
Genetic Variants, Bioactive Compounds, and PCSK9 Inhibitors in Hyper-LDL-Cholesterolemia: A GWAS and In Silico Study on Cardiovascular Disease Risk



Abstract
1. Introduction
2. Methods
2.1. Participants
2.2. Participant Characteristics and Biochemical Measurements
2.3. Dietary Assessment and Analysis
2.4. Hyper-LDL-Cholesterolemia Definition
2.5. Genetic Variants Affecting Hyper-LDL-Cholesterolemia in Koreans
2.6. Genotyping and Quality Control
2.7. PRS Affects Hyper-LDL-Cholesterolemia
2.8. Interactions Between Genetic Variants and Lifestyle Factors Regarding Hyper-LDL-Cholesterolemia
2.9. Collection and Screening of Bioactive Compounds with Low Binding Energy from Foods with PCSK9
2.10. Molecular Dynamics Simulation (MDS)
2.11. Statistical Analysis
3. Results
3.1. Metabolic Parameters According to Gender and Serum LDL-C Concentrations
3.2. Nutrient Intake of Participants
3.3. Distribution of Genetic Variants for Hyper-LDL-Cholesterolemia Risk in GWAS
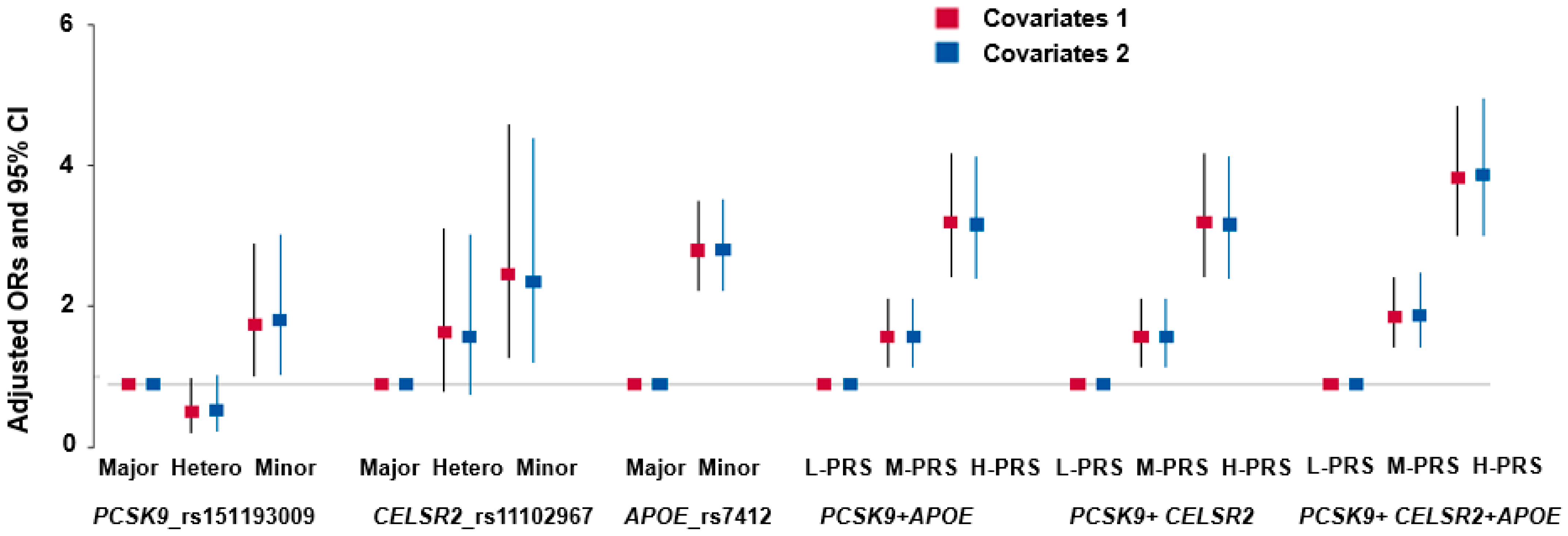
3.4. Genetic Variants Associated with Hyper-LDL-Cholesterolemia
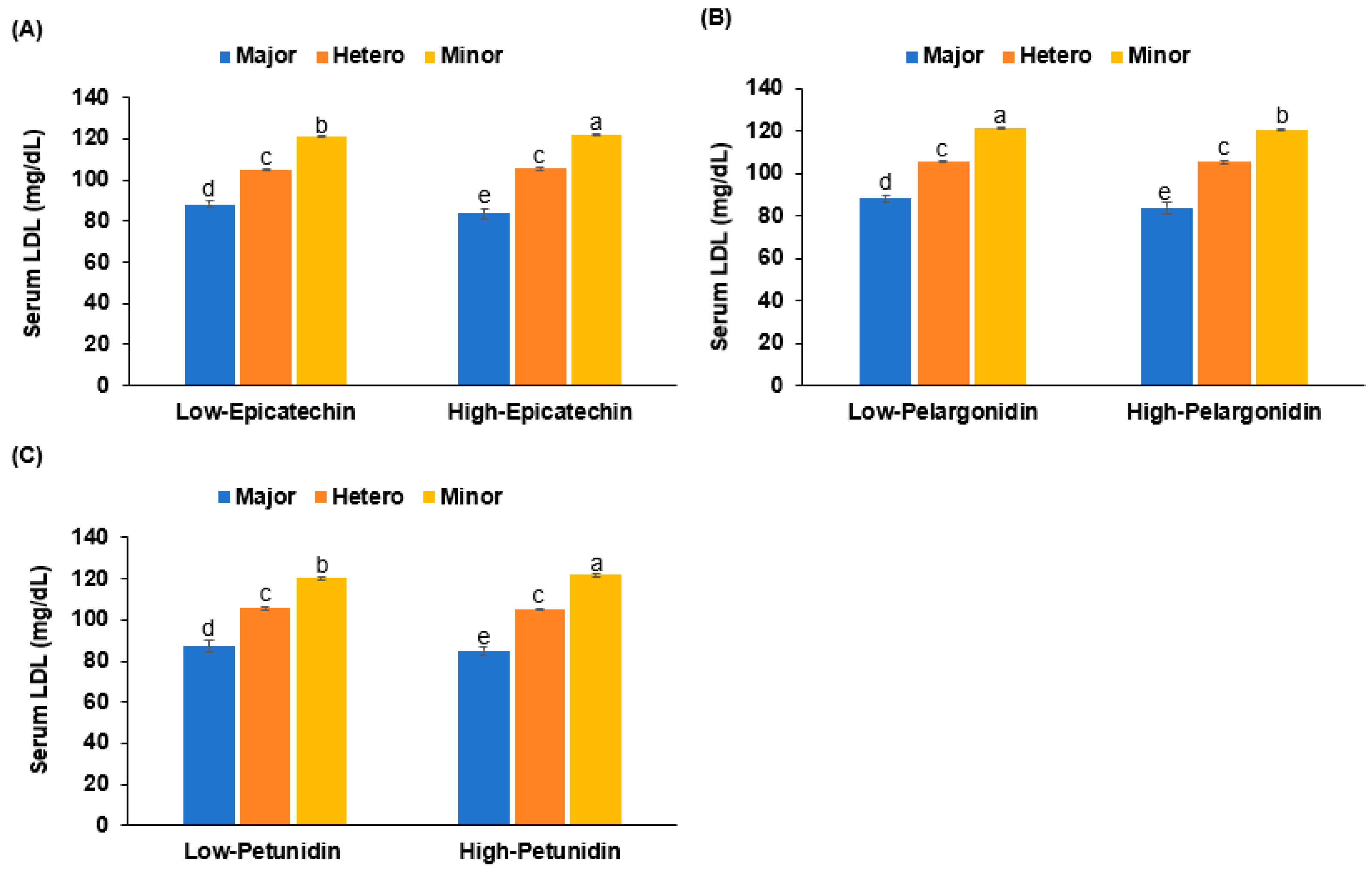
3.5. Interaction of PRS and Nutrient Intake in Hyper-LDL-Cholesterolemia
3.6. Molecular Interaction
3.7. Molecular Dynamics (MD) Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Merćep, I.; Vujević, A.; Strikić, D.; Radman, I.; Pećin, I.; Reiner, Ž. Present and Future of Dyslipidaemia Treatment—A Review. J. Clin. Med. 2023, 12, 5839. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Kehinde, I.O.; Akawa, O.; Adewumi, A.T.; Rabbad, A.H.; Soliman, M.E.S. PCSK9 inhibitors as safer therapeutics for atherosclerotic cardiovascular disease (ASCVD): Pharmacophore design and molecular dynamics analysis. J. Cell. Biochem. 