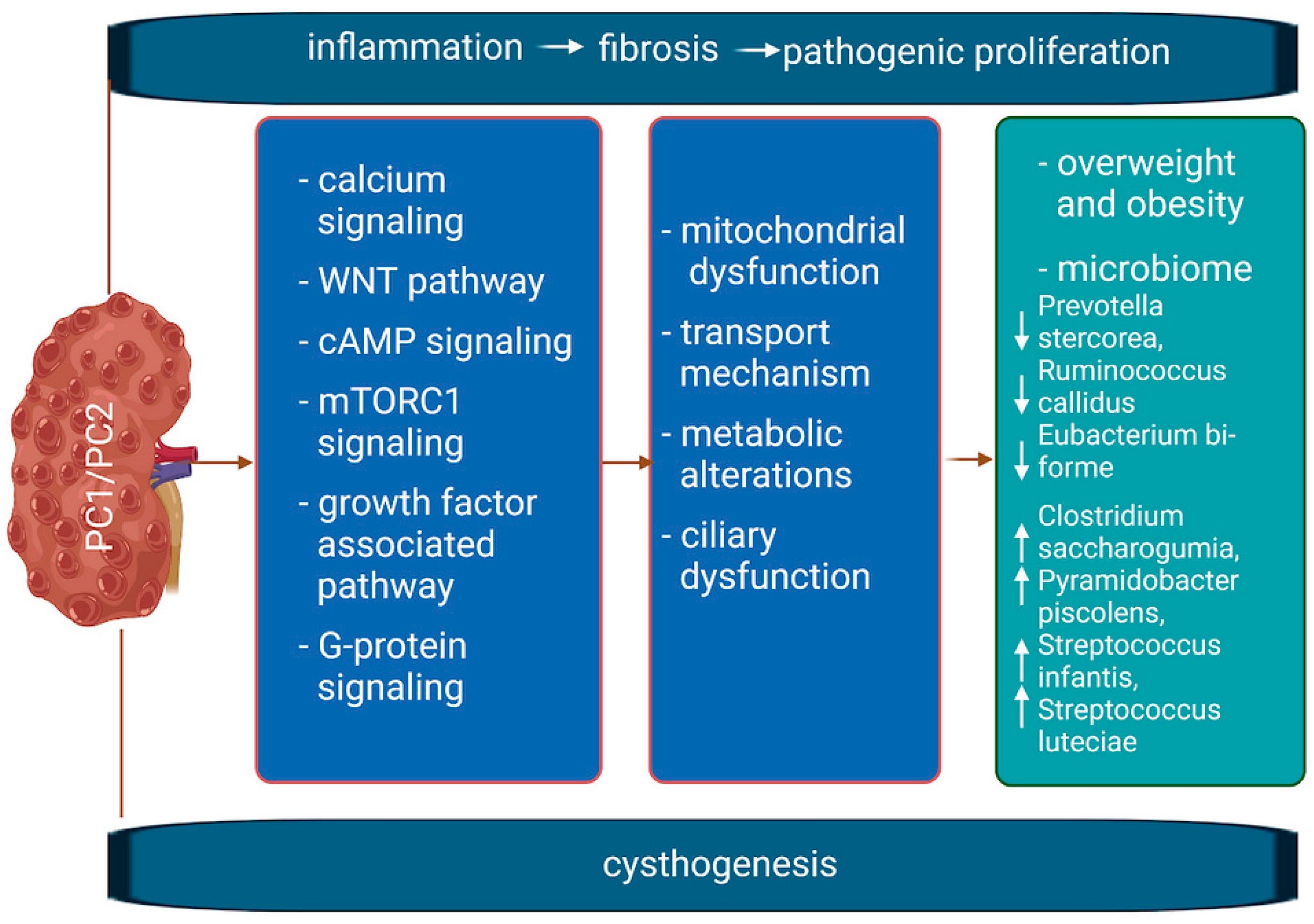
1.1. Molecular Mechanism of the Disease
1.2. Supplementation, Diet, Herbs, and Treatment in ADPKD
2. Materials and Methods
3. ADPKD, a Disease with Dysregulated Metabolism: In Vitro and Animal Model Studies
4. Metabolic Reprogramming: A Common Feature in Human ADPKD
4.1. Metabolic Pathway Changes in Overweight and Obesity: Parallel Metabolic Disturbances in ADPKD
4.2. Dietary Strategies to Address Metabolic Abnormalities
4.3. Nutritional Approaches Using Non-PKD Rodent Models
4.4. Calorie Restriction and Fasting Trials in Humans without ADPKD
4.5. Calorie Restriction and Nutrient Availability in ADPKD
5. Pharmacological Alternatives to Diet Changes Modulating Metabolic Disorders in ADPKD
5.1. Tolvaptan
5.2. AMPK Activators
5.3. Sodium-Glucose Cotransporter-2 Inhibitors
5.4. Niacinamide/Nicotinamide
5.5. Thiazolidinediones
5.6. Analogues and Agonists of Gut Hormones
5.6.1. Dual Agonists of GLP-1 and Glucagon Receptors
5.6.2. Dual Agonist for GLP-1 and GIP Receptors (Tirzepatide)
5.7. m TOR Inhibitors
6. Polycystic Kidney Disease and Microbiota
7. Recommendations
8. Conclusions, Limitations, and Future Directions
- ADPKD is linked to metabolic defects that contribute to cyst growth, with overlaps seen between ADPKD, obesity, and related conditions. Dietary and pharmacological strategies targeting these issues are explored as potential therapies.
