Abstract
Immunochemical methods for mycotoxin analysis require antigens with well-defined structures and antibodies with outstanding binding properties. Immunoreagents for the mycotoxins alternariol and/or alternariol monomethyl ether have typically been obtained with chemically uncharacterized haptens, and antigen conjugates have most likely been prepared with mixtures of functionalized molecules. For the first time, total synthesis was performed, in the present study, to obtain two haptens with opposite linker attachment locations. The functionalized synthetic haptens were purified and deeply characterized by different spectrometric methods, allowing the preparation of bioconjugates with unequivocal structures. Direct and indirect competitive enzyme-linked immunosorbent assays, using homologous and heterologous conjugates, were employed to extensively evaluate the generated immunoreagents. Antibodies with high affinity were raised from conjugates of both haptens, and a structure-activity relationship between the synthetic haptens and the specificity of the generated antibodies could be established. These results pave the way for the development of novel highly sensitive immunoassays selective of one or two of these Alternaria mycotoxins.
Key Contribution:
Two pure regioisomeric alternariol haptens were prepared. Alternariol immunoreagents were generated and characterized.
1. Introduction
Alternaria sp. fungi, particularly A. alternata, are ubiquitous plant pathogens and saprophytes that infect economically relevant crops such as cereals, vegetables, oilseeds, and fruits. Moreover, these microorganisms can contaminate these commodities after harvest even under refrigeration conditions. They are known to produce a wide variety of toxic secondary metabolites [1], and some of them have been identified by the EFSA Panel on Contaminants in the Food Chain (CONTAM) as a potential risk to human and animal health due to their toxicity and occurrence in food and feed. Surprisingly, there are no specific international regulations for any of the Alternaria mycotoxins, and the available data on toxicity, occurrence, and dietary exposure are still limited. In 2011, EFSA carried out the first assessment of the risk of these mycotoxins to human and animal health, based on government and published data [2]. More recently, EFSA conducted a survey on the dietary exposure of European consumers to Alternaria toxins [3]. This study found that 8% of these mycotoxins are present in food, with infants and other children being the most exposed population group, and fruit and fruit-based products contributing most to dietary exposure. It is therefore expected that the European Commission will soon set maximum levels for the most common Alternaria mycotoxins in foodstuffs.
Alternariol (AOH) and alternariol monomethyl ether (AME), two of the most important compounds belonging to the group of Alternaria mycotoxins, appear to be responsible for the teratogenic effects observed in animals. They have also been shown to inhibit in vitro the catalytic topoisomerase activity, which may be associated with human colon and oesophageal cancer [4]. Frequently, these two mycotoxins are found together in samples because they share most of the biosynthetic pathway [5]. AOH and AME have been detected in a wide variety of products, including lentils, carrots, tomatoes, berries, apples, pears, beer, wines, juices, and various grains and flours [6]. To evaluate the relative hazard level of these toxins to human health, a threshold of toxicological concern (TTC) for AOH and AME in adults of 2.5 ng/kg body weight per day was established as a reference parameter by the CONTAM Panel [2]. With limited data available, a 2016 German survey concluded that the percentage of TTC reached by the average adult daily exposure was 1400% and 280% for AOH and AME, respectively [7].
A variety of analytical techniques have been developed for monitoring Alternaria toxins in food, including liquid and gas chromatographic methods coupled to several detection systems, as well as different types of immunochemical assays [8,9]. Molecular affinity techniques nowadays represent alternative strategies for rapid, economical, and/or on-site monitoring of mycotoxins. The first antibodies and immunoassays for AOH were reported in 2011 [10,11]. Since then, a few immunoassays have been described using either polyclonal or monoclonal antibodies specific to AOH [12,13], and only one study has been published using an antibody specific to AME [14]. Additionally, Wang et al. have reported a generic antibody for both mycotoxins [15]. In all these studies, the immunogens used to generate antibodies against AOH were made by attaching the mycotoxin to the carrier protein, either directly by a Mannich-type reaction or by carbodiimide-mediated chemistry after nonselective carboxymethylation of the hydroxyl groups. Neither of these methods can be used to guide the position of attachment of the AOH molecular scaffold to the carrier proteins, so the antibodies were actually generated from an undefined mixture of functionalized haptens. More recently, Yao et al. published an immunoassay for AOH using a carboxymethyl hapten to generate polyclonal antibodies [16]. Disappointingly, no synthetic details and spectrometric data of the prepared hapten were provided in that work.
AOH and AME are the two most representative mycotoxins with a tricyclic benzochromenone chemical backbone. Moreover, these compounds contain hydroxyl, methyl, and other substituents in their chemical structure. It is well known that the orientation of the molecule, i.e., the way the hapten is displayed to the immune system, strongly influences the specificity of the generated antibodies. Thus, the synthesis of haptens with the optimal linker tethering site is critical, although it is hard to predict and challenging to perform. Surprisingly, deeply characterized haptens for AOH or AME with unambiguous chemical structures have not been published so far. The aim of the present study was to prepare, purify, and characterize two rationally-designed synthetic haptens of these Alternaria mycotoxins with a functionalized linker located at precise sites of the molecule. The ability of these novel immunoreagents to elicit a potent immune response, ultimately leading to high-affinity antibodies, was investigated. In addition, these bioconjugates also allowed the study of the relationship between the functionalization position in the hapten and the specificity of the resulting antibodies.
2. Results and Discussion
2.1. Hapten Design and Synthesis
The generation of antibodies to AOH has so far been based on the preparation of the required immunogens from AOH itself, which does not allow fine control over the specific position of the mycotoxin framework where the functionalized linker is introduced. In this study, we have synthesized two regioisomeric haptens of AOH from scratch. One of them, hapten ALa, incorporates a five-atom carboxylated aliphatic spacer arm through the hydroxyl group at C-9, whereas the other one, hapten ALb, incorporates the same linker via the hydroxyl group at C-3 (Figure 1). In contrast to previous strategies, these two haptens allowed the preparation of bioconjugates with well-defined compositions.
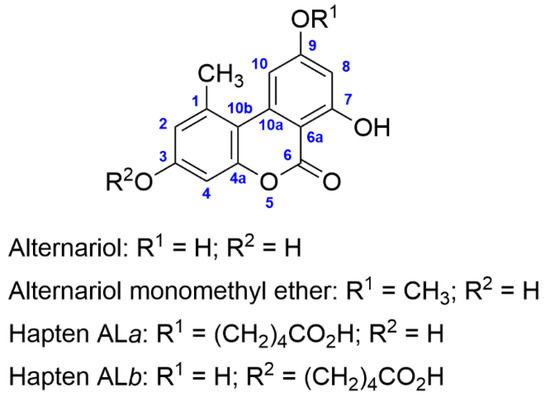
Figure 1.
Chemical structures of alternariol, alternariol monomethyl ether, and the synthetic haptens.
The synthetic strategy for preparing hapten ALa was based on a convergent methodology previously used by several research groups to synthesize AOH and other structurally related molecules [17,18,19,20]. A key step in this synthesis is a Pd(0)-catalyzed cross-coupling reaction between an aryl triflate (4), which already contained the spacer arm, and an appropriately functionalized arylboronic acid (6) (Scheme 1). The aryl triflate 4 was prepared in two steps from the readily available 1,3-benzodioxinone 1 [21,22]. First, an O-alkylation reaction with methyl 5-bromovalerate was performed under standard Williamson ether synthesis conditions. The alkylation process produced a 6:1 mixture of di- and mono-O-alkylation products, 2 and 3, respectively, which were easily separated by column chromatography to provide the product resulting from the selective O-alkylation of the less hindered hydroxyl group, i.e., 3, with a 75% yield. The free hydroxyl group of 3 was then converted to the required triflate group by reaction with triflic anhydride in pyridine, giving the triflate 4 in 91% yield. The additional required coupling reactant, aryl boronic acid 6, was prepared from the orcinol-derived bromide 5 [23] by halogen-metal exchange using butyllithium and reaction of the resulting lithiated derivative with triisopropyl borate.
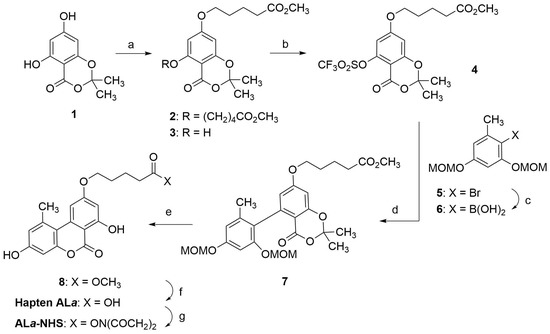
Scheme 1.