2024, 125, e30581. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Melchionda, E.; Morotti, A.; Russo, I. PCSK9 Biology and Its Role in Atherothrombosis. Int. J. Mol. Sci. 2021, 22, 5880. [Google Scholar] [CrossRef]
- Brousseau, M.E.; Clairmont, K.B.; Spraggon, G.; Flyer, A.N.; Golosov, A.A.; Grosche, P.; Amin, J.; Andre, J.; Burdick, D.; Caplan, S.; et al. Identification of a PCSK9-LDLR disruptor peptide with in vivo function. Cell Chem. Biol. 2022, 29, 249–258.e5. [Google Scholar] [CrossRef]
- Xia, X.D.; Peng, Z.S.; Gu, H.M.; Wang, M.; Wang, G.Q.; Zhang, D.W. Regulation of PCSK9 Expression and Function: Mechanisms and Therapeutic Implications. Front. Cardiovasc. Med. 2021, 8, 764038. [Google Scholar] [CrossRef]
- Noguchi, T.; Katsuda, S.; Kawashiri, M.-A.; Tada, H.; Nohara, A.; Inazu, A.; Yamagishi, M.; Kobayashi, J.; Mabuchi, H. The E32K variant of PCSK9 exacerbates the phenotype of familial hypercholesterolaemia by increasing PCSK9 function and concentration in the circulation. Atherosclerosis 2010, 210, 166–172. [Google Scholar] [CrossRef]
- Yang, L.; Pu, T.; Zhang, Y.; Yan, H.; Yu, H.; Gao, W. The R93C Variant of PCSK9 Reduces the Risk of Premature MI in a Chinese Han Population. Front. Genet. 2022, 13, 875269. [Google Scholar] [CrossRef]
- Wu, L.; Kong, Q.; Huang, H.; Xu, S.; Qu, W.; Zhang, P.; Yu, Z.; Luo, X. Effect of PCSK9 inhibition in combination with statin therapy on intracranial atherosclerotic stenosis: A high-resolution MRI study. Front. Aging Neurosci. 2023, 15, 1127534. [Google Scholar] [CrossRef]