- Metabolic reprogramming therapies show promise in slowing ADPKD progression, but more research is needed, especially for treatments such as GLP-1 analogs and dual agonists that target specific pathways.
- Dietary interventions have limitations, such as long-term adherence challenges, and weight loss is not suitable for everyone, requiring careful nutrition management.
- Combining metabolic reprogramming therapies with drugs such as tolvaptan could enhance treatment, reduce side effects, and minimize dietary restrictions, but further research is needed, especially regarding use in children.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Correction Statement
Abbreviations
References
- Ma, M. Cilia and polycystic kidney disease. Semin. Cell Dev. Biol. 2021, 110, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Ong, A.; Torra, A. Can ketogenic dietary interventions slow disease progression in ADPKD: What we know and what we don’t. Clin. Kidney J. 2022, 15, 1034–1036. [Google Scholar] [CrossRef] [PubMed]
- Rangan, G.K.; Tchan, M.C.; Tong, A.; Wong, A.T.Y.; Nankivell, B.J. Recent advances in autosomal-dominant polycystic kidney disease. Intern. Med. J. 2016, 46, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Carriazo, S.; Perez-Gomez, M.V.; Cordido, A.; García-González, M.A.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Dietary care for ADPKD patients: Current status and future directions. Nutrients 2019, 1, 1576. [Google Scholar] [CrossRef]
- Shibazaki, S.; Yu, Z.; Nishio, S.; Tian, X.; Thomson, R.B.; Mitobe, M.; Louvi, A.; Velazquez, H.; Ishibe, S.; Cantley, L.G.; et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum. Mol. Genet. 2008, 17, 1505–1516. [Google Scholar] [CrossRef]
- Liu, X.; Tang, J.; Chen, X.Z. Role of PKD2 in the endoplasmic reticulum calcium homeostasis. Front. Physiol. 2022, 13, 962571. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.X.; Ding, H.; Torres, V.E.; Yu, C.; Li, X. Ferroptosis Promotes Cyst Growth in Autosomal Dominant Polycystic Kidney Disease Mouse Models. J. Am. Soc. Nephrol. 2021, 32, 2759–2776. [Google Scholar] [CrossRef]
- Torres, J.A.; Kruger, S.L.; Broderick, C.; Amarlkhagva, T.; Agrawal, S.; Dodam, J.R.; Mrug, M.; Lyons, L.A.; Weimbs, T. Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease. Cell Metab. 2019, 30, 1007–1023.e5. [Google Scholar] [CrossRef]
- Sousa, M.V.; Amaral, A.G.; Freitas, J.A.; Murata, G.M.; Watanabe, E.H.; Balbo, B.E.; Tavares, M.D.; Hortegal, R.A.; Rocon, C.; Souza, L.E.; et al. Smoking accelerates renal cystic disease and worsens cardiac phenotype in Pkd1-deficient mice. Sci. Rep. 2021, 11, 14443. [Google Scholar] [CrossRef]
- Meijer, E.; Gansevoort, R.T. Emerging non-pharmacological interventions in ADPKD: An update on dietary advices for clinical practice. Curr. Opin. Nephrol. Hypertens. 2021, 30, 482–492. [Google Scholar] [CrossRef]
- Eroglu, E.; Kocyigit, I.; Cetin, M.; Zararsiz, G.; Imamoglu, H.; Bayramov, R.; Tastan, S.; Sipahioglu, M.H.; Tokgoz, B.; Oymak, O. Multiple urinary tract infections are associated with genotype and phenotype in adult polycystic kidney disease. Clin. Exp. Nephrol. 2019, 23, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Winterbottom, J.; Simms, R.J.; Caroli, A.; Gall, E.C.; Demoulin, N.; Furlano, M.; Meijer, E.; Devuyst, O.; Gansevoort, R.T.; Le-Meur, Y.; et al. Flank pain has a significant adverse impact on quality of life in ADPKD: The CYSTic-QoL study. Clin. Kidney J. 2022, 15, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Santoro, D.; Satta, E.; Messina, S.; Costantino, G.; Savica, V.; Bellinghieri, G. Pain in end-stage renal disease: A frequent and neglected clinical problem. Clin. Nephrol. 2013, 79 (Suppl. S1), S2–S11. [Google Scholar] [CrossRef]
- Yacoub, R.; Nadkarni, G.N.; McSkimming, D.I.; Chaves, L.D.; Abyad, S.; Bryniarski, M.A.; Honan, A.M.; Thomas, S.A.; Gowda, M.; He, J.C.; et al. Fecal microbiota analysis of polycystic kidney disease patients according to renal function: A pilot study. Exp. Biol. Med. 2019, 244, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Cukoski, S.; Lindemann, C.H.; Arjune, S.; Todorova, P.; Brecht, T.; Kühn, A.; Oehm, S.; Strubl, S.; Becker, I.; Kämmerer, U.; et al. Feasibility and impact of ketogenic dietary interventions in polycystic kidney disease: KETO-ADPKD—A randomized controlled trial. Cell Rep. Med. 2023, 4, 101283. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Hopp, K. Metabolic reprogramming in autosomal dominant polycystic kidney disease evidence and therapeutic potential. Clin. J. Am. Soc. Nephrol. 2020, 15, 577–584. [Google Scholar] [CrossRef]
