mRNA as a Tool for Gene Transfection in 3D Cell Culture for Future Regenerative Therapy

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
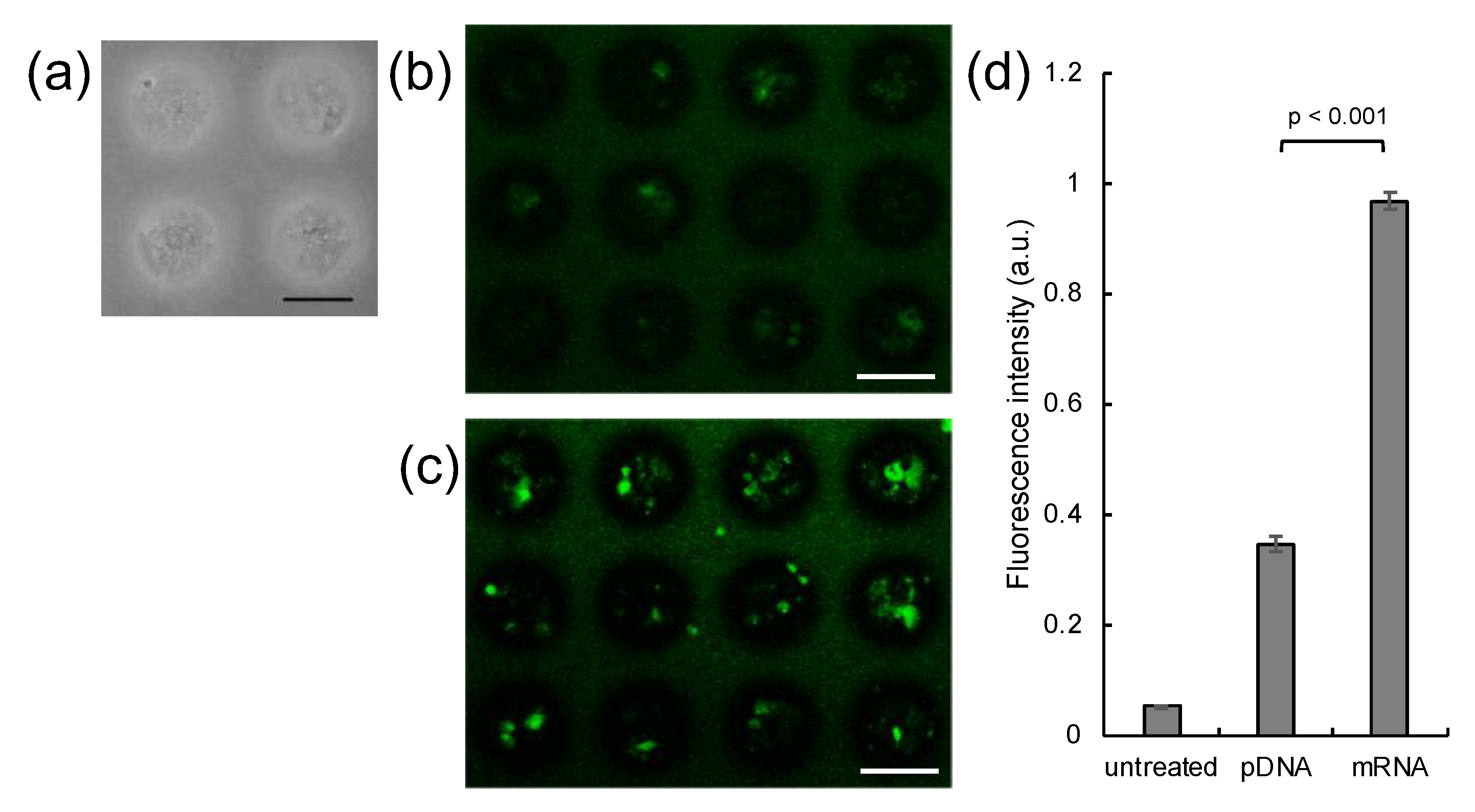
2.1. Spheroid Preparation
2.2. Preparation of mRNA and pDNA
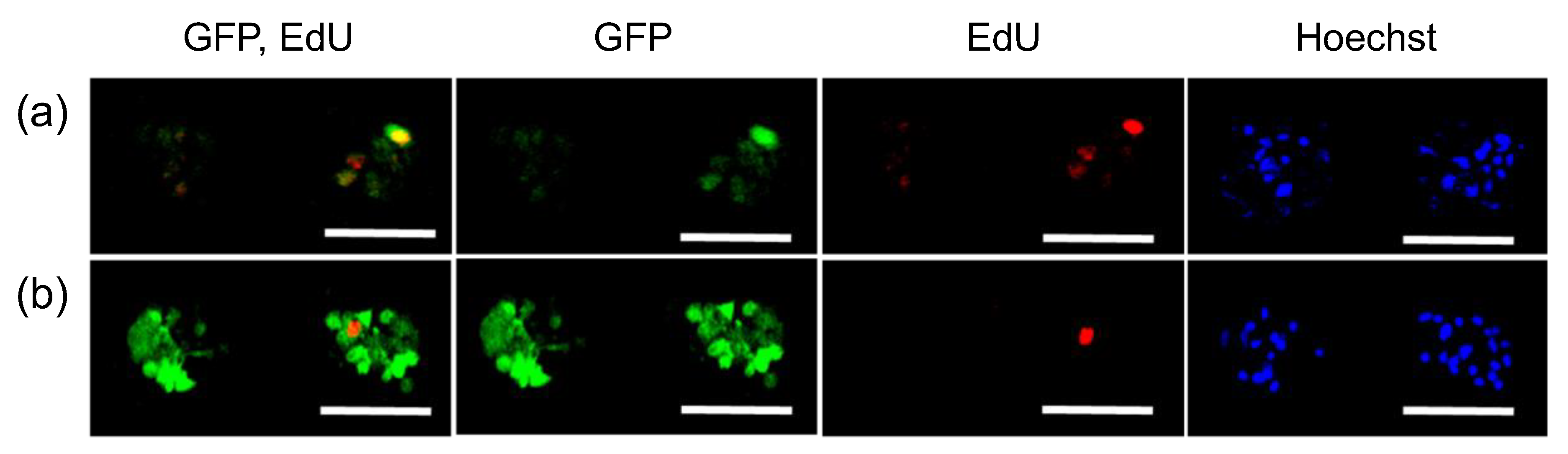
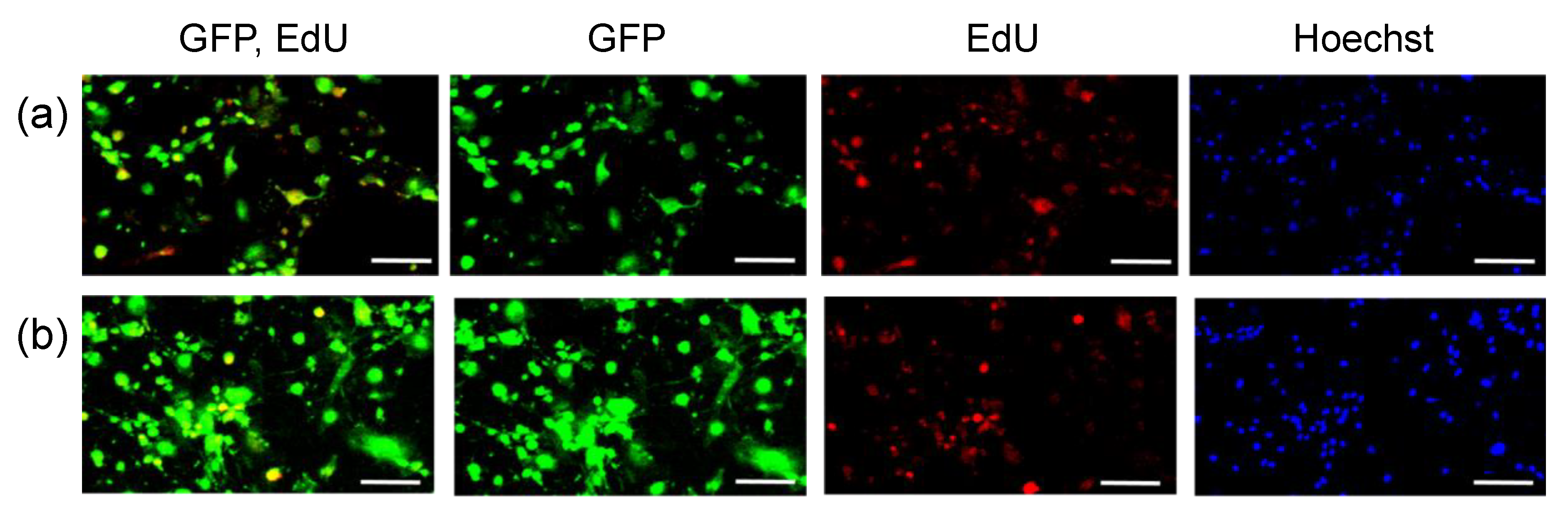
2.3. Transfection of MSCs with GFP mRNA and pDNA
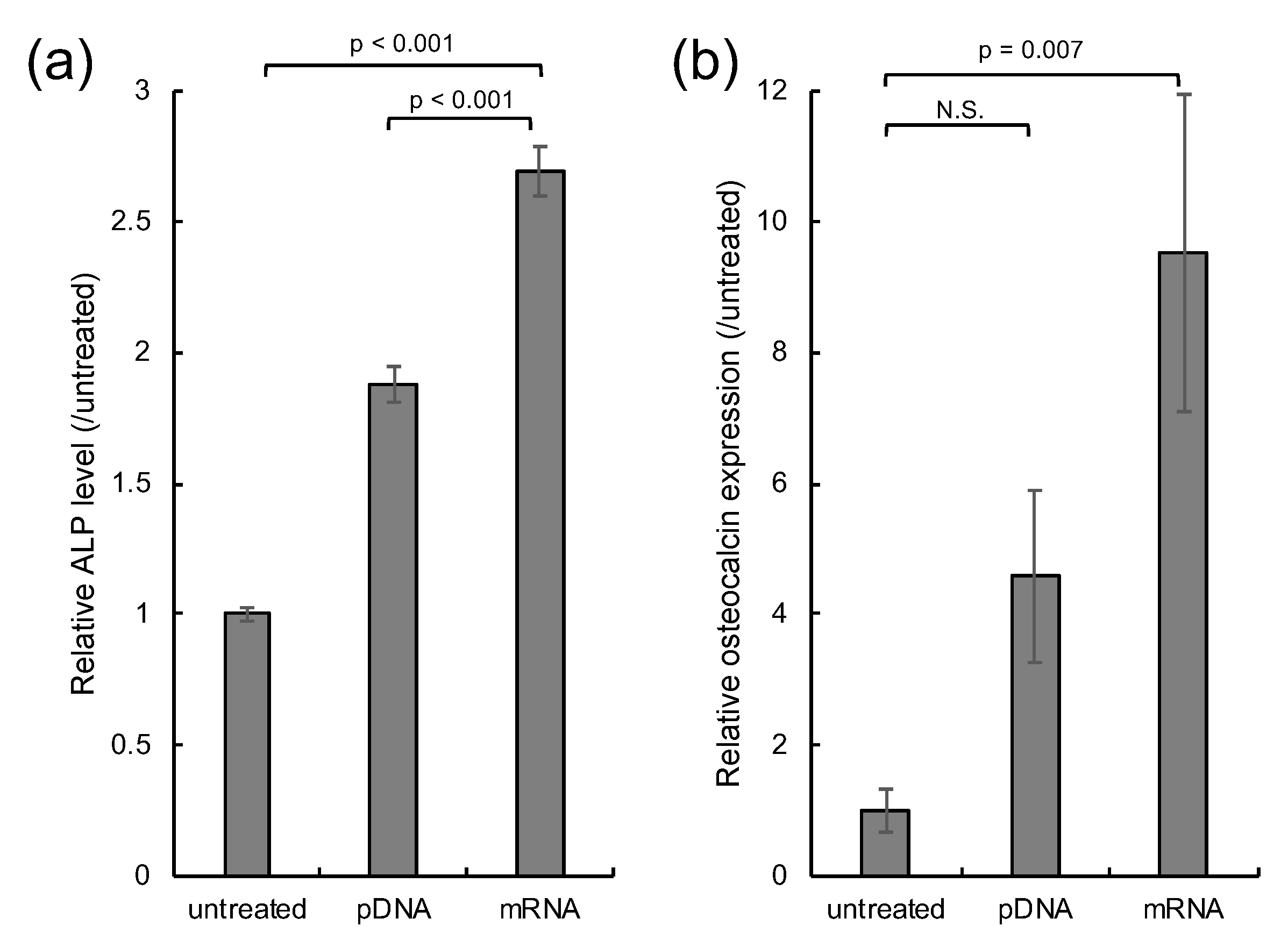
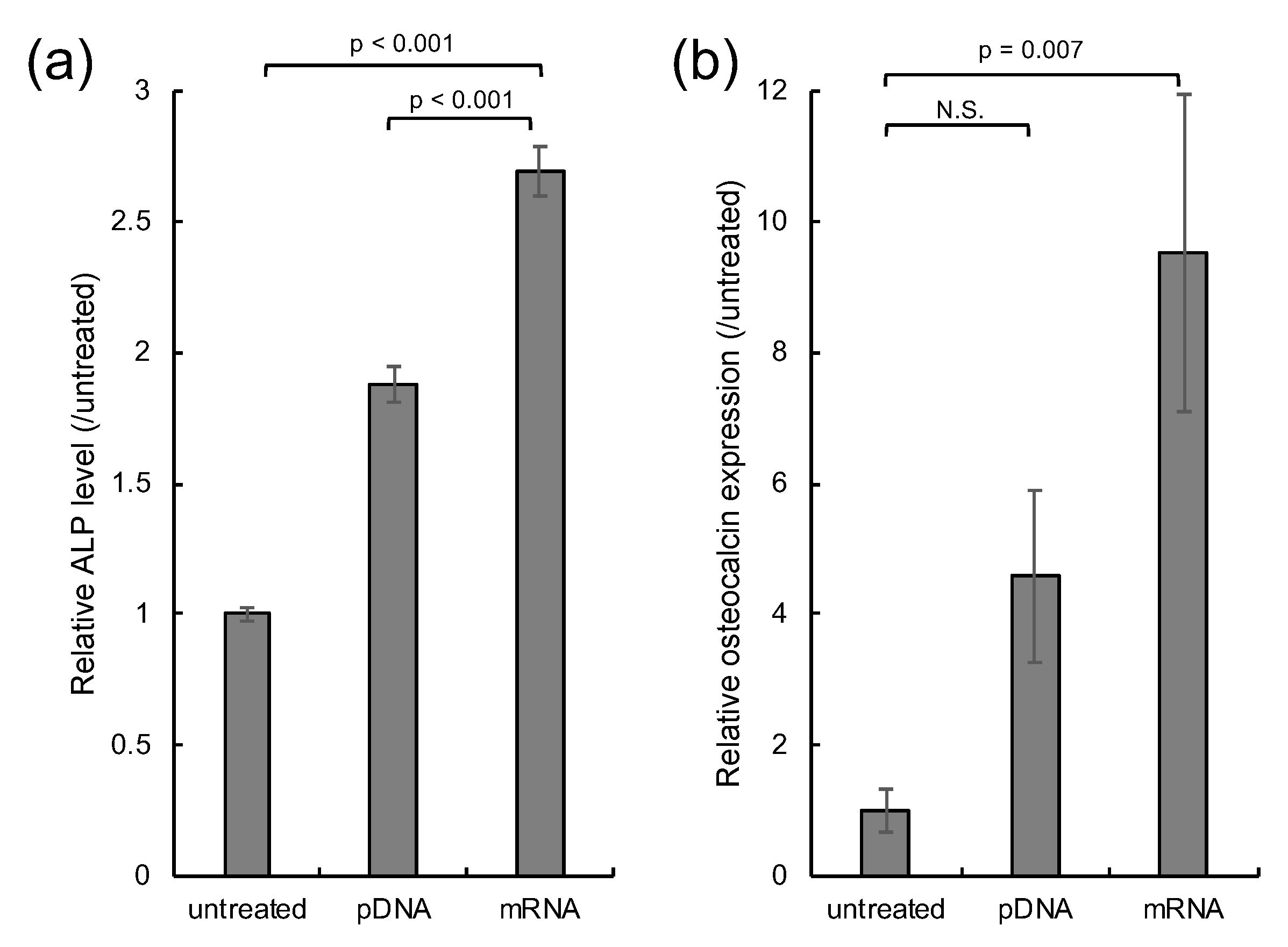
2.4. In Vitro Osteogenic Differentiation
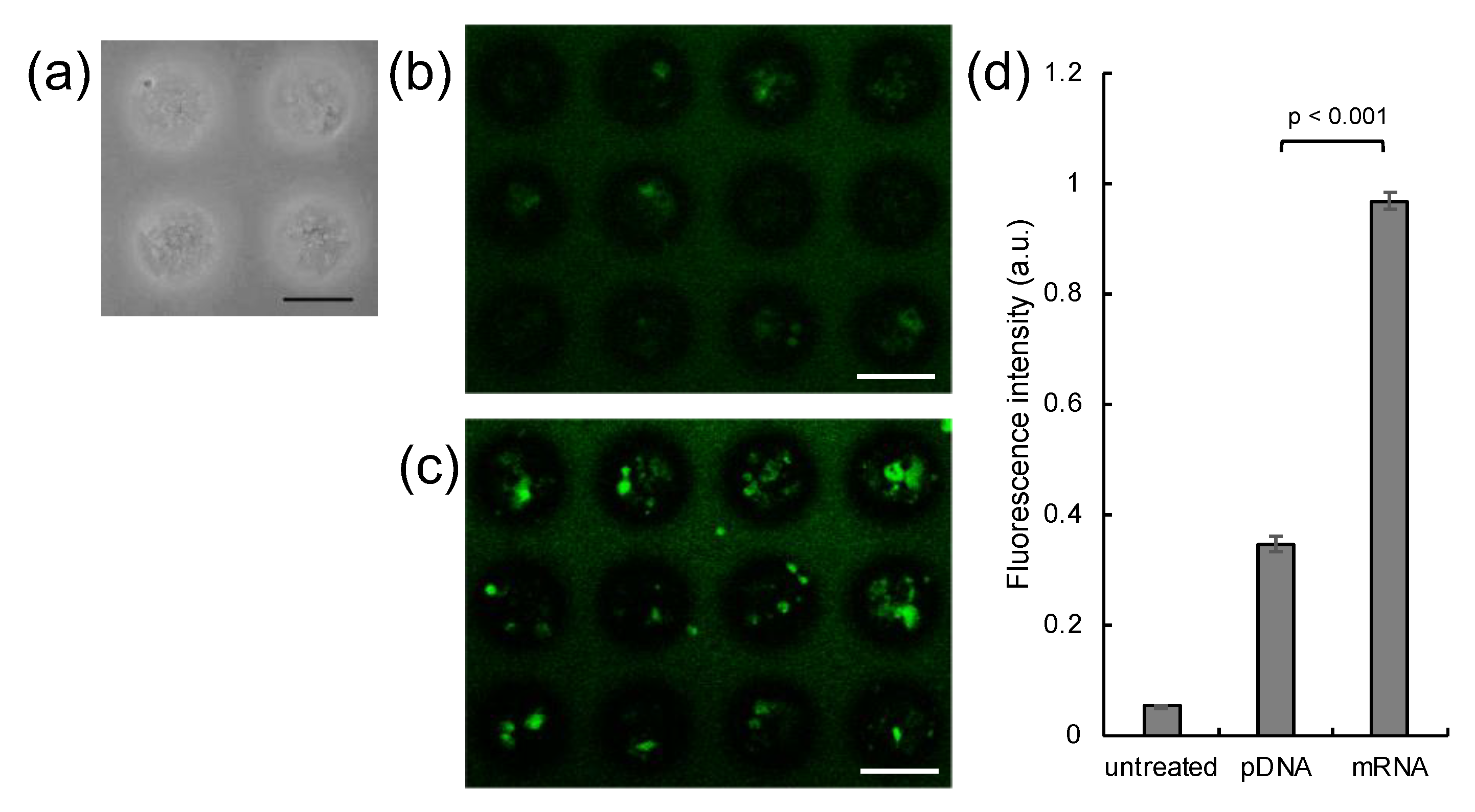
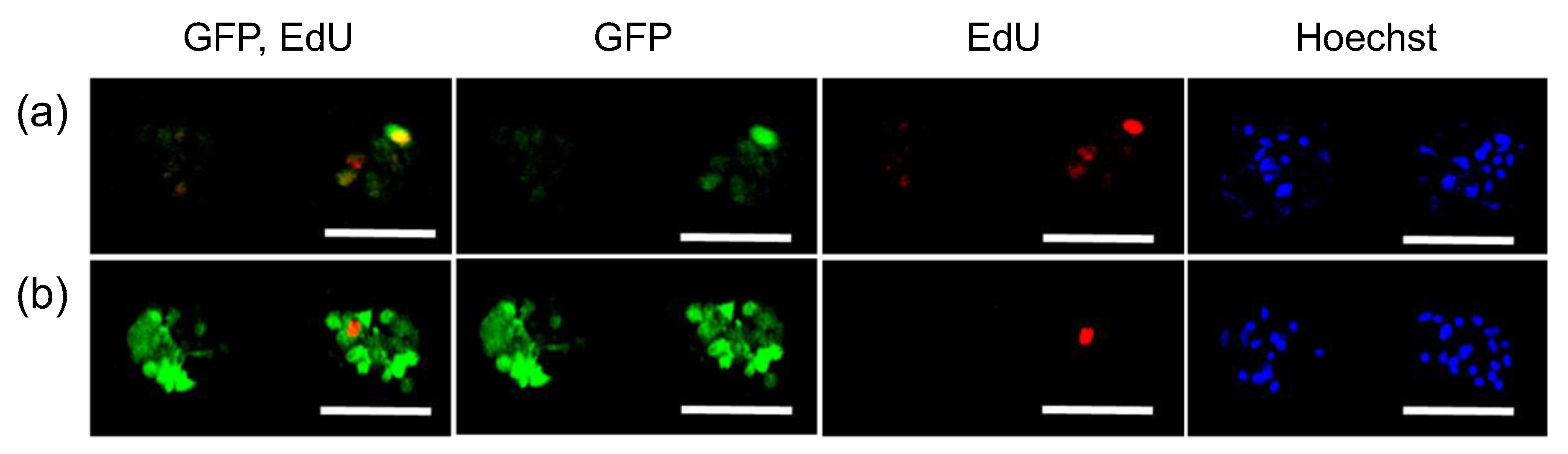
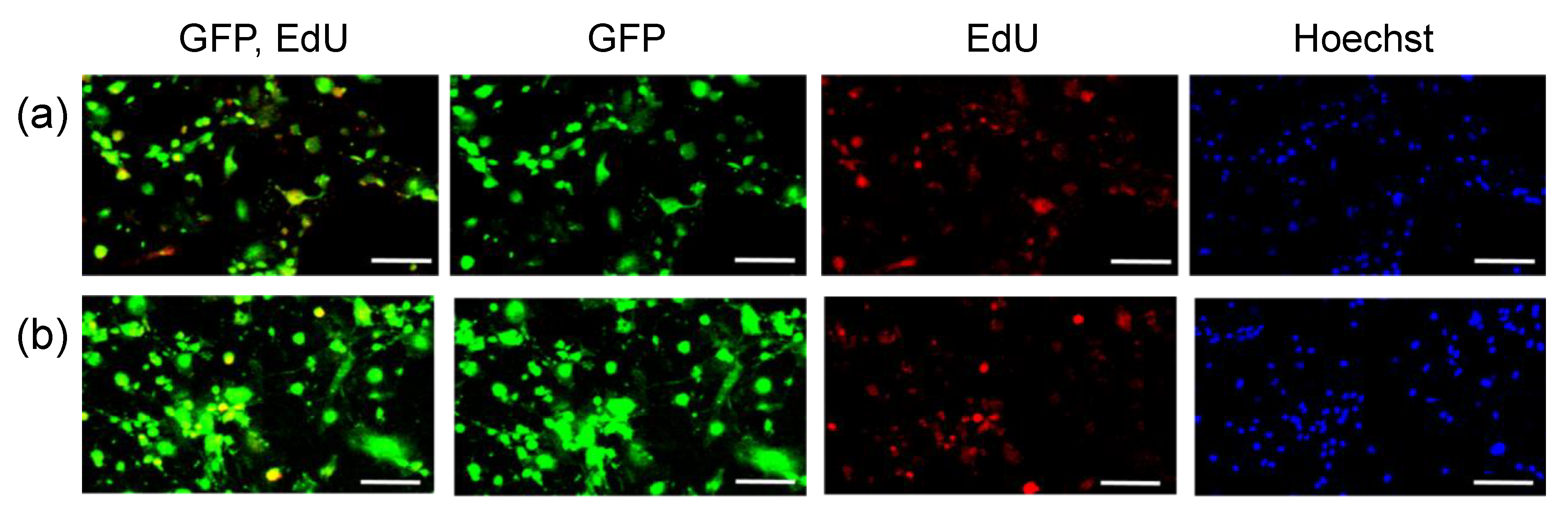
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Trounson, A.; McDonald, C. Stem Cell Therapies in Clinical Trials: Progress and Challenges. Cell Stem Cell 2015, 17, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Dimmeler, S.; Ding, S.; Rando, T.A.; Trounson, A. Translational strategies and challenges in regenerative medicine. Nat. Med. 2014, 20, 814–821. [Google Scholar] [CrossRef]
- Clarke, G.; Harley, P.; Hubber, E.L.; Manea, T.; Manuelli, L.; Read, E.; Watt, F.M. Bench to bedside: Current advances in regenerative medicine. Curr. Opin. Cell Biol. 2018, 55, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, I.J.; Daley, G.Q.; Goldman, S.A.; Huard, J.; Kamp, T.J.; Trucco, M. Stem cell therapy. Use of differentiated pluripotent stem cells as replacement