- Ejlskov, L.; Plana-Ripoll, O. Income in epidemiological research: A guide to measurement and analytical treatment with a case study on mental disorders and mortality. J. Epidemiol. Community Health 2025. [Google Scholar] [CrossRef]
- Park, S.; Liu, M.; Kang, S. Alcohol intake interacts with CDKAL1, HHEX, and OAS3 genetic variants, associated with the risk of type 2 diabetes by lowering insulin secretion in Korean adults. Alcohol. Clin. Exp. Res. 2018, 42, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Han, B.-G.; Group, K. Cohort profile: The Korean genome and epidemiology study (KoGES) consortium. Int. J. Epidemiol. 2017, 46, e20. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shan, Y.; Dai, L.; Jiang, X.; Song, C.; Chen, B.; Zhang, J.; Li, J.; Zhang, Y.; Xu, J.; et al. Sex-specific equations to estimate body composition: Derivation and validation of diagnostic prediction models using UK biobank. Clin. Nutr. 2023, 42, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, C.; Wu, X. Development and validation of an insulin resistance predicting model using a machine-learning approach in a population-based cohort in Korea. Diagnostics 2022, 12, 212. [Google Scholar] [CrossRef]
- Park, S.; Kang, S. A Western-style diet interacts with genetic variants of the LDL receptor to hyper-LDL cholesterolemia in Korean adults. Public Health Nutr. 2021, 24, 2964–2974. [Google Scholar] [CrossRef]
- Committee of KDRI. Korean Dietary Reference Intakes; Ministry of Health & Welfare: Seoul, Republic of Korea, 2015; Volume 1, pp. 54–56.
