Scanning Electron Microscopy of Circulating Tumor Cells and Tumor-Derived Extracellular Vesicles

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cell Preparation for SEM Imaging
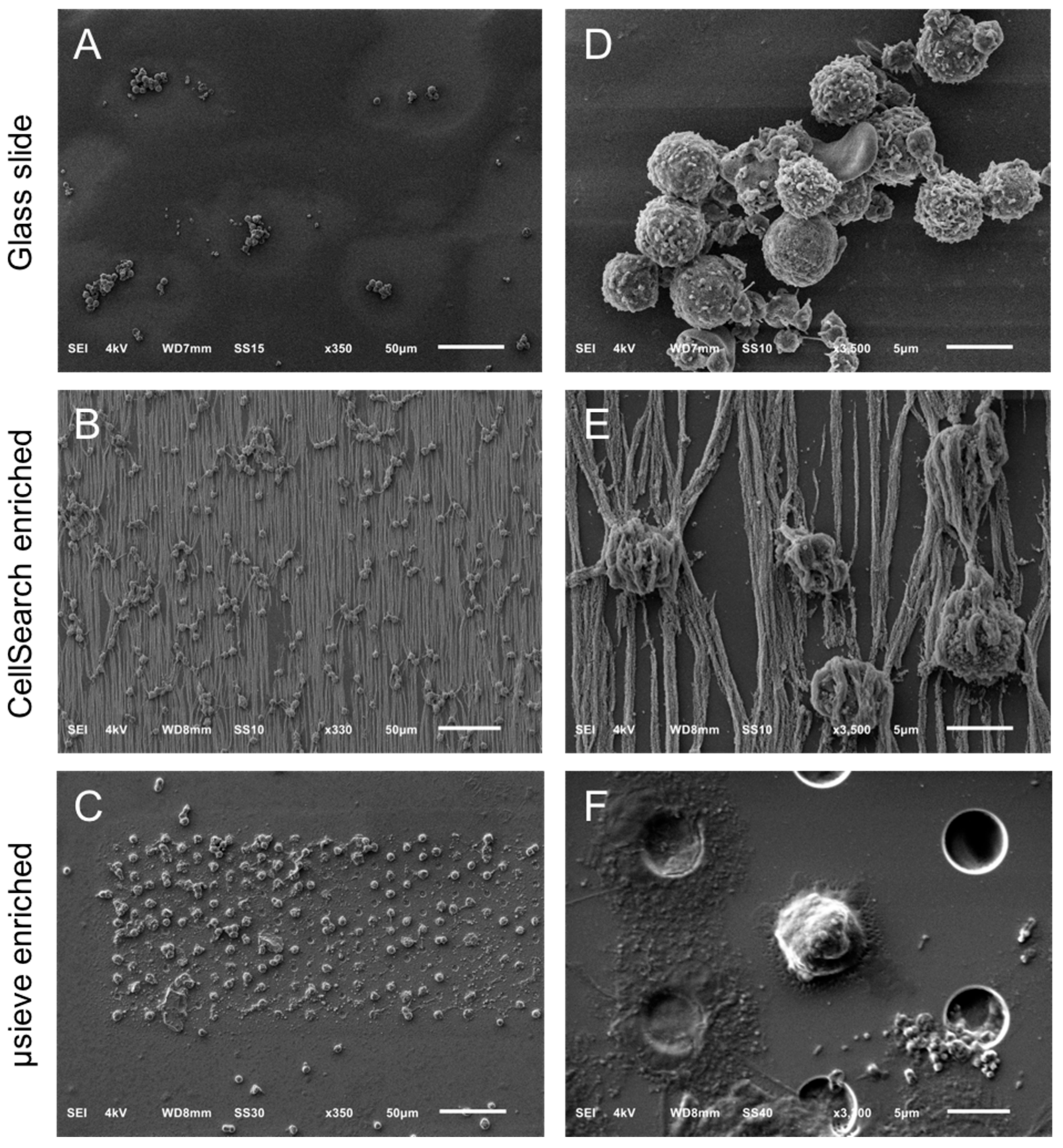
2.2. Overview of Cells by SEM Imaging on Glass Slides, CellSearch Cartridges, and 5 μm Pore Microsieves
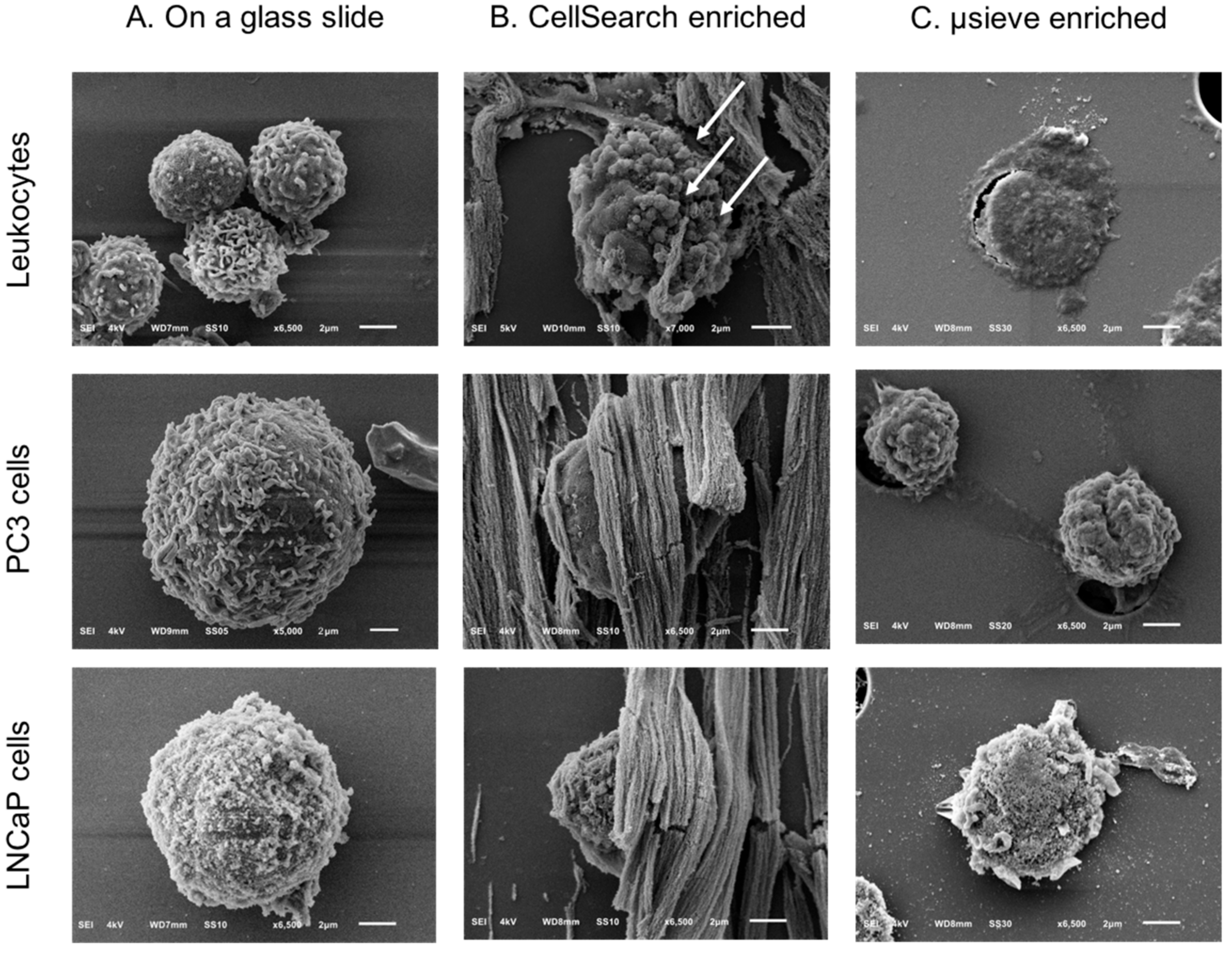
2.3. SEM Imaging of Isolated PC3 and LNCaP Cells Spiked in Blood by the CellSearch System and Microsieves
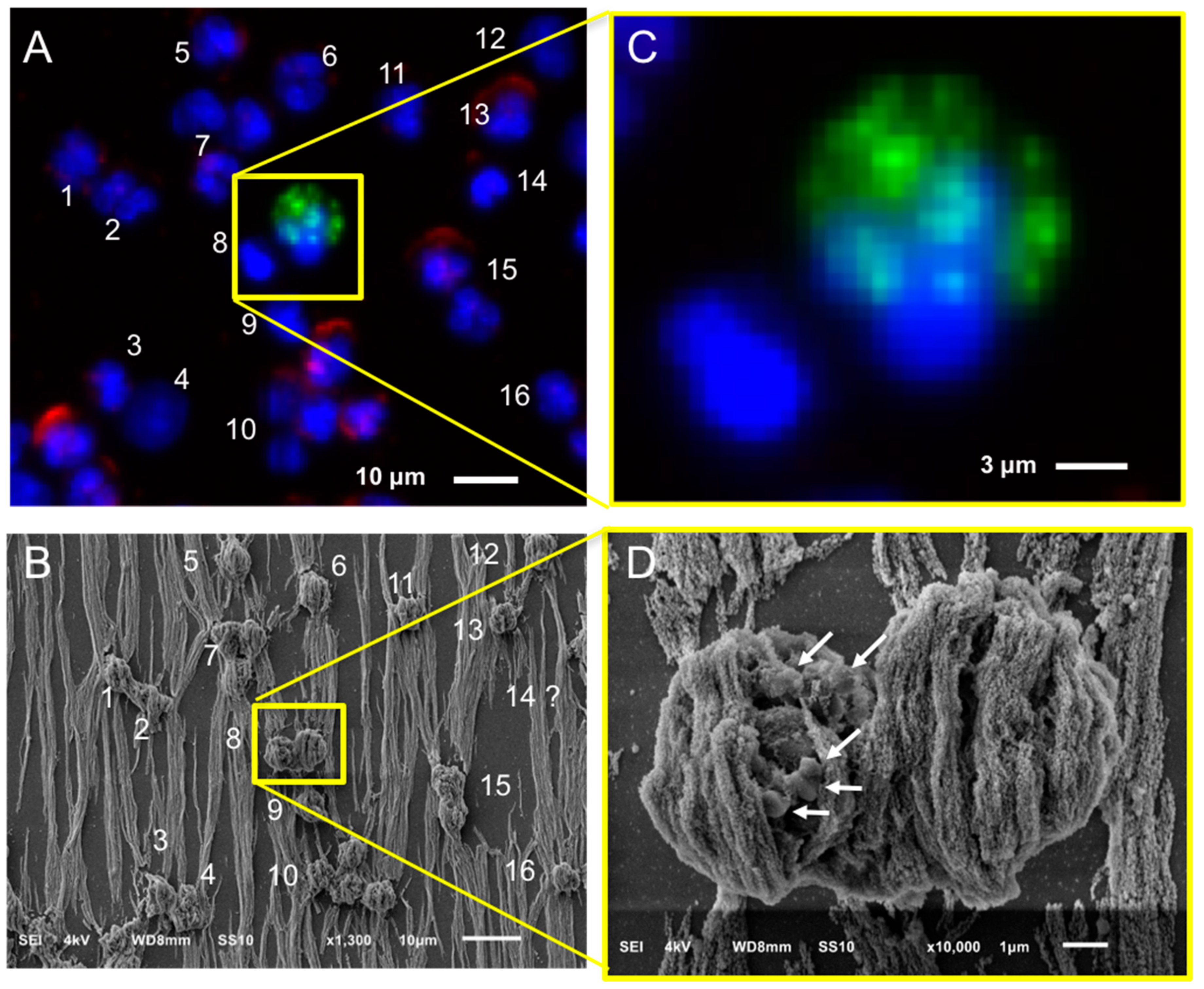
2.4. Relocation and Correlated SEM-Fluorescence Images of CTCs and tdEVs of CRPC Patients Isolated by the CellSearch
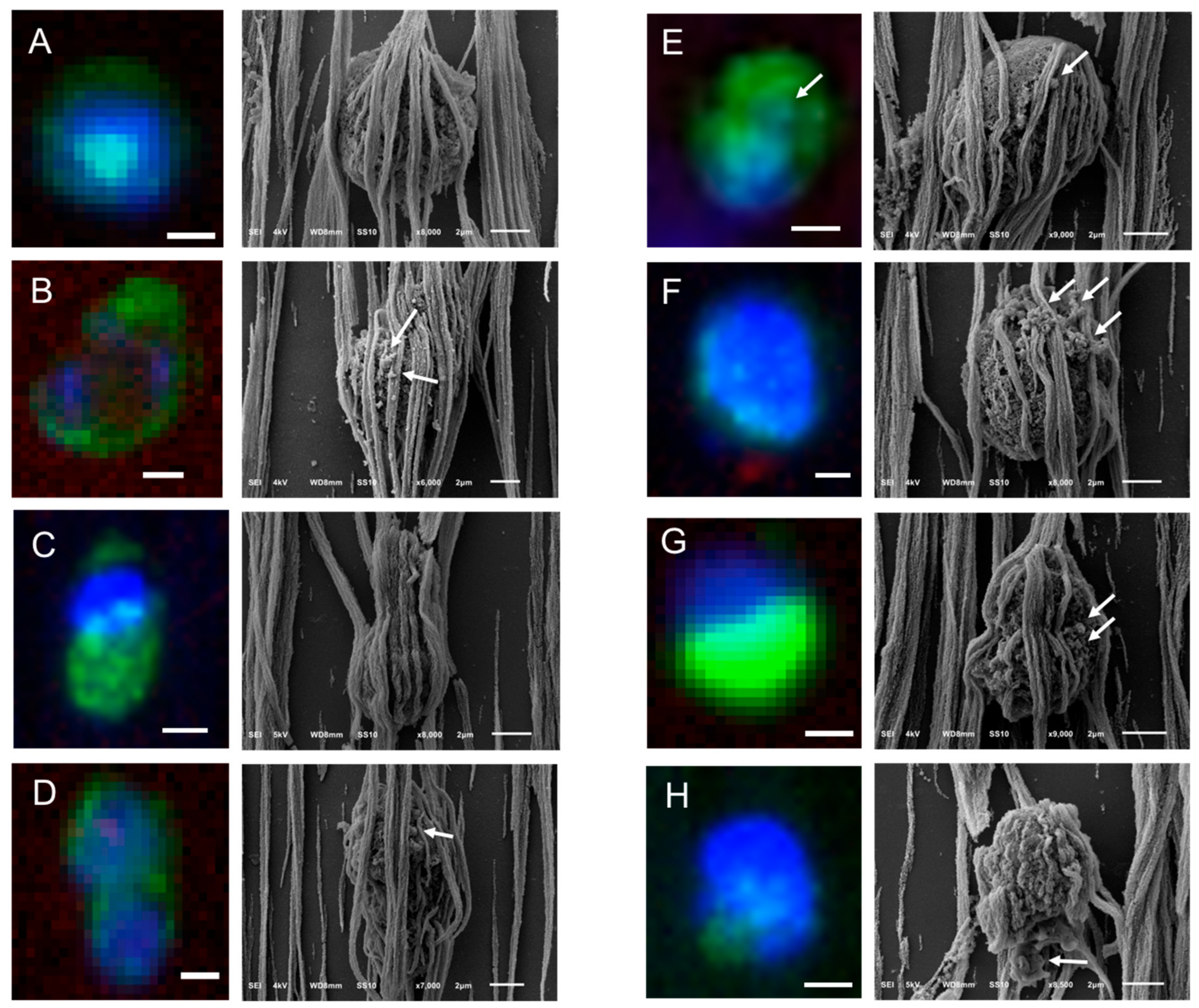
2.5. Relocation and Correlated SEM-Fluorescence Images of tdEVs of CRPC Patients Isolated by the CellSearch
2.6. Relocation and Correlated SEM-Fluorescence Images of CTCs of CRPC Patients Isolated by Microsieves
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. CRPC Patient and Healthy Donor Blood Samples
4.3. Immunomagnetic CTC and tdEV Isolation
4.4. Size-Based CTC Isolation
4.5. Specimen Preparation for Scanning Electron Microscopy (SEM)
4.6. Scanning Electron Microscopy (SEM)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, M.; Mimori, K.; Ueo, H.; Karimine, N.; Barnard, G.F.; Sugimachi, K.; Akiyoshi, T. Molecular detection of circulating solid carcinoma cells in the peripheral blood: The concept of early systemic disease. Int. J. Cancer 1996, 68, 739–743. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.G.; Miller, M.C.; Gross, S.; Allard, W.J.; Gomella, L.G.; Terstappen, L.W. Circulating tumor cells predict survival in patients with metastatic prostate cancer. Urology 2005, 65, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.C.; Peeters, D.J.; Fehm, T.; Nole, F.; Gisbert-Criado, R.; Mavroudis, D.; Grisanti, S.; Generali, D.; Garcia-Saenz, J.A.; Stebbing, J.; et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: A pooled analysis of individual patient data. Lancet Oncol. 2014, 15, 406–414. [Google Scholar] [CrossRef]
- Cohen, S.J.; Punt, C.J.A.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.V.; Terstappen, L.W.W.M.; Pienta, K.J.; Raghavan, D. Circulating Tumor Cells Predict Survival Benefit from Treatment in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2008, 14, 6302–6309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, M.G.; Sloane, R.; Priest, L.; Lancashire, L.; Hou, J.M.; Greystoke, A.; Ward, T.H.; Ferraldeschi, R.; Hughes, A.; Clack, G.; et al. Evaluation and Prognostic Significance of Circulating Tumor Cells in Patients with Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2011, 29, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Hiltermann, T.J.N.; Pore, M.M.; van den Berg, A.; Timens, W.; Boezen, H.M.; Liesker, J.J.W.; Schouwink, J.H.; Wijnands, W.J.A.; Kerner, G.S.M.A.; Kruyt, F.A.E.; et al. Circulating tumor cells in small-cell lung cancer: A predictive and prognostic factor. Ann. Oncol. 2012, 23, 2937–2942. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.M.; Ramani, V.C.; Jeffrey, S.S. Circulating tumor cell technologies. Mol. Oncol. 2016, 10, 374–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, M.G.; Metcalf, R.L.; Carter, L.; Brady, G.; Blackhall, F.H.; Dive, C. Molecular analysis of circulating tumour cells-biology and biomarkers. Nat. Rev. Clin. Oncol. 