- Chebib, F.T.; Nowak, K.L.; Chonchol, M.B.; Bing, K.; Ghanem, A.; Rahbari-Oskoui, F.F.; Dahl, N.K.; Mrug, M. Polycystic Kidney Disease Diet: What is Known and What is Safe. Clin. J. Am. Soc. Nephrol. 2024, 19, 664–682. [Google Scholar] [CrossRef]
- Grahammer, F.; Wanner, N.; Huber, T.B. mTOR controls kidney epithelia in health and disease. Nephrol. Dial. Transplant. 2014, 29 (Suppl. S1), i9–i18. [Google Scholar] [CrossRef]
- Di Iorio, B.R.; Cupisti, A.; D’Alessandro, C.; Bellasi, A.; Barbera, V.; Di Lullo, L. Nutritional therapy in autosomal dominant polycystic kidney disease. J. Nephrol. 2018, 31, 635–643. [Google Scholar] [CrossRef]
- Vendramini, L.C.; Dalboni, M.A.; de Carvalho, J.T.G., Jr.; Batista, M.C.; Nishiura, J.L.; Heilberg, I.P. Association of Vitamin D Levels With Kidney Volume in Autosomal Dominant Polycystic Kidney Disease (ADPKD). Front. Med. 2019, 6, 112. [Google Scholar] [CrossRef]
- Rangan, G.K.; Harris, D.C. Rationale and design of an observational study to determine the effects of cholecalciferol on hypertension, proteinuria and urinary MCP-1 in ADPKD. Curr. Hypertens. Rev. 2013, 9, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, J.; Yang, X.; Li, T.; Yang, B.; Aili, A. Combination of curcumin and ginkgolide B inhibits cystogenesis by regulating multiple signaling pathways. Mol. Med. Rep. 2021, 23, 195. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Farmer-Bailey, H.; Wang, W.; You, Z.; Steele, C.; Cadnapaphornchai, M.A.; Klawitter, J.; Patel, N.; George, D.; Jovanovich, A.; et al. Curcumin Therapy to Treat Vascular Dysfunction in Children and Young Adults with ADPKD: A Randomized Controlled Trial. Clin. J. Am. Soc. Nephrol. 2022, 17, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Xu, D.; Gu, J.; Xue, C.; Yang, B.; Fu, L.; Song, S.; Liu, D.; Zhou, W.; Lv, J.; et al. Saikosaponin-d inhibits proliferation by up-regulating autophagy via the CaMKKbeta-AMPK-mTOR pathway in ADPKD cells. Mol. Cell Biochem. 2018, 449, 219–226. [Google Scholar] [CrossRef]
- Capelli, I.; Lerario, S.; Aiello, V.; Provenzano, M.; Di Costanzo, R.; Squadrani, A.; Vella, A.; Vicennati, V.; Poli, C.; La Manna, G.; et al. Diet and Physical Activity in Adult Dominant Polycystic Kidney Disease: A Review of the Literature. Nutrients 2023, 15, 2621. [Google Scholar] [CrossRef]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Tsakiridis, E.; Steinberg, G.R.; Pei, Y. Targeting AMP-activated protein kinase (AMPK) for treatment of autosomal dominant polycystic kidney disease. Cell Signal. 2020, 73, 109704. [Google Scholar] [CrossRef]
- Li, F.; Dai, X.Q.; Li, Q.; Wu, Y.; Chen, X.Z. Inhibition of polycystin-L channel by the Chinese herb Sparganum stoloniferum Buch.-Ham. Can. J. Physiol. Pharmacol. 2006, 84, 923–927. [Google Scholar] [CrossRef]
- Miettinen, H.E.; Piippo, K.; Hannila-Handelberg, T.; Paukku, K.; Hiltunen, T.P.; Gautschi, I.; Schild, L.; Kontula, K. Licorice-induced hypertension and common variants of genes regulating renal sodium reabsorption. Ann. Med. 2010, 42, 465–674. [Google Scholar] [CrossRef]
- Posadzki, P.; Watson, L.K.; Ernst, E. Adverse effects of herbal medicines: An overview of systematic reviews. Clin. Med. 2013, 13, 7–12. [Google Scholar] [CrossRef]
- Menezes, L.F.; Lin, C.C.; Zhou, F.; Germino, G.G. Fatty Acid Oxidation is Impaired in An Orthologous Mouse Model of Autosomal Dominant Polycystic Kidney Disease. EBioMedicine 2016, 5, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Chiaravalli, M.; Rowe, I.; Mannella, V.; Quilici, G.; Canu, T.; Bianchi, V.; Gurgone, A.; Antunes, S.; D’Adamo, P.; Esposito, A.; et al. 2-Deoxy-D-glucose ameliorates PKD progression. J. Am. Soc. Nephrol. 2016, 27, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Warner, G.; Hein, K.Z.; Nin, V.; Edwards, M.; Chini, C.C.S.; Hopp, K.; Harris, P.C.; Torres, V.E.; Chini, E.N. Food restriction ameliorates the development of polycystic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Schley, G.; Kunzelmann, K.; Schreiber, R.; Peters, D.J.M.; Stadler, R.; Eckardt, K.U.; Buchholz, B. Glucose promotes secretion-dependent renal cyst growth. J. Mol. Med. 2016, 94, 107–117. [Google Scholar] [CrossRef]
- Sas, K.M.; Yin, H.; Fitzgibbon, W.R.; Baicu, C.F.; Zile, M.R.; Steele, S.L.; Amria, M.; Saigusa, T.; Funk, J.; Bunni, M.A.; et al. Hyperglycemia in the absence of cilia accelerates cystogenesis and induces renal damage. Am. J. Physiol. Ren. Physiol. 2015, 309, F79–F87. [Google Scholar] [CrossRef]