- Zainab, R.; Kaleem, A.; Ponczek, M.B.; Abdullah, R.; Iqtedar, M.; Hoessli, D.C. Finding inhibitors for PCSK9 using computational methods. PLoS ONE 2021, 16, e0255523. [Google Scholar] [CrossRef]
- Park, S.; Liu, M.; Huang, S. Association of polygenic variants involved in immunity and inflammation with duodenal ulcer risk and their interaction with irregular eating habits. Nutrients 2023, 15, 296. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Liu, M.; Jin, H.S.; Park, S. Protein and fat intake interacts with the haplotype of PTPN11_rs11066325, RPH3A_rs886477, and OAS3_rs2072134 to modulate serum HDL concentrations in middle-aged people. Clin. Nutr. 2020, 39, 942–949. [Google Scholar] [CrossRef]
- Wang, Z.; Fan, Z.; Yang, L.; Liu, L.; Sheng, C.; Song, F.; Huang, Y.; Chen, K. Higher risk of cardiovascular mortality than cancer mortality among long-term cancer survivors. Front. Cardiovasc. Med. 2023, 10, 1014400. [Google Scholar] [CrossRef]
- Jiang, Y.; Yu, W.; Zhou, J.; Dong, X. Bidirectional causal relationship between hypercholesterolemia and ischemic heart disease: A Mendelian randomization study. Front. Cardiovasc. Med. 2023, 10, 1302282. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005, 37, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Zeng, P.; Li, X.; Zhang, Z.; Pan, B.; Peng, Z.Y.F.; Li, Y.; Ma, Y.; Leng, Y.; Chen, R. What is the impact of PCSK9 rs505151 and rs11591147 polymorphisms on serum lipids level and cardiovascular risk: A meta-analysis. Lipids Health Dis. 2017, 16, 111. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, H.; Su, C.; Wang, Z.; Du, W.; Huang, F.; Zhang, J.; Jia, X.; Jiang, H.; Ouyang, Y.; et al. Trajectories of Energy Intake Distribution and Risk of Dyslipidemia: Findings from the China Health and Nutrition Survey (1991–2018). Nutrients 2021, 13, 3488. [Google Scholar] [CrossRef]
- Tanisawa, K.; Ito, T.; Kawakami, R.; Usui, C.; Kawamura, T.; Suzuki, K.; Sakamoto, S.; Ishii, K.; Muraoka, I.; Oka, K.; et al. Association between alcohol dietary pattern and prevalence of dyslipidemia: WASEDA’S Health Study. Br. J. Nutr. 2022, 127, 1712–1722. [Google Scholar] [CrossRef]
- Zhou, J.Y.; Park, S. Regular exercise, alcohol consumption, and smoking interact with the polygenetic risk scores involved in insulin sensitivity and secretion for the risk of concurrent hyperglycemia, hypertension, and dyslipidemia. Nutrition 2021, 91–92, 111422. [Google Scholar] [CrossRef]