2014, 11, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Zhuang, R.; Long, M.; Pavlovic, M.; Kang, Y.Q.; Ilyas, A.; Asghar, W. Circulating tumor cell isolation, culture, and downstream molecular analysis. Biotechnol. Adv. 2018, 36, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, D.R.; Dracopoli, N.; Petty, B.G.; Compton, C.; Cristofanilli, M.; Deisseroth, A.; Hayes, D.F.; Kapke, G.; Kumar, P.; Lee, J.S.H.; et al. Considerations in the development of circulating tumor cell technology for clinical use. J. Transl. Med. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Lin, H.; Liu, J.Q.; Balic, M.; Datar, R.; Cote, R.J.; Tai, Y.C. Membrane microfilter device for selective capture, electrolysis and genomic analysis of human circulating tumor cells. J. Chromatogr. A 2007, 1162, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.K.; Zheng, S.; Williams, A.J.; Balic, M.; Groshen, S.; Scher, H.I.; Fleisher, M.; Stadler, W.; Datar, R.H.; Tai, Y.C.; et al. Portable filter-based microdevice for detection and characterization of circulating tumor cells. Clin. Cancer Res. 2010, 16, 5011–5018. [Google Scholar] [CrossRef] [PubMed]
- Sarioglu, A.F.; Aceto, N.; Kojic, N.; Donaldson, M.C.; Zeinali, M.; Hamza, B.; Engstrom, A.; Zhu, H.; Sundaresan, T.K.; Miyamoto, D.T.; et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods 2015, 12, 685–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, C.J.; Moreno, J.G.; Pienta, K.J.; Gross, S.; Repollet, M.; O’Hara, S.M.; Russell, T.; Terstappen, L.W. Apoptosis of circulating tumor cells in prostate cancer patients. Cytom. A 2004, 62, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coumans, F.A.W.; Doggen, C.J.M.; Attard, G.; de Bono, J.S.; Terstappen, L.W.M.M. All circulating EpCAM+CK+CD45-objects predict overall survival in castration-resistant prostate cancer. Ann. Oncol. 