Synthesis of ALa-NHS ester. Reagents and conditions: (a) Br(CH2)4CO2CH3, K2CO3, KI, Bu4NBr, acetone, reflux, 16 h, 75% of 3. (b) Tf2O, pyridine, 0 °C to rt, 20 h, 91%. (c) i. n-BuLi, THF, −78 °C, 40 min; ii. B(OiPr)3, −78 °C to 0 °C, 1.5 h, 93%. (d) Pd(PPh3)4, K2CO3, DMF, 93 °C, 24 h, 75%. (e) i. HCl, MeOH, rt, 22 h; ii. TFA, CH2Cl2, rt, 20 h, 97%. (f) Lipase acrylic resin, THF-PB 100 mM, rt, 20 h, 93%. (g) EDC·HCl, NHS, DMF, rt, overnight, 99% of crude product.
The subsequent palladium-catalyzed Suzuki-Miyaura coupling between the aryl triflate 4 and the labile boronic acid 6 gave the biaryl 7 in 75% yield. Hydrolysis of the methoxymethyl ether (MOM) groups by treatment with methanolic HCl, followed by intramolecular transesterification promoted by trifluoroacetic acid, completed the synthesis of the tricyclic benzochromenone backbone and afforded the methyl ester of hapten ALa, compound 8, in 97% yield. To complete the synthesis of hapten ALa, only the hydrolysis of the methyl ester moiety of 8 was required, which was initially carried out under basic conditions (LiOH in THF-H2O at room temperature, rt). However, under these conditions, the central lactone group of the benzochromenone core was partially opened, requiring acid treatment of the reaction crude to reconstruct the tricyclic ring system. It proved more convenient to carry out this transformation using enzymatic hydrolysis, so a lipase from Candida antarctica immobilized on an acrylic resin was used to hydrolyze the methyl ester group, providing hapten ALa in practically quantitative yield.
Upon completion of the synthesis of hapten ALa, its carboxylic group was activated by forming the corresponding N-hydroxysuccinimidyl ester. This transformation was carried out under conventional activation conditions, with N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) and N-hydroxisuccinimide (NHS) in N,N-dimethylformamide (DMF) at rt, yielding the corresponding N-hydroxysuccinimidyl ester, ALa-NHS, in good yield. The activated hapten was extracted essentially pure from the reaction as judged by 1H NMR, so it was further used without additional purification by column chromatography. NMR spectra of all of the intermediates and the hapten can be found in the Supplementary Materials file.
Hapten ALb was synthesized following a similar procedure as hapten ALa, except that in this case the tricyclic benzochromenone core was built first, with the hydroxyl groups appropriately protected to allow subsequent incorporation of the spacer arm at the required C-3 position. As shown in Scheme 2, the synthesis of the benzochromenone ring system began with the palladium-catalyzed cross-coupling reaction between the aryl boronic acid 6 and the previously reported bromobenzaldehyde 9 [24,25]. This coupling was carried out under conditions similar to those previously used for the conversion of 4 and 6 into 7, obtaining the biphenyl-2-carbaldehyde 10 in 77% yield. The formyl group was further oxidized to the carboxylic group under Pinnick oxidation conditions to afford the biphenyl-2-carboxylic acid 11, which was then treated with methanolic HCl at 55 °C to promote deprotection of the MOM groups and further intramolecular esterification, thus completing the formation of the tricyclic benzochromenone system. Under these conditions, both sequential processes worked extremely well, affording 12 in practically quantitative yield. O-alkylation of the phenol-like hydroxyl group at the C-3 position of 12 with methyl 5-bromovalerate, using Cs2CO3 in DMF as base, gave the O-alkylated derivative 13 in 94% yield. The methyl ester of 13 was further converted to the corresponding carboxylic group under enzymatic hydrolytic conditions, yielding 14 also in high yield. The hapten ALb was first obtained by hydrogenolysis of both benzyl ether groups of 14 using 5% Pd on activated carbon as catalyst. With hapten ALb in hand, we activated the carboxylic group using the carbodiimide-NHS procedure as was done for hapten ALa. However, the overall yield from these two processes was low, most likely motivated by an intermolecular esterification reaction between a hydroxyl group and the aliphatic active ester that resulted in the spontaneous formation of a transparent thin film, a polyester polymer, on the flask walls. By reversing the order of these steps, i.e., by activating the carboxylic group first and then releasing the hydroxyl groups, a much better result was obtained. Thus, treatment of carboxylic acid 14 with EDC and NHS as before, followed by hydrogenolysis of the benzyl ether moieties of the resulting N-hydroxysuccinimidyl ester 15 with 5% Pd on activated carbon in acetone, gave the desired N-hydroxysuccinimidyl ester of hapten ALb, ALb-NHS ester, with a very high overall yield. As in the case of the active ester of hapten ALa, the ALb-NHS ester was extracted essentially pure from the reaction as judged by 1H NMR, so it was further used without additional purification by column chromatography. NMR spectra of all of the intermediates and the hapten can be found in the Supplementary Materials file.
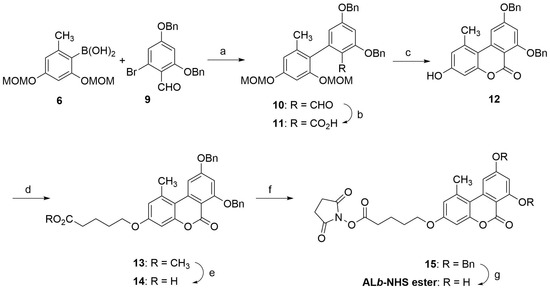
Scheme 2.
Synthesis of ALb-NHS ester. Reagents and conditions: (a) Pd(PPh3)4, K2CO3, DMF, 95 °C, 19 h, 77%. (b) NaH2PO4·H2O, NaClO2, tBuOH-H2O (5:1), rt, 5 h, 96%. (c) iPrOH, THF, conc HCl, 55 °C, 24 h, 98%. (d) Br(CH2)4CO2CH3, Cs2CO3, DMF, 94%. (e) Lipase acrylic resin, THF-PB 100 mM, rt, 20 h, 99%. (f) EDC·HCl, NHS, DMF, rt, overnight. (g) 5% Pd/C, acetone, H2 (1.5 atm), rt, 19 h, 95% of crude product from 14.
2.2. Bioconjugate Preparation
Bioconjugates of haptens ALa and ALb were prepared by the active ester method. The activated haptens were dissolved in dimethyl sulfoxide (DMSO) instead of DMF to improve the solubility. Moreover, the number of hapten equivalents required for efficiently labelling the studied proteins was higher than usual. Commonly, 20-fold hapten-to-protein molar excess for bovine serum albumin (BSA), and 10-fold excess for ovalbumin (OVA) and horseradish peroxidase (HRP) are usually employed in our laboratory. For these haptens, 40-fold and 15-fold excess was used for BSA and HRP conjugates, respectively. Moreover, extremely slow addition of the hapten over the protein solution was required. These concentrations and procedures were necessary, probably due to low hapten solubility in buffer and potential intermolecular polymerization reactions that inactivate the hapten. The obtained bioconjugates were purified by size-exclusion chromatography and characterized by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF/MS) analysis. The two BSA conjugates had similar hapten densities, with hapten-to-protein molar ratios of 15.2 and 18.6 for BSA-ALa and BSA-ALb, respectively, which is considered optimal for immunogens—excessive molar ratios could lead to low protein solubility, and higher or lower hapten densities could be counter-productive for high-affinity antibody generation. Regarding ovalbumin (OVA) conjugates, molar ratios were lower than those of BSA conjugates—around 3 for both haptens –, as it is desirable for coating conjugates to enhance the competitive reaction with the target analyte. Finally, the hapten densities of the enzyme tracers were estimated to be 2.0 and 2.2 for haptens ALa and ALb, respectively, which is within the expected range for HRP conjugates. The MALDI spectra of the prepared bioconjugates can be seen in Figure 2.
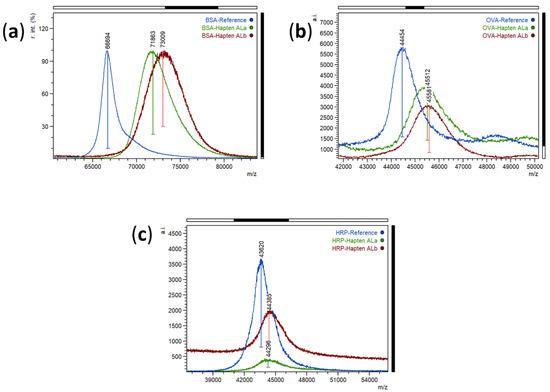
Figure 2.