- Yamamoto, T.; Harada-Shiba, M.; Nakatani, M.; Wada, S.; Yasuhara, H.; Narukawa, K.; Sasaki, K.; Shibata, M.-A.; Torigoe, H.; Yamaoka, T.; et al. Cholesterol-lowering action of BNA-based antisense oligonucleotides targeting PCSK9 in atherogenic diet-induced hypercholesterolemic mice. Mol. Ther. Nucleic Acids 2012, 1, e22. [Google Scholar] [CrossRef]
- Denis, M.; Marcinkiewicz, J.; Zaid, A.; Gauthier, D.; Poirier, S.; Lazure, C.; Prat, A. Gene inactivation of proprotein convertase subtilisin/kexin type 9 reduces atherosclerosis in mice. Circulation 2012, 125, 894–901. [Google Scholar] [CrossRef]
- Schulz, R.; Schlüter, K.-D.; Laufs, U. Molecular and cellular function of the proprotein convertase subtilisin/kexin type 9 (PCSK9). Basic Res. Cardiol. 2015, 110, 1–19. [Google Scholar] [CrossRef]
- Sun, S.; Ma, J.; Zuo, T.; Shi, J.; Sun, L.; Meng, C.; Zhang, Z. Inhibition of PCSK9: A Promising Enhancer for Anti-PD-1/PD-L1 Immunotherapy. Research 2024, 7, 0488. [Google Scholar] [CrossRef]
- Tan, J.; Che, Y.; Liu, Y.; Hu, J.; Wang, W.; Hu, L.; Zhou, Q.; Wang, H.; Li, J. CELSR2 deficiency suppresses lipid accumulation in hepatocytes by impairing the UPR and elevating ROS level. FASEB J. 2021, 35, e21908. [Google Scholar] [CrossRef] [PubMed]
- Samani, N.J.; Braund, P.S.; Erdmann, J.; Götz, A.; Tomaszewski, M.; Linsel-Nitschke, P.; Tobin, M.D. The novel genetic variant predisposing to coronary artery disease in the region of the PSRC1 and CELSR2 genes on chromosome 1 associates with serum cholesterol. J. Mol. Med. 2008, 86, 1233–1241. [Google Scholar] [CrossRef]
- Chai, T.; Wang, Z.; Yang, X.; Qiu, Z.; Chen, L. PSRC1 may affect coronary artery disease risk by altering CELSR2, PSRC1, and SORT1 gene expression and circulating granulin and Apolipoprotein B protein levels. Front. Cardiovasc. Med. 2022, 9, 763015. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.D. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology 2019, 51, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aziz, T.A.; Mohamed, R.H. LDLR, ApoB and ApoE genes polymorphisms and classical risk factors in premature coronary artery disease. Gene 2016, 590, 263–269. [Google Scholar] [CrossRef]
- Yuan, L.; Liu, J.; Dong, L.; Cai, C.; Wang, S.; Wang, B.; Xiao, R. Effects of APOE rs429358, rs7412 and GSTM1/GSTT1 polymorphism on plasma and erythrocyte antioxidant parameters and cognition in old Chinese adults. Nutrients 2015, 7, 8261–8273. [Google Scholar] [CrossRef]
- Liu, H.-W.; Zhang, F.; Fan, P.; Bai, H.; Zhang, J.-X.; Wang, Y. Effects of apolipoprotein E genotypes on metabolic profile and oxidative stress in south-west Chinese women with polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 170, 146–151. [Google Scholar] [CrossRef]