therapy for treating disease. Science 2014, 345, 1247391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyerrose, T.; Olson, S.; Pontow, S.; Kalomoiris, S.; Jung, Y.; Annett, G.; Bauer, G.; Nolta, J.A. Mesenchymal stem cells for the sustained in vivo delivery of bioactive factors. Adv. Drug Deliv. Rev. 2010, 62, 1167–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, S.; Shimizu, T. Three-dimensional cardiac tissue fabrication based on cell sheet technology. Adv. Drug Deliv. Rev. 2016, 96, 103–109. [Google Scholar] [CrossRef]
- Vijayavenkataraman, S.; Yan, W.C.; Lu, W.F.; Wang, C.H.; Fuh, J.Y.H. 3D bioprinting of tissues and organs for regenerative medicine. Adv. Drug Deliv. Rev. 2018, 132, 296–332. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- De Ravin, S.S.; Li, L.; Wu, X.; Choi, U.; Allen, C.; Koontz, S.; Lee, J.; Theobald-Whiting, N.; Chu, J.; Garofalo, M.; et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med. 2017, 9, eaah3480. [Google Scholar] [CrossRef]
- Sheyn, D.; Mizrahi, O.; Benjamin, S.; Gazit, Z.; Pelled, G.; Gazit, D. Genetically modified cells in regenerative medicine and tissue engineering. Adv. Drug Deliv. Rev. 2010, 62, 683–698. [Google Scholar] [CrossRef]
- Zhang, K.; Fang, H.; Qin, Y.; Zhang, L.; Yin, J. Functionalized Scaffold for in Situ Efficient Gene Transfection of Mesenchymal Stem Cells Spheroids toward Chondrogenesis. ACS Appl. Mater. Interfaces 2018, 10, 33993–34004. [Google Scholar] [CrossRef] [PubMed]
