Propranolol Promotes Glucose Dependence and Synergizes with Dichloroacetate for Anti-Cancer Activity in HNSCC


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
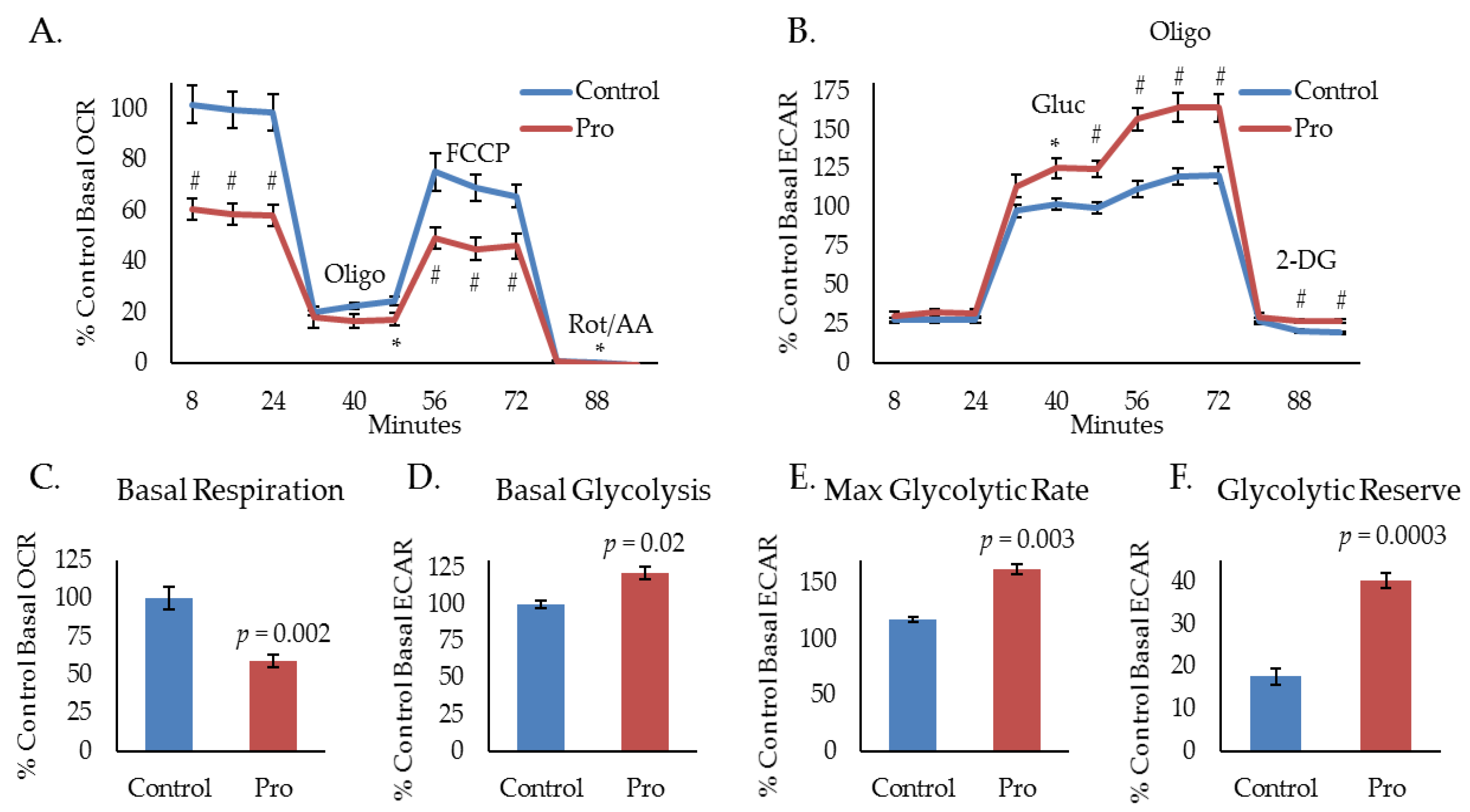
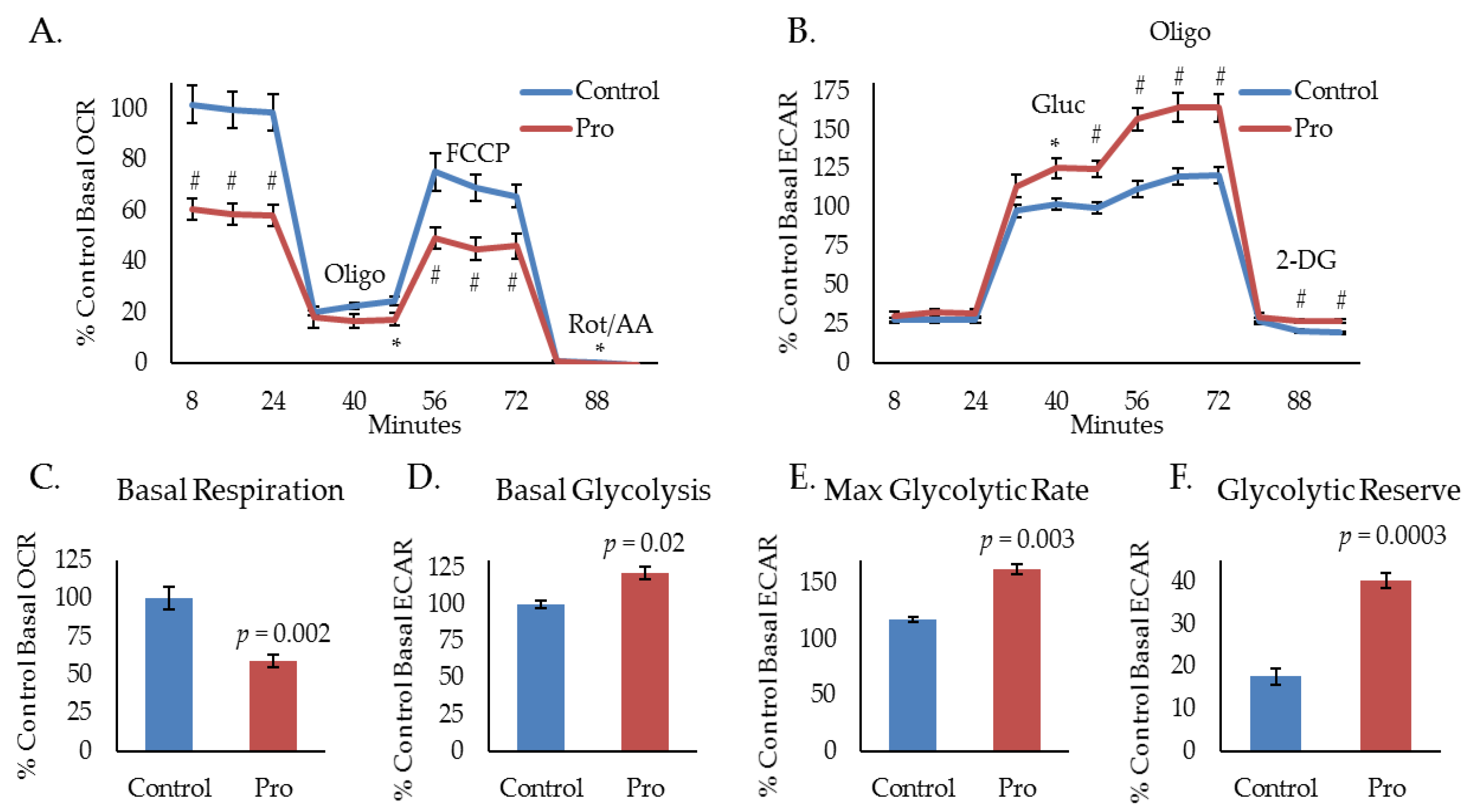
2.1. Propranolol Alters Tumor Cell Metabolism.