- Tejedor, M.T.; Garcia-Sobreviela, M.P.; Ledesma, M.; Arbones-Mainar, J.M. The apolipoprotein E polymorphism rs7412 associates with body fatness independently of plasma lipids in middle-aged men. PLoS ONE 2014, 9, e108605. [Google Scholar] [CrossRef]
- Carré, C.; Carluer, J.B.; Chaux, C.; Estoup-Streiff, C.; Roche, N.; Hosy, E.; Mas, A.; Krouk, G. Next-Gen GWAS: Full 2D epistatic interaction maps retrieve part of missing heritability and improve phenotypic prediction. Genome Biol. 2024, 25, 76. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Men (n = 20,293) | Women (n = 38,408) | |||
|---|---|---|---|---|
| Low-LDL (n = 17,845) | High-LDL (n = 2448) | Low-LDL (n = 31,890) | High-LDL (n = 6518) | |
| Age (years) | 56.2 ± 0.06 b | 57.3 ± 0.15 a | 51.9 ± 0.04 d | 55.3 ± 0.09 c***+++### |
| Education (N, %) | ||||
| ≤Middle school | 1582 (14.0) | 171 (12.1) † | 5340 (21.4) | 1398 (25.7) ††† |
| High school | 8361 (75.6) | 1073 (76.0) | 18,085 (72.5) | 3786 (69.7) |
| ≥Collage | 1123 (10.2) | 168 (11.9) | 1534 (6.15) | 248 (4.57) |
| Income (dollar/month) | ||||
| ≤2000 | 1442 (8.49) | 164 (7.07) †† | 3289 (11.0) | 883 (14.4) ††† |
| 2000–4000 | 7273 (42.8) | 934 (40.3) | 13,058 (43.5) | 2927 (47.8) |
| >4000 | 8278 (48.7) | 1222 (52.7) | 13,661 (45.5) | 2309 (37.7) |
| BMI (kg/m2) | 24.3 ± 0.02 b | 25 ± 0.06 a | 23.5 ± 0.02 c | 24.3 ± 0.04 b***+++ |
| Waist circumference (cm) | 84.5 ± 0.04 b | 85 ± 0.1 a | 78.6 ± 0.03 d | 79.2 ± 0.06 c***+++ |
| SMI (kg/m) | 7.14 ± 0.005 a | 7.13 ± 0.011 a | 6.14 ± 0.003 b | 6.03 ± 0.007 c***+++### |
| Fat mass (%) | 22.7 ± 0.01 d | 23 ± 0.02 c | 31.2 ± 0.01 b | 31.8 ± 0.01 a***+++### |
| Serum glucose (mg/dL) | 98.1 ± 0.19 b | 100.5 ± 0.42 a | 92.7 ± 0.13 d | 96.6 ± 0.25 c***+++## |
| HbA1c (%) | 5.71 ± 0.01 b | 5.9 ± 0.02 a | 5.65 ± 0.01 c | 5.9 ± 0.01 a*+++# |
| Insulin resistance (yes, %) | 1943 (10.9) | 369 (15.1) ††† | 1634 (5.12) | 658 (10.1) ††† |
| Total-C (mg/dL) | 186 ± 0.31 d | 222 ± 0.69 b | 194 ± 0.21 c | 237 ± 0.42 a***+++### |
| HDL-C (mg/dL) | 49.3 ± 0.12 d | 50.8 ± 0.27 c | 55.9 ± 0.08 b | 56.5 ± 0.16 a**+++## |
| Triglyceride (mg/dL) | 134 ± 0.78 a | 135 ± 1.76 a | 118 ± 0.53 c | 128 ± 1.06 b***+++### |
| LDL-C (mg/dL) | 110 ± 0.28 d | 144 ± 0.63 b | 114 ± 0.19 c | 155 ± 0.38 a***+++### |
| Lipid-lowering medication (N, %) | 0 (0) | 941(40.5) ††† | 0 (0) | 1788 (30.7) ††† |
| LDL-C on lipid-lowering medication (mg/dL) | 97.4 ± 1.45 | 105 ± 1.01 †† | ||
| LDL-C on no lipid-lowering medication (mg/dL) | 177 ± 0.70 | 179 ± 0.41 | ||
| SBP (mmHg) | 126 ± 0.13 a | 126 ± 0.3 a | 120 ± 0.09 c | 123 ± 0.18 b***+++### |