- Song, S.Y.; Hong, J.; Go, S.; Lim, S.; Sohn, H.S.; Kang, M.; Jung, G.J.; Yoon, J.K.; Kang, M.L.; Im, G.I.; et al. Interleukin-4 Gene Transfection and Spheroid Formation Potentiate Therapeutic Efficacy of Mesenchymal Stem Cells for Osteoarthritis. Adv. Healthc. Mater. 2020, 9, e1901612. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Hayakawa, K.; Ogata, T.; Tanaka, S.; Kataoka, K.; Itaka, K. Treatment of spinal cord injury by an advanced cell transplantation technology using brain-derived neurotrophic factor-transfected mesenchymal stem cell spheroids. Biomaterials 2016, 109, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, K.; Uchida, S.; Ohba, S.; Kataoka, K.; Itaka, K. Treatment of Bone Defects by Transplantation of Genetically Modified Mesenchymal Stem Cell Spheroids. Mol. Ther.-Methods Clin. Dev. 2018, 9, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, J. How safe is a popular gene therapy vector? Science 2020, 367, 131. [Google Scholar] [CrossRef]
- Qin, L.; Ding, Y.; Pahud, D.R.; Robson, N.D.; Shaked, A.; Bromberg, J.S. Adenovirus-mediated gene transfer of viral interleukin-10 inhibits the immune response to both alloantigen and adenoviral antigen. Hum. Gene Ther. 1997, 8, 1365–1374. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Freyer, J.P. Role of necrosis in regulating the growth saturation of multicellular spheroids. Cancer Res. 1988, 48, 2432–2439. [Google Scholar]