- Lakhia, R.; Yheskel, M.; Flaten, A.; Quittner-Strom, E.B.; Holland, W.L.; Patel, V. PPARα agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am. J. Physiol. Ren. Physiol. 2018, 314, F122–F131. [Google Scholar] [CrossRef]
- Soomro, I.; Sun, Y.; Li, Z.; Diggs, L.; Hatzivassiliou, G.; Thomas, A.G.; Rais, R.; Slusher, B.S.; Somlo, S.; Skolnik, E.Y. Glutamine metabolism via glutaminase 1 in autosomal-dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2018, 33, 1343–1353. [Google Scholar] [CrossRef]
- Flowers, E.M.; Sudderth, J.; Zacharias, L.; Mernaugh, G.; Zent, R.; DeBerardinis, R.J.; Carroll, T.J. Lkb1 deficiency confers glutamine dependency in polycystic kidney disease. Nat. Commun. 2018, 9, 814. [Google Scholar] [CrossRef]
- Trott, J.F.; Hwang, V.J.; Ishimaru, T.; Chmiel, K.J.; Zhou, J.X.; Shim, K.; Stewart, B.J.; Mahjoub, M.R.; Jen, K.Y.; Barupal, D.K.; et al. Arginine reprogramming in ADPKD results in arginine-dependent cystogenesis. Am. J. Physiol. Ren. Physiol. 2018, 315, F1855–F1868. [Google Scholar] [CrossRef]
- Chou, L.F.; Cheng, Y.L.; Hsieh, C.Y.; Lin, C.Y.; Yang, H.Y.; Chen, Y.C.; Hung, C.C.; Tian, Y.C.; Yang, C.W.; Chang, M.Y. Effect of trehalose supplementation on autophagy and cystogenesis in a mouse model of polycystic kidney disease. Nutrients 2019, 11, 42. [Google Scholar] [CrossRef]
- Zhu, P.; Sieben, C.J.; Xu, X.; Harris, P.C.; Lin, X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 2017, 26, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Belibi, F.; Zafar, I.; Ravichandran, K.; Segvic, A.B.; Jani, A.; Ljubanovic, D.G.; Edelstein, C.L. Hypoxia-inducible factor-1α (HIF-1α) and autophagy in polycystic kidney disease (PKD). Am. J. Physiol. Ren. Physiol. 2011, 300, F1235–F1243. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y.; Brill, A.L.; Lemos, F.O.; Jiang, J.Y.; Falcone, J.L.; Kimmerling, E.P.; Cai, Y.; Dong, K.; Kaplan, D.L.; Wallace, D.P.; et al. Polycystin 2 regulates mitochondrial Ca2+ signaling, bioenergetics, and dynamics through mitofusin 2. Sci. Signal. 2019, 12, eaat7397. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Kurashige, M.; Liu, Y.; Terabayashi, T.; Ishimoto, Y.; Wang, T.; Choudhary, V.; Hobbs, R.; Liu, L.K.; Lee, P.H.; et al. A cleavage product of polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci. Rep. 2018, 8, 2743. [Google Scholar] [CrossRef]
- Padovano, V.; Kuo, I.Y.; Stavola, L.K.; Aerni, H.R.; Flaherty, B.J.; Chapin, H.C.; Ma, M.; Somlo, S.; Boletta, A.; Ehrlich, B.E.; et al. The polycystins are modulated by cellular oxygen-sensing pathways and regulate mitochondrial function. Mol. Biol. Cell 2017, 28, 261–269. [Google Scholar] [CrossRef]
- Kim, K.; Trott, J.F.; Gao, G.; Chapman, A.; Weiss, R.H. Plasma metabolites and lipids associate with kidney function and kidney volume in hypertensive ADPKD patients early in the disease course. BMC Nephrol. 2019, 20, 66. [Google Scholar] [CrossRef]
- Klawitter, J.; Klawitter, J.; McFann, K.; Pennington, A.T.; Abebe, K.Z.; Brosnahan, G.; Cadnapaphornchai, M.A.; Chonchol, M.; Gitomer, B.; Christians, U.; et al. Bioactive lipid mediators in polycystic kidney disease. J. Lipid Res. 2014, 55, 1139–1149. [Google Scholar] [CrossRef]
- Grams, M.E.; Tin, A.; Rebholz, C.M.; Shafi, T.; Köttgen, A.; Perrone, R.D.; Sarnak, M.J.; Inker, L.A.; Levey, A.S.; Coresh, J. Metabolomic alterations associated with cause of CKD. Clin. J. Am. Soc. Nephrol. 2017, 12, 1787–1794. [Google Scholar] [CrossRef]
- Mao, Z.; Xie, G.; Ong, A.C.M. Metabolic abnormalities in autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 197–203. [Google Scholar] [CrossRef]
- Reed, B.; Helal, I.; McFann, K.; Wang, W.; Yan, X.D.; Schrier, R.W. The impact of type II diabetes mellitus in patients with autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2012, 27, 2862–2865. [Google Scholar] [CrossRef]
- Cheungpasitporn, W.; Thongprayoon, C.; Vijayvargiya, P.; Anthanont, P.; Erickson, S.B. The Risk for New-Onset Diabetes Mellitus after Kidney Transplantation in Patients with Autosomal Dominant Polycystic Kidney Disease: A Systematic Review and Meta-Analysis. Can. J. Diabetes 2016, 40, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W.; McFann, K.K.; Johnson, A.M. Epidemiological study of kidney survival in autosomal dominant polycystic kidney disease. Kidney Int. 2003, 63, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; You, Z.; Gitomer, B.; Brosnahan, G.; Torres, V.E.; Chapman, A.B.; Perrone, R.D.; Steinman, T.I.; Abebe, K.Z.; Rahbari-Oskoui, F.F.; et al. Overweight and Obesity Are Predictors of Progression in Early Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Song, Y.; Caballero, B.; Cheskin, L.J. Association between obesity and kidney disease: A systematic review and meta-analysis. Kidney Int. 2008, 73, 19–33. [Google Scholar] [CrossRef]