MALDI-TOF mass spectra (singly charged ions) of proteins (blue) and bioconjugates with hapten ALa (green) and hapten ALb (brick-red). (a) Normalized spectra of BSA and BSA conjugates, (b) Spectra of OVA and OVA conjugates, and (c) Spectra of HRP and HRP conjugates.
2.3. Assessment of the Immune Response
Four polyclonal antibodies were generated in this study, two from each BSA-hapten conjugate. To evaluate the immune response to the prepared synthetic haptens, binding of the antibodies to the homologous conjugate—the conjugate with the same hapten that was used to generate the corresponding antibody—was studied by checkerboard competitive ELISA, using the direct and the indirect assay formats.
Concerning direct assays, the IC50 values for AOH of the obtained antibodies were in the low nanomolar range (Table 1). ALa-type antibodies showed equal or similar IC50 values for AOH and AME. In particular, antibody ALa#1 showed very high affinity—IC50 values were 2.2 nM—for both mycotoxins, and the cross-reactivity (CR) values of antibodies ALa#1 and ALa#2 for AME were 100% and 199%, respectively. These are the first reported polyclonal antibodies with equivalent recognition to both Alternaria toxins. To date, only one monoclonal antibody with such specificity has been published [15]. In contrast, ALb-type antibodies bound AOH with high affinity, but their recognition for AME was negligible—CR values were below 1% (Table 1). The IC50 values to AOH of these specific antibodies were 1.2 nM, an affinity comparable to that of previously published polyclonal antibodies [11,13,16]. The position of the spacer arm in hapten ALa provided a closer mimic of the alkylated hydroxyl group of AME (C-9 position), whereas in hapten ALb the hydroxyl groups at C-7 and C-9 were unsubstituted, as in the molecule of AOH (Figure 1). Therefore, display of the hydroxyl group at C-9 was maximized in hapten ALb, which explains the much lower affinity of ALb-type antibodies for AME compared to AOH.

Table 1.
Antibody characterization by checkerboard direct and indirect competitive ELISA using the corresponding homologous conjugate (n = 3) a.
Regarding the indirect assay format, the four antibodies bound the corresponding homologous coating conjugate. As observed with the direct format, the ALa-derived antibodies recognized AOH and AME, whereas the ALb-derived antibodies were more specific to AOH (Table 1). The IC50 values were consistent with previously published results for indirect competitive ELISA with polyclonal antibodies [10,11,16]. Our strategy to prepare immunizing haptens with opposite linker tethering sites clearly demonstrated that the linker position strongly determines the specificity of antibodies to these Alternaria mycotoxins.
2.4. Assessment of Heterologous Conjugates
Heterologous conjugates constitute a well-known strategy for improving the sensitivity of immunoassays. To further characterize the generated antibodies, competitive assays were carried out using the heterologous conjugate, i.e., assay conjugates of haptens ALa and ALb for ALb- and ALa-type antibodies, respectively. In the direct assay format, low binding to the heterologous tracer—with the linker on the opposite side of the AOH molecule compared to the immunizing conjugate—was observed (Amax values were below 0.6). In contrast, the change in the linker attachment site was not detrimental to hapten recognition in the indirect format, as the four antibodies bound the corresponding heterologous coating conjugate (Table 2). Reasonably, higher antibody and/or conjugate concentrations were required with the heterologous conjugates to reach sufficient signal. The obtained IC50 values using the heterologous coating conjugate were mostly lower than those obtained with the homologous assays. Anyway, CR values did not significantly change with heterologous conjugates.
Table 2.
Antibody characterization by checkerboard indirect competitive ELISA using the corresponding heterologous coating conjugate (n = 3) a.
3. Conclusions
In this study, two de novo synthesized and purified AOH haptens were comprehensively characterized by spectrometric methods, and bioconjugates with unique structure and composition were prepared for the first time. In this perspective, it is worth noting the challenges of obtaining stable enzyme tracers with high activity. This matter was most likely caused by the chemical characteristics of Alternaria toxins and their haptens, which could explain why no direct competitive immunoassays for these mycotoxins have been reported up to now. Once this issue was overcome, the resultant immunoreagents were thoroughly investigated utilizing both direct and indirect competitive ELISA, as well as homologous and heterologous conjugates. Remarkably, antibodies capable of binding AOH and AME with affinities in the low nanomolar range were eventually generated from both haptens. Given that the levels of these mycotoxins are not yet regulated, both specific and generic antibodies are relevant. Our findings showed that hapten ALa, with the linker at the methylated hydroxyl group in AME (C-9 position), was particularly well-suited for producing antibodies that recognized similarly both toxins, whereas antibodies generated from hapten ALb, with the spacer arm at the hydroxyl group in C-3 position, primarily bound AOH. In contrast to previous one-pot hapten synthesis and bioconjugation procedures, the strategy described here for producing AOH haptens with alternative linker tethering sites not only enabled high-affinity antibodies with different specificities, but it may also help to improve the sensitivity of immunoassays to Alternaria mycotoxins by using site heterologous haptens.
4. Materials and Methods
4.1. Reagents and Instruments
Standard AOH [3,7,9-trihydroxy-1-methyl-benzo[c]chromen-6-one, CAS registry number 641-38-3, Mw 258.23] and AME [3,7-dihydroxy-9-methoxy-1-methyl-benzo[c]chromen-6-one, CAS registry number 23452-05-3, Mw 272.25] from Alternaria sp. were purchased from Merck (Darmstadt, Germany). Mycotoxins were dissolved in anhydrous N,N-dimethylformamide (DMF), and the stock solutions were stored at −20 °C. Phosphate buffered saline (PBS) 10× solution (Fisher BioReagents BP399-20) was from Thermo Fisher Scientific (Waltham, MA, USA). Immunizing bioconjugates were prepared with BSA, fraction V, obtained from Roche Applied Science (Mannheim, Germany). OVA, HRP, Freund’s adjuvants, and adult bovine serum, were acquired from Merck (Darmstadt, Germany). Polyclonal goat anti-rabbit (GAR) immunoglobulins antibody and polyclonal goat anti-rabbit immunoglobulins antibody conjugated to peroxidase (GAR-HRP) were purchased from Rockland Immunochemicals Inc. (Pottstown, PA, USA) and BioRad (Madrid, Spain), respectively. 3,3′,5,5′-Tetramethylbenzidine (TMB) liquid substrate for ELISA was obtained from Biopanda Reagents Ltd. (Belfast, UK). Other reagents, materials, and instruments employed for bioconjugate preparation and ELISA experiments are described in the Supplementary Materials file.
4.2. Synthesis of the N-hydroxysuccinimidyl Ester of Hapten ALa
4.2.1. Preparation of methyl 5-((5-hydroxy-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)pentanoate (3)
Methyl 5-bromovalerate (186 µL, 266 mg, 1.36 mmol, 1.1 equiv) was added to a solution of 1,3-benzodioxinone 1 (260 mg, 1.237 mmol), KI (83 mg, 0.500 mmol, 0.4 equiv), Bu4NBr (0.5 mg, 1.6 µmol) y K2CO3 (188 mg, 1.36 mmol, 1 equiv) in dry acetone (9 mL) under nitrogen. After heating the mixture at reflux for 16 h, the acetone was eliminated at reduced pressure and the resulting brownish residue was diluted with water and extracted with Et2O. The combined organic layers were washed with water and brine, dried over anhydrous MgSO4 and concentrated under vacuum. The obtained crude product was purified by chromatography on silica gel, using hexane-EtOAc mixtures from 9:1 to 7:3 as eluent, to afford, in order of elution, dialkylated derivative 2 (67.7 mg, 12.5%) and monoalkylated compound 3 (300 mg, 75%) as a white solid. Mp 97.3–98.2 °C (crystallized from hexane-EtOAc) IR (ATR) νmax (cm−1) 3017 (w), 1740 (s), 1672 (s), 1251 (s), 1159 (s), 840 (s), 794 (s); 1H NMR (300 MHz, CDCl3) δ 10.42 (s, 1H, OH), 6.11 (d, J = 2.2 Hz, 1H, H-6), 5.97 (d, J = 2.2 Hz, 1H, H-8), 3.98 (t, J = 5.8 Hz, 2H, H2-5), 3.68 (s, 3H, OCH3), 2.39 (t, J = 7.0 Hz, 2H, H2-2), 1.81 (m, 4H, H2-3 and H2-4), 1.72 (s, 6H, 2×CH3); 13C NMR (75 MHz, CDCl3) δ 173.8 (CO2CH3), 167.2 (CO), 165.3 (C-7), 163.2 (C-8a), 156.9 (C-5), 107.0 (C-2), 96.3 (CH-6), 95.1 (CH-8), 93.1 (C-4a), 68.1 (CH2-5), 51.7 (OCH3), 33.7 (CH2-2), 28.4 (CH2-4), 25.8 (2×CH3), 21.6 (CH2-3); HRMS (TOF MS ES+) m/z calculated for C16H20O7 [M + H]+ 325.1282, found 325.1283.