- Onozato, Y.; Kaida, A.; Harada, H.; Miura, M. Radiosensitivity of quiescent and proliferating cells grown as multicellular tumor spheroids. Cancer Sci. 2017, 108, 704–712. [Google Scholar] [CrossRef]
- Capecchi, M.R. High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell 1980, 22, 479–488. [Google Scholar] [CrossRef]
- Grosse, S.; Thevenot, G.; Monsigny, M.; Fajac, I. Which mechanism for nuclear import of plasmid DNA complexed with polyethylenimine derivatives? J. Gene Med. 2006, 8, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Scarfo, K.; Nantz, M.H.; Hecker, J.G. Lipid-mediated delivery of RNA is more efficient than delivery of DNA in non-dividing cells. Int. J. Pharm. 2010, 389, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Otsuka, H.; Hirano, A.; Nagasaki, Y.; Okano, T.; Horiike, Y.; Kataoka, K. Two-dimensional multiarray formation of hepatocyte spheroids on a microfabricated PEG-brush surface. Chembiochem 2004, 5, 850–855. [Google Scholar] [CrossRef]
- Uchida, S.; Itaka, K.; Nomoto, T.; Endo, T.; Matsumoto, Y.; Ishii, T.; Kataoka, K. An injectable spheroid system with genetic modification for cell transplantation therapy. Biomaterials 2014, 35, 2499–2506. [Google Scholar] [CrossRef]