- Navaneethan, S.D.; Yehnert, H.; Moustarah, F.; Schreiber, M.J.; Schauer, P.R.; Beddhu, S. Weight loss interventions in chronic kidney disease: A systematic review and meta-analysis. Clin. J. Am. Soc. Nephrol. 2009, 4, 1565–1574. [Google Scholar] [CrossRef]
- Moore, T.; Beltran, L.; Carbajal, S.; Strom, S.; Traag, J.; Hursting, S.D.; DiGiovanni, J. Dietary energy balance modulates signaling through the Akt/mammalian target of rapamycin pathways in multiple epithelial tissues. Cancer Prev. Res. 2008, 1, 65–76. [Google Scholar] [CrossRef]
- Dann, S.G.; Selvaraj, A.; Thomas, G. mTOR Complex1-S6K1 signaling: At the crossroads of obesity, diabetes and cancer. Trends Mol. Med. 2007, 13, 252–259. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Kemp, B.E. AMPK in health and disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118. [Google Scholar] [CrossRef]
- Lempiäinen, J.; Finckenberg, P.; Mervaala, E.E.; Sankari, S.; Levijoki, J.; Mervaala, E.M. Caloric restriction ameliorates kidney ischaemia/reperfusion injury through PGC-1α-eNOS pathway and enhanced autophagy. Acta Physiol. 2013, 208, 410–421. [Google Scholar] [CrossRef]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Le, H.D.; Melkani, G.C.; Panda, S. Time-restricted feeding attenuates age-related cardiac decline in Drosophila. Science 2015, 347, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Chaix, A.; Zarrinpar, A.; Miu, P.; Panda, S. Time-restricted feeding is a preventative and therapeutic intervention against diverse nutritional challenges. Cell Metab. 2014, 20, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Hatori, M.; Vollmers, C.; Zarrinpar, A.; DiTacchio, L.; Bushong, E.A.; Gill, S.; Leblanc, M.; Chaix, A.; Joens, M.; Fitzpatrick, J.A.J.; et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012, 15, 848–860. [Google Scholar] [CrossRef]
- Rickman, A.D.; Williamson, D.A.; Martin, C.K.; Gilhooly, C.H.; Stein, R.I.; Bales, C.W.; Roberts, S.; Das, S.K. The CALERIE Study: Design and methods of an innovative 25% caloric restriction intervention. Contemp. Clin. Trials 2011, 32, 874–881. [Google Scholar] [CrossRef]
- Hoddy, K.K.; Kroeger, C.M.; Trepanowski, J.F.; Barnosky, A.; Bhutani, S.; Varady, K.A. Meal timing during alternate day fasting: Impact on body weight and cardiovascular disease risk in obese adults. Obesity 2014, 22, 2524–2531. [Google Scholar] [CrossRef]
- Ravussin, E.; Redman, L.M.; Rochon, J.; Das, S.K.; Fontana, L.; Kraus, W.E.; Romashkan, S.; Williamson, D.A.; Meydani, S.N.; Villareal, D.T.; et al. A 2-year randomized controlled trial of human caloric restriction: Feasibility and effects on predictors of health span and longevity. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 1097–1104. [Google Scholar] [CrossRef]
- Varady, K.A.; Bhutani, S.; Church, E.C.; Klempel, M.C. Short-term modified alternate-day fasting: A novel dietary strategy for weight loss and cardioprotection in obese adults. Am. J. Clin. Nutr. 2009, 90, 1138–1143. [Google Scholar] [CrossRef]
- Harvie, M.N.; Pegington, M.; Mattson, M.P.; Frystyk, J.; Dillon, B.; Evans, G.; Cuzick, J.; Jebb, S.A.; Martin, B.; Cutler, R.G.; et al. The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: A randomized trial in young overweight women. Int. J. Obes. 2011, 35, 714–727. [Google Scholar] [CrossRef]
- Mattson, M.P.; Allison, D.B.; Fontana, L.; Harvie, M.; Longo, V.D.; Malaisse, W.J.; Mosley, M.; Notterpek, L.; Ravussin, E.; Scheer, F.A.J.L.; et al. Meal frequency and timing in health and disease. Proc. Natl. Acad. Sci. USA 2014, 111, 16647–16653. [Google Scholar] [CrossRef]
- Kipp, K.R.; Rezaei, M.; Lin, L.; Dewey, E.C.; Weimbs, T. A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 2016, 310, F726–F731. [Google Scholar] [CrossRef] [PubMed]
- Tomobe, K.; Philbrick, D.; Aukema, H.M.; Clark, W.F.; Ogborn, M.R.; Parbtani, A.; Takahashi, H.; Holub, B.J. Early dietary protein restriction slows disease progression and lengthens survival in mice with polycystic kidney disease. J. Am. Soc. Nephrol. 1994, 5, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Ogborn, M.R.; Nitschmann, E.; Weiler, H.A.; Bankovic-Calic, N. Modification of polycystic kidney disease and fatty acid status by soy protein diet. Kidney Int. 2000, 57, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Tolvaptan: A Review in Autosomal Dominant Polycystic Kidney Disease. Drugs 2019, 79, 303–313. [Google Scholar] [CrossRef]