4.2.2. Preparation of methyl 5-((2,2-dimethyl-4-oxo-5-(((trifluoromethyl)sulfonyl)oxy)-4H-benzo[d][1,3]dioxin-7-yl)oxy)pentanoate (4)
Triflic anhydride (230 µL, 1.369 mmol, 1.5 equiv) was added to a solution of phenol 3 (296 mg, 0.913 mmol) in anhydrous pyridine (4.5 mL) at 0 °C under nitrogen. The reaction mixture was allowed to warm to rt and stirred for 20 h, then cooled down to 0 °C and treated with a saturated aqueous solution of NaHCO3, stirred for a few minutes at rt and then extracted with Et2O. The organic layers were washed with water, a 1% (w/v) aqueous solution of CuSO4 and brine, dried over anhydrous MgSO4 and concentrated at reduced pressure. The obtained residue was chromatographed on silica gel, using hexane-EtOAc mixtures from 9:1 to 8:2 as eluent, to give aryl triflate 4 (378.8 mg, 91%) as a white semisolid. IR (ATR) νmax (cm−1) 3114 (w), 1746 (s), 1733 (s), 1381 (s), 1228 (s), 1167 (s), 869 (s); 1H NMR (300 MHz, CDCl3) δ 6.51 (d, J = 2.3 Hz, 1H, H-6), 6.45 (d, J = 2.3 Hz, 1H, H-8), 4.02 (t, J = 5.8 Hz, 2H, H2-5), 3.68 (s, 3H, CO2CH3), 2.40 (t, J = 6.9 Hz, 2H, H2-2), 1.85 (m, 4H, H2-3 and H2-4), 1.73 (s, 6H, 2xCH3); 13C NMR (75 MHz, CDCl3) δ 173.7 (CO2CH3), 165.0 (CO), 158.9 (C-7), 157.2 (C-8a), 150.1 (C-5), 106.7 (C-2), 105.7 (CH-6), 101.6 (CH-8), 101.0 (C-4a), 68.9 (CH2-5), 51.8 (OCH3), 33.6 (CH2-2), 28.3 (CH2-4), 25.7 (2×CH3), 21.5 (CH2-3); 19F NMR (282 MHz, CDCl3) δ 73.1 (s, CF3); HRMS (TOF, ES+) m/z calculated for C17H23F3NO9S [M + NH4]+ 474.1040; found 474.1027.
4.2.3. Preparation of methyl 5-((5-(2,4-bis(methoxymethoxy)-6-methylphenyl)-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-7-yl)oxy)pentanoate (7)
(i) Preparation of boronic acid 6. A solution of n-BuLi in hexane (1.3 M, 336 µL, 0.436 mmol, 1.05 equiv) was dropwise added to a solution of aryl bromide 5 (122.3 mg, 0.420 mmol) in anhydrous THF (2.5 mL) at −78 °C under nitrogen. The reaction mixture was stirred at this temperature for 40 min, B(OiPr)3 (322 µL, 1.386 mmol, 3.3 equiv) was then added and the mixture stirred for 1.5 h. After this time, the dry ice bath was replaced by an ice bath and the mixture treated with an aqueous saturated solution of NH4Cl (0.7 mL), then diluted with water and extracted with Et2O. The organic layers were washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give boronic acid 6 (100.0 mg, 93%) as a thick oil that was immediately used in the next reaction without further purification since it is relatively prone to protodeboronation [26]. 1H NMR (300 MHz, DMSO-d6) δ 7.91 (s, 1H, BOH), 6.49 (d, J = 2.1 Hz, 1H, H-4), 6.47 (d, J = 2.10 Hz, 1H, H-6), 5.12 and 5.09 (each s, 2H each, 2×OCH2O), 3.37 and 3.34 (each s, 3H each, 2×OCH3), 2.20 (s, 3H, CH3).
(ii) Coupling reaction between aryl triflate 4 and boronic acid 6. A mixture of the above obtained boronic acid 6 (47.4 mg, 0.185 mmol), aryl triflate 4 (41.6 mg, 0.091 mmol), powdered K2CO3 (43.2 mg, 0.312 mmol) and Pd(PPh3)4 (11.4 mg, 9.9 µmol) under nitrogen was dissolved in anhydrous DMF (1.2 mL), previously degassed by three freeze-vacuum-thaw cycles. The mixture was heated at 93 °C and stirred at this temperature for 24 h. The mixture was cooled to rt, quenched with water and extracted with EtOAc. The combined organic layers were successively washed with water, a 1.5% (w/v) aqueous solution of LiCl and brine, and dried over anhydrous MgSO4. The obtained residue after evaporation of the solvent was chromatographed on silica gel, using hexane-EtOAc 8:2 as eluent, to afford biaryl compound 7 (35.2 mg, 75%) as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 6.71 (d, J = 2.3 Hz, 1H, H-6), 6.64 (d, J = 2.3 Hz, 1H, H-8), 6.42 (d, J = 2.5 Hz, 1H, H-5), 6.40 (d, J = 2.5 Hz, 1H, H-3), 5.18 (AB system, J = 6.7 Hz, 2H, OCH2O), 4.98 (AB system, J = 6.6 Hz 2H, OCH2O), 3.99 (t, J = 5.6 Hz, 2H, H2-5), 3.67 (s, 3H, CO2CH3), 3.50 and 3.29 (each s, 3H each, 2×OCH3), 2.39 (t, J = 6.8 Hz, 2H, H2-2), 2.04 (s, 3H, CH3 Ph), 1.81 (m, 4H, H2-3 and H2-4), 1.71 (s, 6H, 2×CH3); 13C NMR (75 MHz, CDCl3) δ 173.9 (CO2CH3), 164.1 (CO), 159.2 (C-7), 158. 5 (C-8a), 157.4 (C-4), 154.8 (OC-2), 142.9 (C-6), 136.9 (C-5), 123.9 (C-1), 113.8 (CH-3), 110.6 (CH-8), 106.8 (C-2), 105.1 (CH-5), 101.2 (CH-6), 94.9 and 94.7 (2×OCH2O), 68.1 (CH2-5), 56.3 and 55.9 (2×OCH3), 51.8 (CO2CH3), 33.7 (CH2-2), 28.6 (CH2-4), 26.3 and 25.2 (2×CH3), 21.7 (CH2-3), 20.7 (CH3 Ph). HRMS (TOF, ES+) m/z calculated for C27H34O10 [M + H]+ 519.2225; found 519.2212.
4.2.4. Preparation of methyl 5-((3,7-dihydroxy-1-methyl-6-oxo-6H-benzo[c]chromen-9-yl)oxy)pentanoate (8)
A 3 M solution of HCl in MeOH (150 µL, 0.450 mmol) was added to a solution of biaryl compound 7 (26.1 mg, 0.050 mmol) in anhydrous MeOH (1.5 mL) and the reaction mixture was stirred at rt for 22 h. After concentration under vacuum, the residue was dissolved in anhydrous CH2Cl2 (4 mL) and treated with trifluoroacetic acid (430 µL). Following stirring for 20 h at rt, thin layer chromatography showed the formation of a single compound and all the volatiles were removed under vacuum, using CHCl3 to co-evaporate the last traces of TFA. The obtained residue was purified by chromatography, using CHCl3 as eluent, to give benzochromenone derivative 8 (18.1 mg, 97%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.79 and 10.33 (each s, 1H each, 2×OH), 7.13 (d, J = 2.2 Hz, 1H, H-10), 6.69 (d, J = 2.6 Hz, 1H, H-2), 6.61 (d, J = 2.6 Hz, 1H, H-4), 6.55 (d, J = 2.2 Hz, 1H, H-8), 4.11 (t, J = 5.9 Hz, 2H, H2-5), 3.59 (s, 3H, OCH3), 2.68 (s, 3H, CH3), 2.41 (t, J = 7.0 Hz, 2H, H2-2), 1.85–1.56 (m, 4H, H2-3 and H2-4); 13C NMR (75 MHz, DMSO-d6) δ 173.2 (CO2CH3), 165.5 (CO), 164.6 (C-9), 164.1 (C-7), 158.5 (C-3), 152.6 (C-4a), 138.4 (C-1), 137.7 (C-10a), 117.5 (CH2-2), 108.8 (C-10b), 103.6 (CH-10), 101.6 (CH-4), 99.5 (CH-8), 98.3 (C-6a), 67.8 (CH2-5), 51.2 (CO2CH3), 32.8 (CH2-2), 27.8 (CH2-4), 25.0 (CH3), 21.1 (C-3); HRMS (TOF, ES+) m/z calculated for C20H21O7 [M + H]+ 373.1282; found 373.1278.