- Kanayama, N.; Fukushima, S.; Nishiyama, N.; Itaka, K.; Jang, W.D.; Miyata, K.; Yamasaki, Y.; Chung, U.I.; Kataoka, K. A PEG-based biocompatible block catiomer with high buffering capacity for the construction of polyplex micelles showing efficient gene transfer toward primary cells. ChemMedChem 2006, 1, 439–444. [Google Scholar] [CrossRef]
- Miyata, K.; Oba, M.; Nakanishi, M.; Fukushima, S.; Yamasaki, Y.; Koyama, H.; Nishiyama, N.; Kataoka, K. Polyplexes from poly(aspartamide) bearing 1,2-diaminoethane side chains induce pH-selective, endosomal membrane destabilization with amplified transfection and negligible cytotoxicity. J. Am. Chem. Soc. 2008, 130, 16287–16294. [Google Scholar] [CrossRef]
- Itaka, K.; Ishii, T.; Hasegawa, Y.; Kataoka, K. Biodegradable polyamino acid-based polycations as safe and effective gene carrier minimizing cumulative toxicity. Biomaterials 2010, 31, 3707–3714. [Google Scholar] [CrossRef]
- Matsui, A.; Uchida, S.; Ishii, T.; Itaka, K.; Kataoka, K. Messenger RNA-based therapeutics for the treatment of apoptosis-associated diseases. Sci. Rep. 2015, 5, 15810. [Google Scholar] [CrossRef]
- Galipeau, J.; Sensebe, L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, C.; McCabe, L.R.; Choi, J.Y.; Hiebert, S.W.; Stein, J.L.; Stein, G.S.; Lian, J.B. Runt homology domain proteins in osteoblast differentiation: AML3/CBFA1 is a major component of a bone-specific complex. J. Cell. Biochem. 