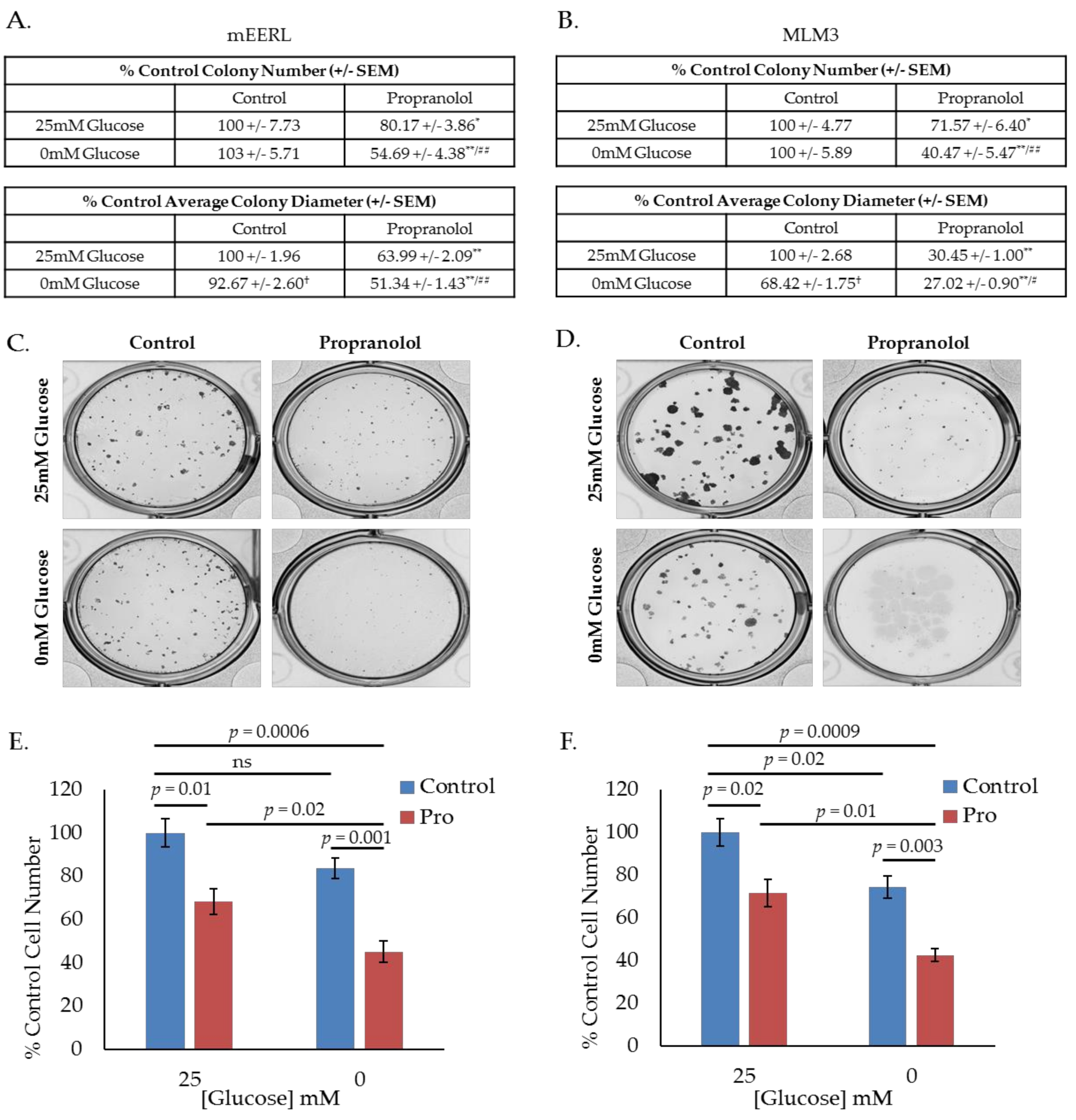
2.2. Propranolol Enhances Glucose Dependence and Synergizes with Glucose Deprivation for Improved Anti-Cancer Activity.