4.2.5. Preparation of 5-((3,7-dihydroxy-1-methyl-6-oxo-6H-benzo[c]chromen-9-yl)oxy)pentanoic acid (Hapten ALa)
Lipase from Candida antarctica immobilized on acrylic resin (23 mg) was added to a solution of methyl ester 8 (16.6 mg, 0.0446 mmol) in a 4:1 mixture of 100 mM sodium phosphate buffer (pH 7.4) and THF (1.5 mL) at 30 °C. The resulting heterogeneous mixture was smoothly stirred for 24 h at rt and then filtered to separate the enzyme. The filtrated and washing THF phases were combined, diluted with EtOAc, washed with brine, dried over anhydrous MgSO4, and concentrated in vacuo to afford hapten ALa (14.9 mg, 93%) as a white amorphous solid. 1H NMR (300 MHz, THF-d8) δ 11.99 and 9.19 (each s, 1H each, 2×OH), 7.27 (d, J = 2.2 Hz, 1H, H-10), 6.67 (d, J = 2.7 Hz, 1H, H-2), 6.61 (d, J = 2.6 Hz, 1H, H-4), 6.56 (d, J = 2.2 Hz, 1H, H-8), 4.13 (t, J = 6.1 Hz, 2H, H2-5), 2.78 (s, 3H, CH3), 2.33 (t, J = 7.1 Hz, 2H, H2-2), 1.91–1.77 (m, 4H, H2-3 and H2-4); 13C NMR (126 MHz, THF-d8) δ 174.5 (CO2H), 167.1 (CO), 166.4 (C-9), 166.2 (C-7), 159.9 (C-3), 154.5 (C-4a), 139.5 (C-1), 139.2 (C-10a), 118.5 (CH-2), 110.7 (C-10b), 105.1 (CH-10), 102.8 (CH-4), 100.3 (CH-8), 100.0 (C-6a), 69.2 (CH2-5), 34.0 (CH2-2), 29.6 (CH2-4), 25.0 (CH3, overlapped with solvent signal), 22.6 (CH2-3); HRMS (TOF, ES+) m/z calculated for C19H18O7 [M + H]+ 359.1125; found 359.1122.
4.2.6. Preparation of 2,5-dioxopyrrolidin-1-yl 5-((3,7-dihydroxy-1-methyl-6-oxo-6H-benzo[c]chromen-9-yl)oxy)pentanoate (ALa-NHS Ester)
A solution of hapten ALa (11.0 mg, 30.7 µmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC∙HCl) (7.0 mg, 36.8 µmol, 1.2 equiv) and N-hydroxisuccinimide (NHS) (5.0 mg, 43.4 µmol, 1.4 equiv) in anhydrous DMF (0.6 mL) was stirred at rt under nitrogen overnight. The reaction mixture was diluted with CH2Cl2, washed with water, a 1.5% (w/v) aqueous solution of LiCl and brine, dried over anhydrous MgSO4 and concentrated under reduced pressure to give the N-hydroxysuccinimidyl ester of hapten ALa, ALa-NHS ester, (13.8 mg, ca. 99% of crude product) as a slightly yellowish oil which was used immediately for the preparation of the corresponding protein bioconjugates. 1H NMR (500 MHz, THF-d8) δ 11.99 and 9.06 (each s, 1H each, 2×OH), 7.28 (d, J = 2.2 Hz, 1H, H-10), 6.66 (d, J = 2.6 Hz, 1H, H-2), 6.60 (d, J = 2.7 Hz, 1H, H-4), 6.57 (d, J = 2.2 Hz, 1H, H-8), 4.17 (t, J = 5.8 Hz, 2H, H2-5), 2.78 (s, 3H, CH3), 2.75 (br s, 4H, COCH2CH2CO), 2.72 (t, J = 7.0 Hz, 2H, H2-2), 1.95 (m, 4H, H2-3 and H2-4).
4.3. Synthesis of the N-hydroxysuccinimidyl Ester of Hapten ALb
4.3.1. Preparation of 3,5-bis(benzyloxy)-2′,4′-bis(methoxymethoxy)-6′-methyl-[1,1′-biphenyl]-2-carbaldehyde (10)
An ampoule containing a mixture of freshly prepared aryl boronic acid 6 (104.5 mg, 0.408 mmol, 2 equiv), 2,4-bis(benzyloxy)-6-bromobenzaldehyde 9 (80.9 mg, 0.204 mmol), K2CO3 (63.6 mg, 0.460 mmol, 2.2 equiv) and Pd(PPh3)4 (26.6 mg, 0.023 mmol, 0.1 equiv) in anhydrous DMF (2 mL) was exhaustively degassed by freeze-thaw cycles. The ampoule was closed under vacuum and heated at 95 °C for 19 h. After cooling, the ampoule was opened and the reaction mixture was poured onto water and extracted with EtOAc. The combined organic extracts were washed with water, a 1.5% (w/v) aqueous solution of LiCl and brine, dried under anhydrous MgSO4 and concentrated under vacuum. The resulting crude reaction mixture was chromatographed on silica gel to give biaryl-2-carbaldehyde 10 (93.6 mg, 77%) as a viscous yellowish oil. 1H NMR (500 MHz, CDCl3) δ 10.02 (s, 1H, CHO), 7.54–7.48 (m, 2H, 2×CH Ph), 7.44–7.36 (m, 6H, 6xCH Ph), 7.36–7.29 (m, 2H, 2×CH Ph), 6.72 (d, J = 2.4 Hz, 1H, H-6), 6.65 (d, J = 2.3 Hz, 1H, H-4), 6.64 (d, J = 2.3 Hz, 1H, H-5′), 6.37 (d, J = 2.3 Hz, 1H, H-3′), 5.21–5.16 (m, two overlapped AB systems, 4H, OCH2O and OCH2Ph), 5.09 and 5.06 (AB system, J = 11.7 Hz, 1H each, OCH2Ph), 5.08 and 4.97 (AB system, J = 6.7 Hz, 1H each, OCH2O), 3.51 and 3.27 (each s, 3H each, 2×OCH3), 1.96 (s, 3H, CH3 Ph); 13C NMR (126 MHz, CDCl3) δ 189.6 (CHO), 163.5 (C-3), 162.1 (C-5), 157.6 (C-4′), 155.1 (C-2′), 144.8 (C-6′), 138.1 (C-1), 136.4 and 136.1 (2×C Ph), 128.9 (2×CH Ph), 128.8 (2×CH Ph), 128.4 (CH Ph), 128.1 CH Ph), 127.7 (2×CH Ph), 127.2 (2×CH Ph), 123.1 (C-1′), 118.6 (C-2), 110.7 (CH-5′), 109.6 (CH-3′), 101.2 (CH-6), 100.1 (CH-4), 94.7 and 94.6 (2×OCH2O), 70.7 and 70.4 (2×OCH2Ph), 56.3 and 56.1 (2×OCH3), 20.6 (CH3 Ph); HRMS (TOF, ES+) m/z calculated for C32H33O7 [M + H]+ 529.2221, found 529.2205.