2010, 21, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Nanou, A.; Coumans, F.A.W.; van Dalum, G.; Zeune, L.L.; Dolling, D.; Onstenk, W.; Crespo, M.; Fontes, M.S.; Rescigno, P.; Fowler, G.; et al. Circulating tumor cells, tumor-derived extracellular vesicles and plasma cytokeratins in castration-resistant prostate cancer patients. Oncotarget 2018, 9, 19283–19293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibbe, A.G.; de Grooth, B.G.; Greve, J.; Dolan, G.J.; Rao, C.; Terstappen, L.W. Magnetic field design for selecting and aligning immunomagnetic labeled cells. Cytometry 2002, 47, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swennenhuis, J.F.; Tibbe, A.G.; Levink, R.; Sipkema, R.C.; Terstappen, L.W. Characterization of circulating tumor cells by fluorescence in situ hybridization. Cytom. A 2009, 75, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligthart, S.T.; Coumans, F.A.; Bidard, F.C.; Simkens, L.H.; Punt, C.J.; de Groot, M.R.; Attard, G.; de Bono, J.S.; Pierga, J.Y.; Terstappen, L.W. Circulating Tumor Cells Count and Morphological Features in Breast, Colorectal and Prostate Cancer. PLoS ONE 2013, 8, e67148. [Google Scholar] [CrossRef] [PubMed]
- Coumans, F.A.W.; Ligthart, S.T.; Uhr, J.W.; Terstappen, L.W.M.M. Challenges in the Enumeration and Phenotyping of CTC. Clin. Cancer Res. 2012, 18, 5711–5718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, S.; van Dalum, G.; Lenferink, A.T.M.; Tibbe, A.G.J.; Hiltermann, T.J.N.; Groen, H.J.M.; van Rijn, C.J.M.; Terstappen, L.W.M.M. The detection of EpCAM(+) and EpCAM(−) circulating tumor cells. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Deutsch, T.M.; Riethdorf, S.; Nees, J.; Hartkopf, A.D.; Schonfisch, B.; Domschke, C.; Sprick, M.R.; Schutz, F.; Brucker, S.Y.; Stefanovic, S.; et al. Impact of apoptotic circulating tumor cells (aCTC) in metastatic breast cancer. Breast Cancer Res. Treat. 2016, 160, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Saroya, B.S.; Kunkel, M.; Dicker, D.T.; Das, A.; Peters, K.L.; Joudeh, J.; Zhu, J.J.; El-Deiry, W.S. Apoptotic circulating tumor cells (CTCs) in the peripheral blood of metastatic colorectal cancer patients are associated with liver metastasis but not CTCs. Oncotarget 2014, 5, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Smerage, J.B.; Budd, G.T.; Doyle, G.V.; Brown, M.; Paoletti, C.; Muniz, M.; Miller, M.C.; Repollet, M.I.; Chianese, D.A.; Connelly, M.C.; et al. Monitoring apoptosis and Bcl-2 on circulating tumor cells in patients with metastatic breast cancer. Mol. Oncol. 2013, 7, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Jansson, S.; Bendahl, P.O.; Larsson, A.M.; Aaltonen, K.E.; Ryden, L. Prognostic impact of circulating tumor cell apoptosis and clusters in serial blood samples from patients with metastatic breast cancer in a prospective observational cohort. BMC Cancer 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Gorges, T.M.; Riethdorf, S.; von Ahsen, O.; Nastal, Y.P.; Rock, K.; Boede, M.; Peine, S.; Kuske, A.; Schmid, E.; Kneip, C.; et al. Heterogeneous PSMA expression on circulating tumor cells: A potential basis for stratification and monitoring of PSMA-directed therapies in prostate cancer. Oncotarget 2016, 7, 34930–34941. [Google Scholar] [CrossRef] [PubMed]