- Chapman, A.B.; Bost, J.E.; Torres, V.E.; Guay-Woodford, L.; Bae, K.T.; Landsittel, D.; Li, J.; King, B.F.; Martin, D.; Wetzel, L.H. Kidney Volume and Functional Outcomes in Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2012, 7, 479–486. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Audrezet, M.-P.; Rousseau, A.; Hourmant, M.; Renaudineau, E.; Charasse, C.; Morin, M.-P.; Moal, M.-C.; Dantal, J.; Wehbe, B. The PROPKD score: A new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 942–951. [Google Scholar] [CrossRef]
- Irazabal, M.V.; Blais, J.D.; Perrone, R.D.; Gansevoort, R.T.; Chapman, A.B.; Devuyst, O.; Higashihara, E.; Harris, P.C.; Zhou, W.; Ouyang, J. Prognostic Enrichment Design in Clinical Trials for Autosomal Dominant Polycystic Kidney Disease: The TEMPO 3:4 Clinical Trial. Kidney Int. Rep. 2016, 1, 213–220. [Google Scholar] [CrossRef]
- Chang, M.Y.; Ma, T.L.; Hung, C.C.; Tian, Y.C.; Chen, Y.C.; Yang, C.W.; Cheng, Y.C. Metformin Inhibits Cyst Formation in a Zebrafish Model of Polycystin-2 Deficiency. Sci. Rep. 2017, 7, 7161. [Google Scholar] [CrossRef]
- Takiar, V.; Nishio, S.; Seo-Mayer, P.; King, J.D.; Li, H.; Zhang, L.; Karihaloo, A.; Hallows, K.R.; Somlo, S.; Caplan, M.J. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 2462–2467. [Google Scholar] [CrossRef]
- Gile, R.D.; Cowley, B.D.; Gattone, V.H.; O’Donnell, M.P.; Swan, S.K.; Grantham, J.J. Effect of lovastatin on the development of polycystic kidney disease in the han:SPRD rat. Am. J. Kidney Dis. 1995, 26, 501–507. [Google Scholar] [CrossRef]
- Cadnapaphornchai, M.A.; George, D.M.; McFann, K.; Wang, W.; Gitomer, B.; Strain, J.D.; Schrier, R.W. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2014, 9, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, S.; Liu, Y.; Spichtig, D.; Kapoor, S.; Koepsell, H.; Mohebbi, N.; Segerer, S.; Serra, A.L.; Rodriguez, D.; et al. Targeting of sodium-glucose cotransporters with phlorizin inhibits polycystic kidney disease progression in Han:SPRD rats. Kidney Int. 2013, 84, 962–968. [Google Scholar] [CrossRef]
- Rodriguez, D.; Kapoor, S.; Edenhofer, I.; Segerer, S.; Riwanto, M.; Kipar, A.; Yang, M.; Mei, C.; Wüthrich, R.P. Inhibition of Sodium-GlucoseCotransporter 2 with Dapagliflozin in Han: SPRD Rats with Polycystic Kidney Disease. Kidney Blood Press. Res. 2015, 40, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S.; Rodriguez, D.; Riwanto, M.; Edenhofer, I.; Segerer, S.; Mitchell, K.; Wüthrich, R.P. Effect of sodium-glucose cotransport inhibition on polycystic kidney disease progression in PCK rats. PLoS ONE 2015, 10, e0125603. [Google Scholar] [CrossRef] [PubMed]
- Leonhard, W.N.; Song, X.; Kanhai, A.A.; Iliuta, I.A.; Bozovic, A.; Steinberg, G.R.; Peters, D.J.M.; Pei, Y. Salsalate, but not metformin or canagliflozin, slows kidney cyst growth in an adult-onset mouse model of polycystic kidney disease. EBioMedicine 2019, 47, 436–445. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Zhou, X.; Fan, L.X.; Sweeney, W.E.; Denu, J.M.; Avner, E.D.; Li, X. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J. Clin. Investig. 2013, 123, 3084–3098. [Google Scholar] [CrossRef]
- Nofziger, C.; Brown, K.K.; Smith, C.D.; Harrington, W.; Murray, D.; Bisi, J.; Ashton, T.T.; Maurio, F.P.; Kalsi, K.; West, T.A.; et al. PPARγ agonists inhibit vasopressin-mediated anion transport in the MDCK-C7 cell line. Am. J. Physiol. Ren. Physiol. 2009, 297, F55–F62. [Google Scholar] [CrossRef]
- Dai, B.; Liu, Y.; Mei, C.; Fu, L.; Xiong, X.; Zhang, Y.; Shen, X.; Hua, Z. Rosiglitazone attenuates development of polycystic kidney disease and prolongs survival in Han:SPRD rats. Clin. Sci. 2010, 119, 323–333. [Google Scholar] [CrossRef]
- Muto, S.; Aiba, A.; Saito, Y.; Nakao, K.; Nakamura, K.; Tomita, K.; Kitamura, T.; Kurabayashi, M.; Nagai, R.; Higashihara, E.; et al. Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum. Mol. Genet. 2002, 11, 1731–1742. [Google Scholar] [CrossRef]
- Müller, T.D.; Finan, B.; Clemmensen, C.; Di Marchi, R.D.; Tschöp, M.H. The new biology and pharmacology of glucagon. Physiol. Rev. 2019, 97, 721–766. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.; Heerspink, H.J.L.; Cuthbertson, D.J.; Wilding, J.P.H.; Sattar, N. Sodium-glucose co-transporter-2 inhibitors and GLP-1 receptor agonists: Established and emerging indications. Lancet 2021, 398, 10296. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Armstrong, M.J. A placebo controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Frías, J.P.; Davies, M.J.; Rosenstock, J.; Pérez Manghi, F.C.; Fernández Landó, L.; Bergman, B.K.; Lapuerta, P. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N. Engl. J. Med. 2021, 385, 503–515. [Google Scholar] [CrossRef]