4.3.2. Preparation of 3,5-bis(benzyloxy)-2′,4′-bis(methoxymethoxy)-6′-methyl-[1,1′-biphenyl]-2-carboxylic Acid (11)
NaH2PO4·H2O (58.6 mg, 0.425 mmol, 2.8 equiv), 2-methylbut-2-ene (322.1 µL, 3.04 mmol, 20 equiv) and NaClO2 (45.3 mg, 0.501 mmol, 3.3 equiv) were successively added to a solution of biaryl-2-carbaldehyde 10 (80.4 mg, 0.152 mmol) in tBuOH (3.2 mL) and milli-Q water (0.4 mL) at 0 °C. The mixture was allowed to warm at rt and stirred for 5 h, then diluted with an aqueous saturated solution of NH4Cl and extracted with EtOAc. The combined organic layers were washed with brine and dried over anhydrous MgSO4. Chromatography on silica gel of the residue left after evaporation of the solvent at reduced pressure, using 8:2 hexane-EtOAc as eluent, gave the biaryl-2-carboxylic acid 11 (79.5 mg, 96%) as a semi solid. 1H NMR (500 MHz, CDCl3) δ 9.32 (s, 1H, CO2H), 7.61–7.32 (m, 10H, 10xCH Ph), 6.68 (d, J = 2.4 Hz, 2H, H-6 and H-4), 6.65 (d, J = 2.4 Hz, 1H, H-5′), 6.44 (d, J = 2.3 Hz, 1H, H-3′), 5.21–5.14 (m, two overlapped AB systems, 4H, OCH2O and OCH2Ph), 5.07 and 5.04 (AB system, J = 11.7 Hz, 1H each, OCH2Ph), 4.99 (br s, 2H, OCH2O), 3.50 and 3.16 (each s, 3H each, 2×OCH3), 2.01 (s, 3H, CH3 Ph); 13C NMR (126 MHz, CDCl3) δ 165.7 (CO2H), 161.1 (C-3), 157.8 (C-5), 157.6 (C-4′), 155.1 (C-2′), 141.3 (C-6′), 138.4 and 135.7 (2×C Ph), 128.9 (2×CH Ph), 128.8 (2×CH Ph), 128.6 (CH Ph), 128.4 (CH Ph), 127.7 (2×CH Ph), 127.6 (2×CH Ph), 125.1 (C-2), 115.9 (C-1′), 111.6 (CH-5′), 109.9 (CH-3′), 102.6 (CH-6), 100.3 (CH-4), 96.1 and 94.7 (2×OCH2O), 71.5 and 70.4 (2×OCH2Ph), 56.3 and 56.1 (2×OCH3), 20.5 (CH3 Ph); HRMS (TOF, ES+) m/z calculated for C32H33O8 [M + H]+ 545.2170, found 545.2156.
4.3.3. Preparation of 7,9-bis(benzyloxy)-3-hydroxy-1-methyl-6H-benzo[c]chromen-6-one (12)
A 50:1 (v/v) mixture of iPrOH and concentrated HCl (1.7 mL) was added to a solution of biaryl-2-carboxylic acid 11 (69.4 mg, 0.127 mmol) in THF (5.1 mL) at rt under nitrogen. The mixture was thermostated at 55 °C in an oil bath and stirred at this temperature for 24 h. After this time, the mixture was cooled to rt, diluted with a concentrated aqueous solution of NaHCO3 and extracted with Et2O. The organic phase was washed with brine, dried over anhydrous MgSO4 and concentrated under vacuum to give 7,9-bis(benzyloxy)alternariol 12 (54.7 mg, 98%) as an amorphous whitish solid. The crude reaction product thus obtained was sufficiently pure, as judged by its NMR spectroscopic data, to be used in the next step without further purification. 1H NMR (500 MHz, DMSO-d6) δ 7.58 (d, J = 7.4 Hz, 2H, 2×CH Ph), 7.48 (d, J = 7.1 Hz, 2H, 2×CH Ph), 7.44–7.31 (m, 6H, 6xCH Ph), 7.28 (d, J = 2.2 Hz, 1H, H-10), 6.90 (d, J = 2.2 Hz, 1H, H-8), 6.63 (d, J = 2.7 Hz, 1H, H-2), 6.53 (d, J = 2.7 Hz, 1H, H-4), 5.31 and 5.29 (each s, 2H each, 2×OCH2Ph), 2.63 (s, 3H, CH3 Ph); 13C NMR (126 MHz, DMSO-d6) δ 163.6 (CO), 162.4 (C-9), 158.4 (C-7), 156.5 (C-3), 153.7 (C-4a), 140.0 (C-1), 138.0 (C-10a), 136.8 and 136.3 (2×CH Ph), 128.7 (2×CH Ph), 128.5 (2×CH Ph), 128.3 (CH Ph), 127.9 (2×CH Ph), 127.7 (CH Ph), 127.0 (2×CH Ph), 116.7 (CH-2), 109.1 (C-10b), 103.2 (C-6a), 102.8 (CH-10), 100.9 (CH-4), 99.8 (CH-8), 70.1 and 69.9 (2×OCH2Ph), 25.0 (CH3 Ph); HRMS (TOF, ES+) m/z calculated for C28H23O5 [M + H]·+ 439.1540, found 439.1530.
4.3.4. Preparation of methyl 5-((7,9-bis(benzyloxy)-1-methyl-6-oxo-6H-benzo[c]chromen-3-yl)oxy)pentanoate (13)
Methyl bromovalerate (29.5 mg, ca. 22 µL, 0.151 mmol, 1.1 equiv) was added via syringe to a stirred suspension of Cs2CO3 (57.8 mg, 0.177 mmol, 1.3 equiv) and phenol 12 (60.1 mg, 0.137 mmol) in anhydrous DMF (2 mL) at rt under nitrogen and the mixture was stirred for 19 h. The resulting pale yellowish reaction mixture was diluted with water and extracted with EtOAc. The combined organic extracts were washed successively with water, a 1.5% (w/v) aqueous solution of LiCl and brine, dried over anhydrous MgSO4 and concentrated under reduced pressure. The crude reaction product was purified by chromatography on silica gel, using CHCl3 as eluent, to afford the O-alkylated product 13 (71.4 mg, 94%) as a pale yellowish semi-solid. 1H NMR (500 MHz, CDCl3) δ 7.59 (m, 2H, CH Ph), 7.43–7.34 (m, 8H, 8×CH Ph), 7.30 (d, J = 2.3 Hz, 1H, H-2), 6.67 (d, J = 2.7 Hz, 1H, H-10), 6.64 (d, J = 4.6 Hz, 2H, H-4), 6.64 (s, 1H, H-8), 5.28 and 5.16 (each s, 2H each, 2×OCH2Ph), 4.00 (t, J = 5.5 Hz, 2H, H2-5), 3.68 (s, 3H, CO2CH3), 2.67 (s, 3H, CH3), 2.41 (t, J = 6.9 Hz, 2H, H2-2), 1.84 (m, 4H, H2-3 and H2-4); 13C NMR (126 MHz, CDCl3) δ 173.9 (CO2CH3), 163.7 (CO), 162.9 (C-9), 159.5 (C-7), 157.8 (C-3), 154.3 (C-4a), 140.7 (C-1), 137.4 (C-10a), 136.5 and 135.9 (2×C Ph), 129.0 (2×CH Ph), 128.8 (2×CH Ph), 128.6 (CH Ph), 127.9 (CH Ph), 127.5 (2×CH Ph), 126.8 (2×CH Ph), 116.7 (CH-2), 111.0 (C-10b), 104.5 (C-6a), 103.9 (CH-10), 100.0 (CH-4), 99.9 (CH-8), 71.0 and 70.5 (each OCH2Ph), 67.7 (CH2-5), 51.7 (OCH3), 33.8 (CH2-2), 28.6 (CH2-4), 25.6 (CH3), 21.7 (CH2-3); HRMS (TOF, ES+) m/z calculated for C34H33O7 [M + H]+ 553.2221, found 553.2112.
4.3.5. Preparation of 5-((7,9-bis(benzyloxy)-1-methyl-6-oxo-6H-benzo[c]chromen-3-yl)oxy)pentanoic Acid (14)
The hydrolysis of the methyl ester moiety of 13 was performed following the same procedure reported for the hydrolysis of ester 8 to obtain hapten ALa. The methyl ester 13 (17.5 mg, 0.032 mmol), lipase from Candida antarctica immobilized on acrylic resin (16 mg) and a 4:1 mixture of 100 mM sodium phosphate buffer (pH 7.4) and THF (1.1 mL). Workup as described for the hydrolysis of 8 yielded acid 14 (17.0 mg, 99%) as a whitish semi-solid. 1H NMR (500 MHz, THF-d8) δ 7.68 (m, 2H, 2×CH Ph), 7.46 (m, 2H, 2×CH Ph), 7.40–7.30 (m, 6H, 4×CH Ph and H-2), 7.25 (br t, J = 7.5 Hz, 1H, CH Ph), 6.85 (d, J = 2.2 Hz, 1H, H-10), 6.71 (d, J = 2.8 Hz, 1H, H-4), 6.69 (d, J = 2.8 Hz, 1H, H-8), 5.26 (s, 4H, 2×OCH2Ph), 4.05 (t, J = 6.2 Hz, 2H, H2-5), 2.71 (s, 3H, Ar-CH3), 2.32 (t, J = 7.2 Hz, 2H, H2-2), 1.78 (m, 4H, H2-3 and H2-4). 13C NMR (126 MHz, THF-d8) δ 174.5 (CO2H), 164.8 (CO), 163.9 (C-9), 160.9 (C-7), 156.7 (C-3), 155.7 (C-4a), 141.4 (C-1), 138.5 (C-10a), 138.3 and 137.9 (each C Ph), 129.5 (2×CH Ph), 129.2 (2×CH Ph), 129.0 (CH Ph), 128.5 (2×CH Ph), 128.2 (CH Ph), 127.6 (2×CH Ph), 117.2 (CH-2), 111.8 (C-10b), 105.4 (C-6a), 104.5 (CH-10), 100.7 (CH-4), 100.5 (CH-8), 71.4 and 71.1 (each OCH2Ph), 68.8 (CH2-5), 34.0 (CH2-2), 29.7 (CH2-4), 26.5 (CH3), 22.6 (CH2-3); HRMS (TOF, ES+) m/z calculated for C33H31O7 [M + H]+ 539.2064, found 539.2073.