- Caulin, C.; Salvesen, G.S.; Oshima, R.G. Caspase cleavage of keratin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J. Cell Biol. 1997, 138, 1379–1394. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.M.; Ling, C.; Zhao, L. Immunofluorescence analysis of cytokeratin 8/18 staining is a sensitive assay for the detection of cell apoptosis. Oncol. Lett. 2015, 9, 1227–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagner, T.; Spinelli, C.; Minciacchi, V.R.; Balaj, L.; Zandian, M.; Conley, A.; Zijlstra, A.; Freeman, M.R.; Demichelis, F.; De, S.; et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell. Vesicles 2018, 7, 1505403. [Google Scholar] [CrossRef] [PubMed]
- Minciacchi, V.R.; You, S.; Spinelli, C.; Morley, S.; Zandian, M.; Aspuria, P.J.; Cavallini, L.; Ciardiello, C.; Reis Sobreiro, M.; Morello, M.; et al. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget 2015, 6, 11327–11341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, Z.; Yuana, Y.; Grootemaat, A.E.; van der Pol, E.; Gollwitzer, C.; Krumrey, M.; Nieuwland, R. Towards traceable size determination of extracellular vesicles. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Boing, A.N.; Gool, E.L.; Nieuwland, R. Recent developments in the nomenclature, presence, isolation, detection and clinical impact of extracellular vesicles. J. Thromb. Haemost. 2016, 14, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Deng, W.T.; Klinke, D.J. Exosomes: Improved methods to characterize their morphology, RNA content, and surface protein biomarkers. Analyst 2015, 140, 6631–6642. [Google Scholar] [CrossRef] [PubMed]