- Körner, M.; Stöckli, M.; Waser, B.; Reubi, J.C. GLP-1 receptor expression in human tumors and human normal tissues: Potential for in vivo targeting. J. Nucl. Med. 2007, 48, 736–743. [Google Scholar] [CrossRef]
- Pocai, A.; Carrington, P.E.; Adams, J.R.; Wright, M.; Eiermann, G.; Zhu, L.; Tschöp, M.H. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 2009, 58, 2258–2266. [Google Scholar] [CrossRef]
- Day, J.W.; Gelfanov, V.; Smiley, D.; Carrington, P.E.; Eiermann, G.; Chicchi, G.; Dimarchi, R.D. Optimization of co-agonism at GLP-1 and glucagon receptors to safely maximize weight reduction in DIO-rodents. Biopolymers 2012, 98, 443–450. [Google Scholar] [CrossRef]
- Welles, J.E.; Hatting, M.; van der Most, P.J.; Berbée, J.F.P. AMPK activation protects against diet-induced obesity through inhibition of ectopic lipid accumulation. J. Lipid Res. 2020, 61. [Google Scholar] [CrossRef]
- Pocai, A. Action and therapeutic potential of oxyntomodulin. Mol. Metab. 2012, 1, 241–251. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Watkins, P.B. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2019, 367, 2407–2418. [Google Scholar] [CrossRef]
- Knol, A.; Caroli, A.; Messa, P.; Remuzzi, G. Endogenous glucagon levels and ADPKD progression: A retrospective observational study. Nephrol. Dial. Transplant. 2021, 36, 2582–2590. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Sattar, N.; Pavo, I.; Haupt, A.; Duffin, K.L.; Yang, Z.; Wiese, R.J.; Tuttle, K.R.; Cherney, D.Z.I. Effects of tirzepatide versus insulin glargine on kidney outcomes in type 2 diabetes in the SURPASS-4 trial: Post-hoc analysis of an open-label, randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2022, 10, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Walz, G.; Budde, K.; Mannaa, M.; Nürnberger, J.; Wanner, C.; Sommerer, C.; Kunzendorf, U.; Banas, B.; Hörl, W.H.; Obermüller, N.; et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2010, 363, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.L.; Poster, D.; Kistler, A.D.; Krauer, F.; Raina, S.; Young, J.; Rentsch, K.M.; Spanaus, K.S.; Senn, O.; Kristanto, P.; et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2010, 363, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Perico, N.; Antiga, L.; Caroli, A.; Ruggenenti, P.; Fasolini, G.; Cafaro, M.; Ondei, P.; Rubis, N.; Diadei, O.; Gherardi, G.; et al. Sirolimus therapy to halt the progression of ADPKD. J. Am. Soc. Nephrol. 2010, 21, 1031–1040. [Google Scholar] [CrossRef]
- Suwabe, T.; Morita, H.; Khasnobish, A.; Araoka, H.; Hoshino, J. Microbiome of infected cysts, feces and saliva in patients with autosomal dominant polycystic kidney disease. CEN Case Rep. 2023, 12, 304–310. [Google Scholar] [CrossRef]
- Zimmermann, P.; Curtis, N. The effect of antibiotics on the composition of the intestinal microbiota—A systematic review. J. Infect. 2019, 79, 471–489. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Mechanism | Description | Model | Author/Year |
|---|---|---|---|
| Dysregulated glucose metabolism | Studies on embryonic fibroblasts from Pkd1−/−mice have shown that altered, reprogrammed cells favor aerobic glycolysis (Warburg effect), increase the mammalian target of rapamycin complex 1 (mTORC1) levels, inhibit AMPK activation, increase proliferation, decrease apoptosis, and cause defective autophagy. Changes in glucose metabolism have been described using in vivo models evidenced by increased expression of key glycolytic genes in the kidneys of patients with cystic epithelium and kidneys of mice with polycystic kidney disease (PKD) | Mouse embryonic fibroblasts | [26] (Rowe et al., 2013) [31] (Menezes et al., 2016) |
| Dysregulated glucose metabolism | Blocking glycolysis with 2-deoxyglucose, a glucose analog that cannot be metabolized, has been shown to reduce cell proliferation in human PKD cells and slow kidney cyst formation in mice. Based on these studies, higher glucose concentrations increase kidney cyst growth, promote cystogenesis, and cause structural and functional kidney damage in the rodent PKD model. | Mouse kidneys (N = 6) Mice (N = 25) Mouse embryonic fibroblasts Mouse kidneys (N = 12) Mice (N = 15) | [32] (Chiaravalli et al., 2016) [33] (Warner et al., 2016) [34] (Kraus et al., 2016) [31] (Menezes et al., 2016) [26] (Rowe et al., 2013) [35] (Sas et al., 2015) |