4.3.6. Preparation of 2,5-dioxopyrrolidin-1-yl 5-((7,9-dihydroxy-1-methyl-6-oxo-6H-benzo[c]chromen-3-yl)oxy)pentanoate (ALb-NHS Ester)
The acid 14 obtained in the above step (15.1 mg, 28 µmol) was transformed into the corresponding N-hydroxysuccinimidyl ester 15 (17.2 mg) following the same procedure previously described for the transformation of hapten ALa into ALa-NHS ester, using EDC∙HCl (6.4 mg, 33.6 µmol, 1.2 equiv) and NHS (4.2 mg, 36.5 µmol, 1.3 equiv) in anhydrous DMF (1 mL). 1H NMR (300 MHz, CDCl3) δ 7.59 (m, 2H, 2×CH Ph), 7.45–7.32 (m, 8H, 8xCH Ph), 7.31 (d, J = 2.3 Hz, 1H, H-2), 6.69 (d, J = 2.7 Hz, 1H, H-10), 6.67 (d, J = 2.7 Hz, 1H, H-4), 6.65 (d, J = 2.2 Hz, 1H, H-8), 5.29 and 5.17 (each s, 2H each, 2×OCH2Ph), 4.04 (t, J = 5.6 Hz, 2H, H2-5), 2.85 (br s, 4H, COCH2CH2CO), 2.72 (t, J = 6.9 Hz, 1H, H2-2), 2.68 (s, 3H, CH3), 1.96 (m, 4H, H2-3 and H2-4).
Thereafter, a suspension of 5% Pd/C (8 mg) and 15 in acetone (3 mL) was degassed and purged with hydrogen by several cycles of freeze-pump-thaw using a water aspirator pump. The hydrogen pressure was adjusted to 1.5 atm and the mixture was stirred vigorously overnight at rt. The reaction mixture was filtered through a disposable Teflon membrane filter (0.45 µm), and the filtrate and washing THF phases were combined and concentrated at reduced pressure to give the N-hydroxysuccinimidyl ester of hapten ALb, ALb-NHS ester, (12.2 mg, 95% of crude product from 14) as a viscous colorless oil which was used immediately for the preparation of the corresponding protein bioconjugates. 1H NMR (500 MHz, THF-d8/DMSO-d6) δ 11.92 and 9.67 (each s, 1H each, 2×OH), 7.24 (d, J = 2.1 Hz, 1H, H-2), 6.83 (d, J = 2.7 Hz, 1H, H-4), 6.82 (d, J = 2.6 Hz, 1H, H-10), 6.36 (d, J = 2.1 Hz, 1H, H-8), 4.10 (t, J = 5.6 Hz, 1H, H2-5), 2.79 (s, 3H, CH3), 2.76 (br s, 4H, COCH2CH2CO), 2.71 (t, J = 6.9 Hz, 2H, H2-2), 1.93 (m, 4H, H2-3 and H2-4).
4.4. Immunoreagent Preparation
Protein conjugates of the two haptens were obtained by the active ester method. A 50 mM solution of the activated hapten was prepared in DMSO. BSA and OVA solutions were prepared at 15 mg/mL in 50 mM carbonate buffer, pH 9.6. The activated hapten was added to the protein solution at 40-fold molar excess for BSA conjugates. Conjugation reactions to OVA were done with 8- or 11-fold excess for ALa and ALb, respectively. Concerning HRP conjugates, the activated hapten solution (5 mM) was added over a 3 mg/mL enzyme solution in the same carbonate buffer to reach a hapten molar excess of 15. The activated haptens were added slowly to the protein solution (ca. 15–20 μL per hour), and the mixtures were incubated overnight at rt, protected from light, and with gentle stirring. Then, they were centrifuged for 5 min at 6700× g, and the conjugates were purified from the supernatant by size-exclusion chromatography using 100 mM phosphate buffer, pH 7.4, as eluent. Fractions containing BSA or OVA conjugates were pooled and diluted with elution buffer to a final concentration of 1 mg/mL. BSA conjugate solutions were passed through 0.45 μm sterile filters. BSA and OVA conjugate solutions were stored at −20 °C. HRP conjugate solutions were 1:1 diluted with PBS containing 1% BSA (w/v) and 0.02% (w/v) thimerosal and stored at 4 °C. The hapten-to-protein molar ratio of the prepared conjugates was determined by MALDI-TOF-MS and running BSA, OVA, and HRP for reference in the same plate, as previously described [27].
Animal manipulation was performed according to Spanish laws (RD1201/2005 and law 32/2007) and the European Directive 2010/63EU regarding the protection of experimental animals. Polyclonal antibodies to AOH and AME were obtained from the sera of immunized animals. Briefly, two female New Zealand white rabbits—weighing 2 kg at the beginning of the experiment—were immunized by four periodic subcutaneous injections of a 1:1 water-in-oil emulsion containing 300 μg of the BSA-hapten conjugate. The inoculum was prepared with complete Freund’s adjuvant for the first injection and with incomplete Freund’s adjuvant for subsequent injections. Boosts were applied with 21-day intervals. Animals were exsanguinated by intracardiac puncture 10 days after the last injection, and the blood was left overnight in the refrigerator at 4 °C for coagulation. Sera were separated from cells by centrifugation (3000× g, 20 min). Finally, immunoglobulins were partially purified by precipitation twice with one volume of cold saturated (3.9 M) ammonium sulphate solution. Antibodies were stored at 4 °C as precipitates.
4.5. Competitive ELISA Procedures
Immunoassays were carried out by competitive ELISA using the capture antibody-coated direct format and the conjugate-coated indirect format. After each incubation step, plates were washed three times with a 150 mM NaCl solution containing 0.05% (v/v) Tween 20. For direct assays, microplates were coated by overnight incubation at 4 °C with 100 μL per well of GAR solution (1 μg/mL) in 50 mM carbonate-bicarbonate buffer, pH 9.6. For indirect assays, microwells were coated with 100 μL per well of OVA-hapten conjugate solution in the same coating buffer, and overnight incubation at rt. The competitive reaction was performed by mixing in each well 50 μL of analyte solution in PBS with 50 μL of antibody dilution or enzyme tracer solution in PBS containing 0.05% (v/v) of Tween-20, and incubating 1 h at rt. For indirect assays, 100 μL per well of GAR-HRP diluted 1/10,000 in PBS containing 0.05% (v/v) of Tween-20 and 10% (v/v) of adult bovine serum was added. Signal was obtained using 100 μL per well of TMB as the chromogenic enzyme substrate and incubation at rt during 10 min. Finally, 100 μL of 1 M H2SO4 was added and the absorbance was read at 450 nm using 650 nm as reference wavelength.
Standard mycotoxin solutions were obtained by serially diluting in buffer the most concentrated standard solution, which was prepared from a concentrated stock solution in DMF. Eight-point standard curves were built using those solutions and a blank sample. SigmaPlot software, version 14.0 from Systat Software Inc. (San Jose, CA, USA), was employed to fit the experimental values to a standard four-parameter logistic equation. The half-maximal inhibition concentration (IC50) and the maximum absorbance (Amax) values were considered in order to compare antibody performance. CR was calculated according to Formula (1):
CR (%) = IC50 (AOH)/IC50 (AME) × 100
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/toxins13120883/s1: General procedures, materials, and equipment; NMR spectra of all of the intermediates and the final activated haptens.