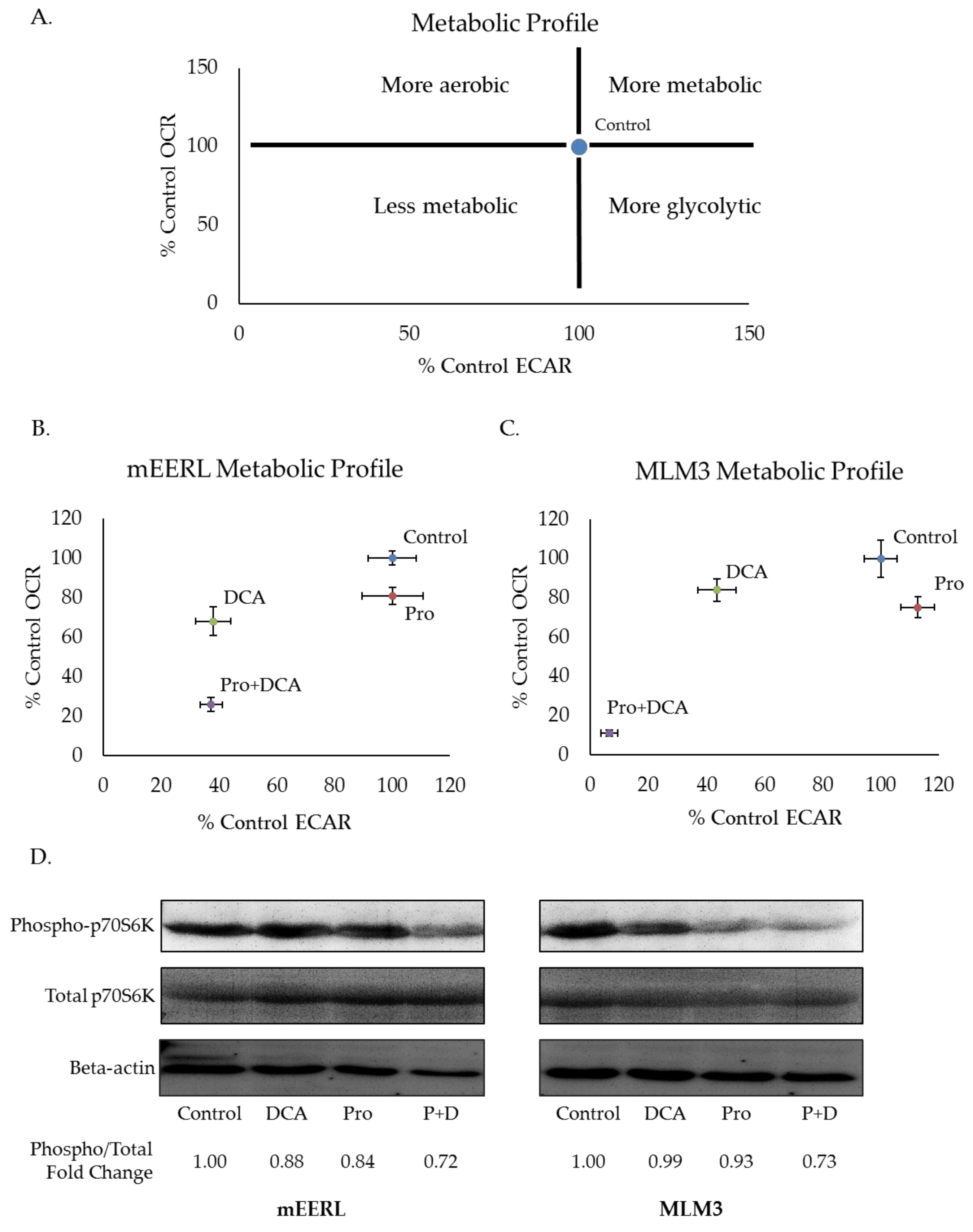
2.3. Propranolol Synergizes with the Glycolytic Inhibitor DCA to Dramatically Attenuate Tumor Cell Metabolism and mTOR Signaling.