1997, 66, 1–8. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhao, M.; Xiao, G.; Franceschi, R.T. Gene transfer of the Runx2 transcription factor enhances osteogenic activity of bone marrow stromal cells in vitro and in vivo. Mol. Ther. 2005, 12, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.M.; Lee, E.H. Transcriptional regulatory cascades in Runx2-dependent bone development. Tissue Eng Part B Rev 2013, 19, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akita, H.; Kurihara, D.; Schmeer, M.; Schleef, M.; Harashima, H. Effect of the Compaction and the Size of DNA on the Nuclear Transfer Efficiency after Microinjection in Synchronized Cells. Pharmaceutics 2015, 7, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, S.; Kataoka, K. Design concepts of polyplex micelles for in vivo therapeutic delivery of plasmid DNA and messenger RNA. J. Biomed. Mater. Res. A 2019, 107, 978–990. [Google Scholar] [CrossRef]
- Endo, T.; Itaka, K.; Shioyama, M.; Uchida, S.; Kataoka, K. Gene transfection to spheroid culture system on micropatterned culture plate by polyplex nanomicelle: A novel platform of genetically-modified cell transplantation. Drug Deliv Transl Res 2012, 2, 398–405. [Google Scholar] [CrossRef]
- Masago, K.; Itaka, K.; Nishiyama, N.; Chung, U.I.; Kataoka, K. Gene delivery with biocompatible cationic polymer: Pharmacogenomic analysis on cell bioactivity. Biomaterials 2007, 28, 5169–5175. [Google Scholar] [CrossRef]