| DBP (mmHg) | 78.3 ± 0.09 a | 78.5 ± 0.2 a | 74.1 ± 0.06 c | 75.4 ± 0.12 b |
| Men (n = 20,293) | Women (n = 38,408) | |||
|---|---|---|---|---|
| Low-LDL (n = 17,845) | High-LDL (n = 2448) | Low-LDL (n = 31,890) | High-LDL (n = 6518) | |
| Energy intake (EER %) | 90 ± 0.29 c | 89.8 ± 0.66 c | 99.1 ± 0.2 b | 101 ± 0.4 a***+## |
| Fiber (g/day) | 14.5 ± 0.07 b | 14.1 ± 0.17 b | 14.8 ± 0.05 a | 15 ± 0.1 a***## |
| Ca (mg/day) | 391 ± 1.84 c | 396 ± 4.17 c | 467 ± 1.25 b | 483 ± 2.52 a***+++# |
| Na (mg/day) | 2428 ± 11 a | 2358 ± 24.9 b | 2433 ± 7.4 a | 2444 ± 15 a*+## |
| Vitamin C (mg/day) | 92.6 ± 0.53 b | 91.8 ± 1.21 b | 112.4 ± 0.36 a | 112.8 ± 0.73 a*** |
| Vitamin D (ug/day) | 5.3 ± 0.05 c | 5.6 ± 0.11 c | 6.9 ± 0.03 b | 7.2 ± 0.06 a***+++ |
| DII | −18.9 ± 0.13 | −18.5 ± 0.3 | −20.4 ± 0.09 | −21.1 ± 0.18***## |
| Flavonoids (mg/day) | 31.4 ± 0.28 b | 31.6 ± 0.63 b | 42.2 ± 0.19 a | 41.8 ± 0.38 a*** |
| KBD (Yes, N, %) | 7148 (40.6) | 953 (38.9) | 9534 (29.9) | 1934 (29.7) |
| PBD (Yes, N, %) | 14,230 (20.3) | 583 (23.8) † | 12,751 (40.0) | 2627 (40.3) |
| WSD (Yes, N, %) | 9096 (51.0) | 1335 (54.5) ++ | 11,078(34.7) | 2043 (31.4) ††† |
| RMD (Yes, N, %) | 5669 (31.8) | 796 (32.5) | 11,015(34.5) | 2091(32.1) ††† |
| Alcohol (g/week) | 28 ± 0.42 a | 23.7 ± 0.95 b | 9.9 ± 0.28 c | 7.8 ± 0.58 d***+++# |
| Exercise (Yes, N, %) | 16,522 (52.0) | 3502 (53.9) ††† | 10,446 (58.7) | 1506 (61.8) †† |
| Former smoking (Yes, N, %) | 7543 (43.0) | 1146 (47.6) ††† | 376 (1.20) | 79 (1.23) |
| Smoking (Yes, N, %) | 4938 (28.1) | 643 (26.7) | 611 (1.94) | 126 (1.96) |
| CHR | SNP | BP | A1 | A2 | MAF | HWE | OR | SE | P | OR | SE | P1 | Location | Gene Names |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | rs151193009 | 55509585 | T | C | 0.015 | 0.208 | 0.330 | 0.132 | 5.97 × 10−17 | 0.158 | 0.453 | 4.82 × 10−5 | missense (Arg93Cys) | PCSK9 |
| 1 | rs11102967 | 109817245 | C | A | 0.060 | 0.286 | 0.689 | 0.049 | 2.66 × 10−14 | 0.692 | 0.093 | 6.90 × 10−5 | 3′ UTR | CELSR2 |
| 19 | rs7412 | 45412079 | T | C | 0.065 | 0.29 | 0.324 | 0.064 | 1.05 × 10−68 | 0.268 | 0.164 | 8.63 × 10−16 | missense (arg176cys) | APOE |
| Low-PRS (n = 14,420) | Medium-PRS (n = 21,641) | High-PRS (n = 4201) | Adjusted OR and 95% CI | |
|---|---|---|---|---|
| LDL-C (mg/dL) 1 | 99 ± 0.82 c | 110.6 ± 0.28 b | 122.3 ± 0.15 a*** | 3.451 (2.771–4.298) |
| BMI (kg/m2) 2 | 23.7 ± 0.07 | 23.9 ± 0.03 | 23.9 ± 0.01 | 1.068 (0.953–1.198) |
| Waist circumference (cm) 3 | 80.6 ± 0.2 | 80.8 ± 0.07 | 80.7 ± 0.04 | 1.077 (0.943–1.23) |
| Total cholesterol (mg/dL) 4 | 181 ± 0.90 c | 190 ± 0.31 b | 200 ± 0.17 a*** | 2.452 (2.084–2.884) |