Author Contributions
Conceptualization, A.A.-F.; Data curation, A.A.-S., A.A.-F. and J.V.M.; Formal analysis, L.G.A.-M., A.A.-S. and C.A.; Funding acquisition, A.A.-S., A.A.-F. and J.V.M.; Investigation, L.G.A.-M. and C.A.; Methodology, A.A.-S., A.A.-F. and J.V.M.; Supervision, A.A.-S., C.A. and J.V.M.; Validation, L.G.A.-M. and C.A.; Writing—original draft, A.A.-S. and J.V.M.; Writing—review & editing, A.A.-S., A.A.-F. and J.V.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the SPANISH MINISTERIO DE ECONOMÍA Y COMPETITIVIDAD, grant numbers AGL2015-64488 and RTI2018-096121, and cofinanced by EUROPEAN REGIONAL DEVELOPMENT FUNDS. Animal manipulation as well as mass, spectrometric, and proteomic analysis was performed at the SCSIE service of the University of Valencia. The proteomics laboratory is a member of Proteored.
Institutional Review Board Statement
This study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board of GENERALITAT VALENCIANA (protocol code 2019/VSC/PEA/0179) on 20 August 2019.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The technical assistance by Paula Peña-Murgui and José V. Gimeno is greatly appreciated.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Escrivá, L.; Oueslati, S.; Font, G.; Manyes, L. Alternaria mycotoxins in food and feed: An overview. J. Food Qual. 2017, 2017, 1569748. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion on the risks for animal and public health related to the presence of Alternaria toxins in feed and food. EFSA J. 2011, 9, 2407. [Google Scholar] [CrossRef]
- Arcella, D.; Eskola, M.; Gómez-Ruiz, J.A. EFSA Scientific Report. Dietary exposure assessment to Alternaria toxins in the European population. EFSA J. 2016, 14, 4654. [Google Scholar] [CrossRef]
- Ostry, V. Alternaria mycotoxins: An overview of chemical characterization, producers, toxicity, analysis and occurrence in foodstuffs. World Mycotoxin J. 2008, 1, 175–188. [Google Scholar] [CrossRef]
- Saha, D.; Fetzner, R.; Burkhardt, B.; Podlech, J.; Metzler, M.; Dang, H.; Lawrence, C.; Fischer, R. Identification of a polyketide synthase required for alternariol (AOH) and alternariol-9-methyl ether (AME) formation in Alternaria alternata. PLoS ONE 2012, 7, e40564. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzo, M. Recent advances on Alternaria mycotoxins. Curr. Opin. Food Sci. 2017, 17, 57–61. [Google Scholar] [CrossRef]
- Hickert, S.; Bergmann, M.; Ersen, S.; Cramer, B.; Humpf, H.-U. Survey of Alternaria toxin contamination in food from the German market, using a rapid HPLC-MS/MS approach. Mycotoxin Res. 2016, 32, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Man, Y.; Liang, G.; Li, A.; Pan, L. Analytical methods for the determination of Alternaria mycotoxins. Chromatographia 2017, 80, 9–22. [Google Scholar] [CrossRef]
- Chen, A.; Mao, X.; Sun, Q.; Wei, Z.; Li, J.; You, Y.; Zhao, J.; Jiang, G.; Wu, Y.; Wang, L.; et al. Alternaria mycotoxins: An overview of toxicity, metabolism, and analysis in food. J. Agric. Food Chem. 2021, 69, 7817–7830. [Google Scholar] [CrossRef]
- Burkin, A.A.; Kononenko, G.P. Enzyme immunassay of alternariol for the assessment of risk of agricultural products contamination. Appl. Biochem. Microbiol. 2011, 47, 72–76. [Google Scholar] [CrossRef]
- Ackermann, Y.; Curtui, V.; Dietrich, R.; Gross, M.; Latif, H.; Märtlbauer, E.; Usleber, E. Widespread occurrence of low levels of alternariol in apple and tomato products, as determined by comparative immunochemical assessment using monoclonal and polyclonal antibodies. J. Agric. Food Chem. 2011, 59, 6360–6368. [Google Scholar] [CrossRef]
- Kong, D.; Xie, Z.; Liu, L.; Song, S.; Zheng, Q.; Kuang, H. Development of an immunochromatographic assay for the detection of alternariol in cereal and fruit juice samples. Food Agric. Immunol. 2017, 28, 1082–1093. [Google Scholar] [CrossRef][Green Version]
- Singh, G.; Velasquez, L.; Brady, B.; Koerner, T.; Huet, A.-C.; Delahaut, P. Development of an indirect competitive ELISA for analysis of alternariol in bread and bran samples. Food Anal. Methods 2018, 11, 1444–1450. [Google Scholar] [CrossRef]
- Man, Y.; Liang, G.; Jia, F.; Li, A.; Fu, H.; Wang, M.; Pan, L. Development of an immunochromatographic strip test for the rapid detection of alternariol monomethyl ether in fruit. Toxins 2017, 9, 152. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Peng, T.; Zhang, X.; Yao, K.; Ke, Y.; Shao, B.; Wang, Z.; Shen, J.; Jiang, H. A novel hapten and monoclonal antibody-based indirect competitive ELISA for simultaneous analysis of alternariol and alternariol monomethyl ether in wheat. Food Control 2018, 94, 65–70. [Google Scholar] [CrossRef]
- Yao, C.-Y.; Xu, Z.-L.; Wang, H.; Zhu, F.; Luo, L.; Yang, J.-Y.; Sun, Y.-M.; Lei, H.-T.; Tian, Y.-X.; Shen, Y.-D. High affinity antibody based on a rationally designed hapten and development of a chemiluminescence enzyme immunoassay for quantification of alternariol in fruit juice, maize and flour. Food Chem. 2019, 283, 359–366. [Google Scholar] [CrossRef]
- Bräse, S.; Gläser, F.; Kramer, C.; Lindner, S.; Linsenmeier, A.M.; Masters, K.-S.; Meister, A.C.; Ruff, B.M.; Zhong, S. The Chemistry of Mycotoxins; Springer: Berlin/Heidelberg, Germany, 2013; Chapter 11; Volume 97, pp. 127–137. [Google Scholar]
- Koch, K.; Podlech, J.; Pfeiffer, E.; Metzler, M. Total synthesis of alternariol. J. Org. Chem. 2005, 70, 3275–3276. [Google Scholar] [CrossRef]
- Liang, D.; Luo, H.; Liu, Y.-F.; Hao, Z.-Y.; Wang, Y.; Zhang, C.-L.; Zhang, Q.J.; Chen, R.-Y.; Yu, D.-Q. Lysilactones A-C, three 6H-dibenzo[b,d]pyran-6-one glycosides from Lysimachia clethroides, total synthesis of lysilactone A. Tetrahedron 2013, 69, 2093–2097. [Google Scholar] [CrossRef]
- Won, M.; Kwon, S.; Kim, T.-H. An efficient synthesis of alternariol. J. Korean Chem. Soc. 2015, 59, 471–474. [Google Scholar] [CrossRef]
- Martinez-Solorio, D.; Belmore, K.A.; Jennings, M.P. Synthesis of the purported ent-pochonin J structure featuring a stereoselective oxocarbenium allylation. J. Org. Chem. 2011, 76, 3898–3908. [Google Scholar] [CrossRef]
- Mallampudi, N.A.; Choudhury, U.M.; Mohapatra, D.K. Total synthesis of (−)-citreoisocoumarin, (−)-citreoisocoumarinol, (−)-12-epi-citreoisocoumarinol, and (−)-mucorisocoumarins A and B using a gold(I)-catalyzed cyclization strategy. J. Org. Chem. 2020, 85, 4122–4129. [Google Scholar] [CrossRef] [PubMed]
- Hinman, A.W.; Davis, D.; Kheifets, V. Preparation of Resorcinol Compounds for Dermatological Use. U.S. Patent 20140256830 A1, 6 March 2014. [Google Scholar]
- Marcyk, P.; Brown, L.E.; Huang, D.; Cowen, L.E.; Whitesell, L. Preparation of Resorcylate Aminopyrazole Compounds as Hsp90 Inhibitors and Uses. Thereof. Patent WO 2020/227368 A1, 6 May 2020. [Google Scholar]
- Roedel, T.; Gerlach, H. Enantioselective synthesis of the polyketide antibiotic (3R,4S)-(−)-citrinin. Liebigs Ann. 1995, 5, 885–888. [Google Scholar] [CrossRef]
- Lozada, J.; Liu, Z.; Perrin, D.M. Base-promoted protodeboronation of 2,6-disubstituted arylboronic acids. J. Org. Chem. 2014, 79, 5365–5368. [Google Scholar] [CrossRef] [PubMed]
- López-Puertollano, D.; Mercader, J.V.; Agulló, C.; Abad-Somovilla, A.; Abad-Fuentes, A. Novel haptens and monoclonal antibodies with subnanomolar affinity for a classical analytical target, ochratoxin A. Sci. Rep. 2018, 8, 9761. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).