- McKinlay, C.J.; Vargas, J.R.; Blake, T.R.; Hardy, J.W.; Kanada, M.; Contag, C.H.; Wender, P.A.; Waymouth, R.M. Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals. Proc. Natl. Acad. Sci. USA 2017, 114, E448–E456. [Google Scholar] [CrossRef] [Green Version]
- Hajj, K.A.; Whitehead, K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017, 2, 17056. [Google Scholar] [CrossRef]
- Zhong, Z.; Mc Cafferty, S.; Combes, F.; Huysmans, H.; De Temmerman, J.; Gitsels, A.; Vanrompay, D.; Portela Catani, J.; Sanders, N.N. mRNA therapeutics deliver a hopeful message. Nano Today 2018, 23, 16–39. [Google Scholar] [CrossRef]
- Li, J.; Wang, W.; He, Y.; Li, Y.; Yan, E.Z.; Zhang, K.; Irvine, D.J.; Hammond, P.T. Structurally Programmed Assembly of Translation Initiation Nanoplex for Superior mRNA Delivery. ACS Nano 2017, 11, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, N.; Cho, E.; Koji, K.; Mochida, Y.; Naito, M.; Osada, K.; Kataoka, K.; Cabral, H.; Uchida, S. Bundling mRNA Strands to Prepare Nano-Assemblies with Enhanced Stability Towards RNase for in Vivo Delivery. Angew. Chem. Int. Ed. 2019, 58, 11360–11363. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, N.; Uchida, S.; Naito, M.; Osada, K.; Cabral, H.; Kataoka, K. Induced packaging of mRNA into polyplex micelles by regulated hybridization with a small number of cholesteryl RNA oligonucleotides directed enhanced in vivo transfection. Biomaterials 2019, 197, 255–267. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EdU (+) 1 | EdU (−) 2 | Total 3 | |