- Kondratov, K.; Petrova, T.; Mikhailovskii, V.Y.; Ivanova, A.; Kostareva, A.; Fedorov, A. A study of extracellular vesicles isolated from blood plasma conducted by low-voltage scanning electron microscopy. Cell Tissue Biol. 2017, 11, 181–190. [Google Scholar] [CrossRef]
- De Wit, S. Circulating Tumor Cells and Beyond. Ph.D. Thesis, University of Twente, Borne, The Netherlands, June 2018; pp. 124–146. [Google Scholar] [CrossRef]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.J.; Uhr, J.W.; Terstappen, L.W.M.M. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nanou, A.; Crespo, M.; Flohr, P.; De Bono, J.S.; Terstappen, L.W.M.M. Scanning Electron Microscopy of Circulating Tumor Cells and Tumor-Derived Extracellular Vesicles. Cancers 2018, 10, 416. https://doi.org/10.3390/cancers10110416
Nanou A, Crespo M, Flohr P, De Bono JS, Terstappen LWMM. Scanning Electron Microscopy of Circulating Tumor Cells and Tumor-Derived Extracellular Vesicles. Cancers. 2018; 10(11):416. https://doi.org/10.3390/cancers10110416
Chicago/Turabian StyleNanou, Afroditi, Mateus Crespo, Penny Flohr, Johann S. De Bono, and Leon W. M. M. Terstappen. 2018. "Scanning Electron Microscopy of Circulating Tumor Cells and Tumor-Derived Extracellular Vesicles" Cancers 10, no. 11: 416. https://doi.org/10.3390/cancers10110416
APA StyleNanou, A., Crespo, M., Flohr, P., De Bono, J. S., & Terstappen, L. W. M. M. (2018). Scanning Electron Microscopy of Circulating Tumor Cells and Tumor-Derived Extracellular Vesicles. Cancers, 10(11), 416. https://doi.org/10.3390/cancers10110416