| Altered lipid metabolism and reduced fatty acid oxidation | This mechanism is believed to involve hepatocyte nuclear factor 4α (Hnf4α) or peroxisome proliferator-activated receptor α (PPARα). In fact, using the PPARα agonist fenofibrate in ADPKD models has been found to enhance fatty acid oxidation and alleviate cyst formation. Conversely, the absence of Hnf4α in PKD models worsens the severity of the cystic disease. | Mouse kidneys (N = 10) human cyst lining cells Mice (N = 25) | [36] (Lakhia et al., 2018); [37] Soomro et al., 2018) [33] Warner et al., 2016 |
| Altered amino acid metabolism | The level of glutaminase 1 in the epithelium lining cysts in human ADPKD kidneys and mice models is increased. Increased levels of glutaminase 1 have been observed in the epithelial lining of cysts in both human ADPKD kidneys and mouse models. Glutamine is essential for the growth of both Pkd1 mutant cells and ADPKD cyst-lining cells, suggesting a dependence on glutamine in PKD. Therefore, inhibiting glutamine metabolism with glutaminase inhibitors such as BPTES or CB839 reduces cyst formation in specific PKD models. However, CB839 proved ineffective in other models, suggesting that cyst growth may also rely on arginine, as argininosuccinate synthetase 1 expression is reduced in ADPKD, and arginine deficiency triggers its upregulation, reducing cystogenesis. | Mouse embryonic kidneys (N = 20) Mouse tubular cell lines | [38] Flowers et al., 2018; [39] Trott et al., 2018 |
| Defects in autophagy and mitochondrial function | Autophagy, a process critical for maintaining cellular energy balance, is typically activated in response to nutrient deprivation. In PKD cells, however, this process is disrupted, primarily due to impaired fusion between autophagosomes and lysosomes, a phenomenon known as defective autophagic flux. Reduced expression of the autophagy-related protein Atg5 has been linked to enhanced cyst formation, while activating autophagy with Beclin-1 has been shown to decrease cyst development in Pkd1 models. Interestingly, while trehalose, a natural autophagy stimulant, failed to mitigate Pkd1 disease, targeting mTORC1, a key autophagy suppressor, with a rapamycin derivative led to both improved autophagy and decreased cyst growth in PKD. | Mice (N = 41) Zebrafish cell lines Han:SPRD rats (N = 6) Cpk mice (N = 7) Pkd2WS25/−mice (N = 4) | [40] Chou et al., 2019; [41] Zhu et al., 2017; [42] Belibi et al., 2011; |
| Mitochondrial dysfunction | Mitochondrial dysfunction in ADPKD is partly due to abnormalities in their structure and biogenesis. Kidney tissue from ADPKD patients and mouse models shows fragmented, swollen mitochondria with irregular movement and reduced mitochondrial DNA. Additionally, mitochondrial function is compromised, with increased reactive oxygen species production, elevated Ca2+ uptake, and diminished cellular respiration. Notably, both polycystin-1 and-2 have been found to influence mitochondrial function directly. | Cell culture Pkd1 human knockout cell lines Human embryonic kidney cells | [43] Kuo et al., 2019; [44] Lin et al., 2018; [45] Padovano et al., 2017 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kędzierska-Kapuza, K.; Łopuszyńska, I.; Niewiński, G.; Franek, E.; Szczuko, M. The Influence of Non-Pharmacological and Pharmacological Interventions on the Course of Autosomal Dominant Polycystic Kidney Disease. Nutrients 2024, 16, 3216. https://doi.org/10.3390/nu16183216
Kędzierska-Kapuza K, Łopuszyńska I, Niewiński G, Franek E, Szczuko M. The Influence of Non-Pharmacological and Pharmacological Interventions on the Course of Autosomal Dominant Polycystic Kidney Disease. Nutrients. 2024; 16(18):3216. https://doi.org/10.3390/nu16183216
Chicago/Turabian StyleKędzierska-Kapuza, Karolina, Inga Łopuszyńska, Grzegorz Niewiński, Edward Franek, and Małgorzata Szczuko. 2024. "The Influence of Non-Pharmacological and Pharmacological Interventions on the Course of Autosomal Dominant Polycystic Kidney Disease" Nutrients 16, no. 18: 3216. https://doi.org/10.3390/nu16183216
APA StyleKędzierska-Kapuza, K., Łopuszyńska, I., Niewiński, G., Franek, E., & Szczuko, M. (2024). The Influence of Non-Pharmacological and Pharmacological Interventions on the Course of Autosomal Dominant Polycystic Kidney Disease. Nutrients, 16(18), 3216. https://doi.org/10.3390/nu16183216