|---|---|---|---|
| GFP pDNA (%) 4 | 75 | 20 | 34 |
| GFP mRNA (%) 4 | 100 | 73 | 77 |
| EdU (+) 1 | EdU (−) 2 | Total 3 | |
|---|---|---|---|
| GFP pDNA (%) 4 | 90 | 32 | 75 |
| GFP mRNA (%) 4 | 97 | 85 | 93 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uchida, S.; Yanagihara, K.; Matsui, A.; Kataoka, K.; Itaka, K. mRNA as a Tool for Gene Transfection in 3D Cell Culture for Future Regenerative Therapy. Micromachines 2020, 11, 426. https://doi.org/10.3390/mi11040426
Uchida S, Yanagihara K, Matsui A, Kataoka K, Itaka K. mRNA as a Tool for Gene Transfection in 3D Cell Culture for Future Regenerative Therapy. Micromachines. 2020; 11(4):426. https://doi.org/10.3390/mi11040426
Chicago/Turabian StyleUchida, Satoshi, Kayoko Yanagihara, Akitsugu Matsui, Kazunori Kataoka, and Keiji Itaka. 2020. "mRNA as a Tool for Gene Transfection in 3D Cell Culture for Future Regenerative Therapy" Micromachines 11, no. 4: 426. https://doi.org/10.3390/mi11040426
APA StyleUchida, S., Yanagihara, K., Matsui, A., Kataoka, K., & Itaka, K. (2020). mRNA as a Tool for Gene Transfection in 3D Cell Culture for Future Regenerative Therapy. Micromachines, 11(4), 426. https://doi.org/10.3390/mi11040426