| HDL-C (mg/dL) 5 | 55.8 ± 0.32 a | 54.7 ± 0.11 b | 53.5 ± 0.06 c*** | 1.439 (1.267–1.634) |
| TG (mg/dL) 6 | 131 ± 1.73 a | 123 ± 0.60 b | 120 ± 0.33 c*** | 0.818 (0.728–0.92) |
| SMI (kg/m) 7 | 6.46 ± 0.01 | 6.46 ± 0 | 6.46 ± 0 | 0.997 (0.882–1.127) |
| Fat mass (%) 8 | 28.3 ± 0.03 | 28.4 ± 0.01 | 28.4 ± 0 | 1.116 (0.997–1.25) |
| Serum glucose (mg/dL) 9 | 95.7 ± 0.5 | 95 ± 0.17 | 95 ± 0.09 | 0.946 (0.809–1.105) |
| HbA1c (%) 10 | 5.7 ± 0.02 | 5.7 ± 0.01 | 5.7 ± 0 | 1.045 (0.803–1.359) |
| Insulin resistance (yes, %) | 383 (7.29) | 2840 (7.70) | 1241 (7.72) | 1.036 (0.841–1.277) |
| SBP (mmHg) 11 | 122 ± 0.36 | 122 ± 0.12 | 122 ± 0.07 | 1.038 (0.924–1.166) |
| DBP (mmHg) 12 | 76.1 ± 0.24 | 75.7 ± 0.08 | 75.7 ± 0.05 | 0.96 (0.802–1.15) |
| PRS3 | Low-PRS (n = 14,420) | Medium-PRS (n = 21,641) | High-PRS (n = 4201) | Gene–Nutrient Interaction p Value |
|---|---|---|---|---|
| Low energy 1 High energy | 1 | 1.761 (1.317–2.356) 1.801 (1.191–2.724) | 3.361 (2.534–4.458) 3.678 (2.460–5.499) | 0.0016 |
| Low exercise 2 High exercise | 1 | 1.995 (1.361–2.924) 1.694 (1.243–2.308) | 4.094 (2.822–5.940) 3.149 (2.331–4.252) | 0.0105 |
| PCSK9 | Non-risk (n = 51,346) | Heterozygotes (n = 7126) | Risk (n = 229) | Gene-nutrient interaction p value |
| Low energy High energy | 1 | 0.999 (0.996–1.001) 0.995 (0.991–0.998) | 2.504 (2.216–2.830) 2.716 (2.292–3.217) | 0.0014 |
| Low exercise High exercise | 1 | 0.996 (0.993–0.999) 0.998 (0.995–1.001) | 2.659 (2.286–3.093) 2.471 (2.169–2.815) | 0.1128 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Zhou, J.; Park, S. Genetic Variants, Bioactive Compounds, and PCSK9 Inhibitors in Hyper-LDL-Cholesterolemia: A GWAS and In Silico Study on Cardiovascular Disease Risk. Nutrients 2025, 17, 1411. https://doi.org/10.3390/nu17091411
Liu M, Zhou J, Park S. Genetic Variants, Bioactive Compounds, and PCSK9 Inhibitors in Hyper-LDL-Cholesterolemia: A GWAS and In Silico Study on Cardiovascular Disease Risk. Nutrients. 2025; 17(9):1411. https://doi.org/10.3390/nu17091411
Chicago/Turabian StyleLiu, Meiling, Junyu Zhou, and Sunmin Park. 2025. "Genetic Variants, Bioactive Compounds, and PCSK9 Inhibitors in Hyper-LDL-Cholesterolemia: A GWAS and In Silico Study on Cardiovascular Disease Risk" Nutrients 17, no. 9: 1411. https://doi.org/10.3390/nu17091411
APA StyleLiu, M., Zhou, J., & Park, S. (2025). Genetic Variants, Bioactive Compounds, and PCSK9 Inhibitors in Hyper-LDL-Cholesterolemia: A GWAS and In Silico Study on Cardiovascular Disease Risk. Nutrients, 17(9), 1411. https://doi.org/10.3390/nu17091411