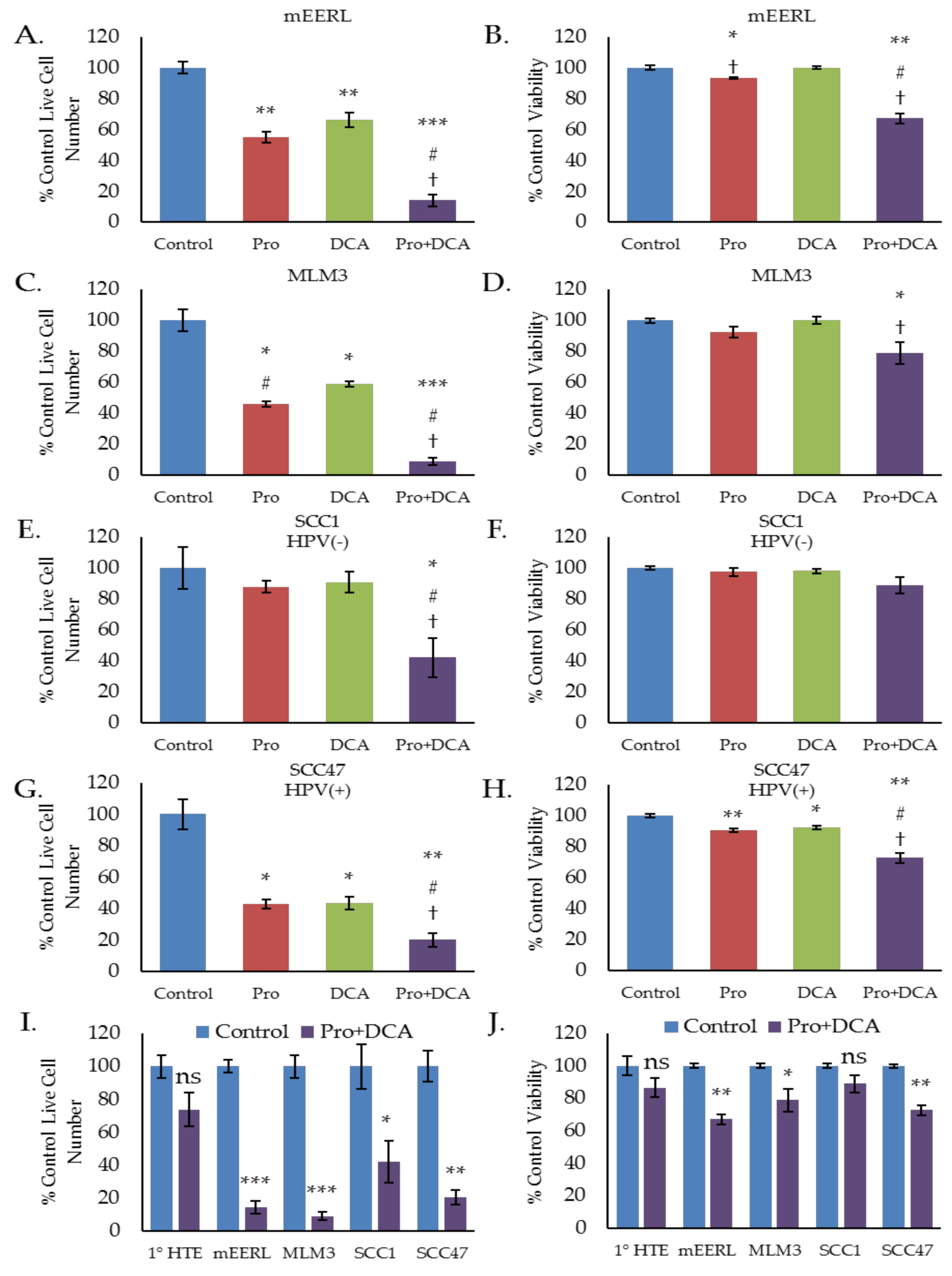
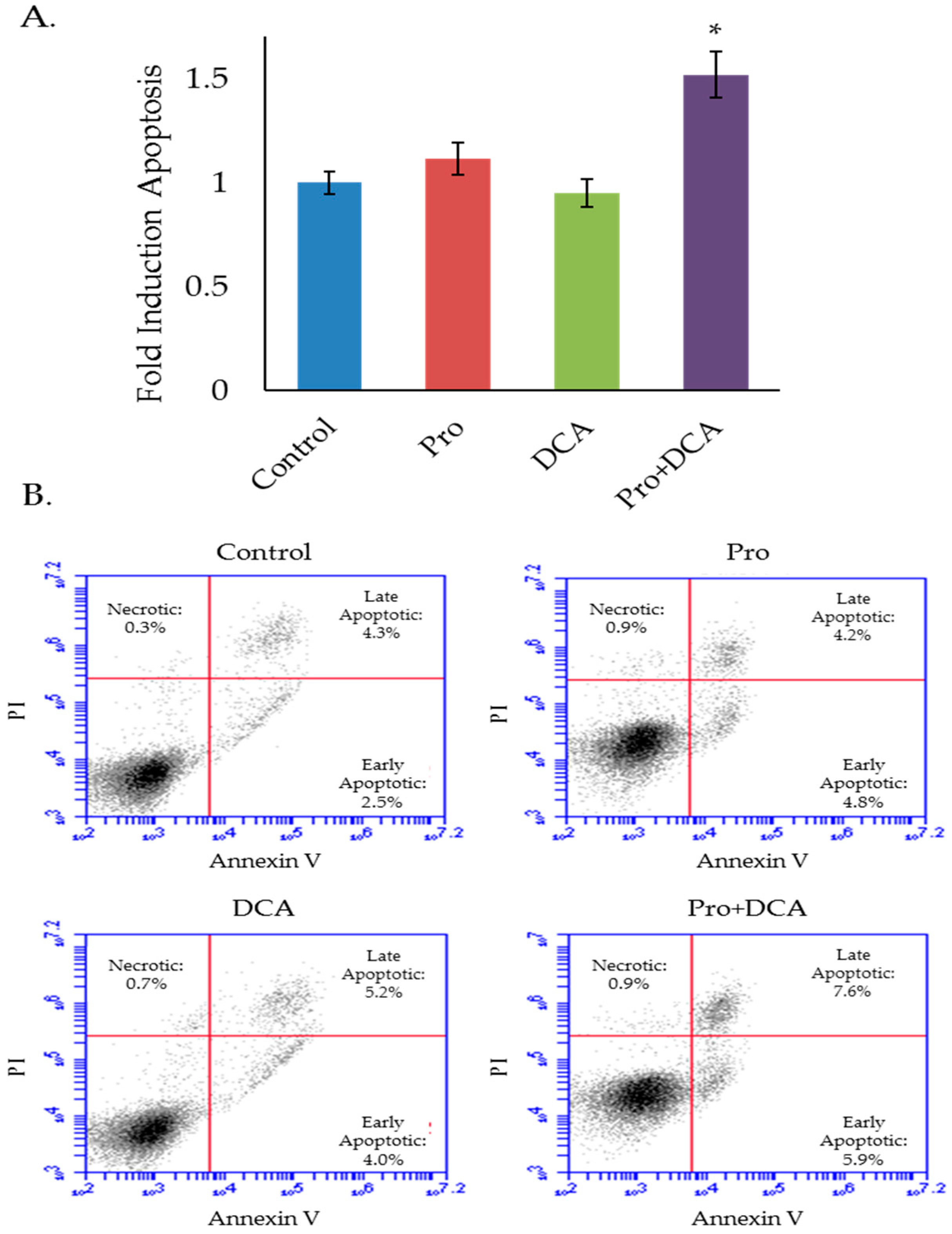
2.4. The Combination of Propranolol and DCA Synergizes for Significant Anti-Cancer Activity in HNSCC, But Has Little Effect on Primary Tonsil Epithelial Cells.
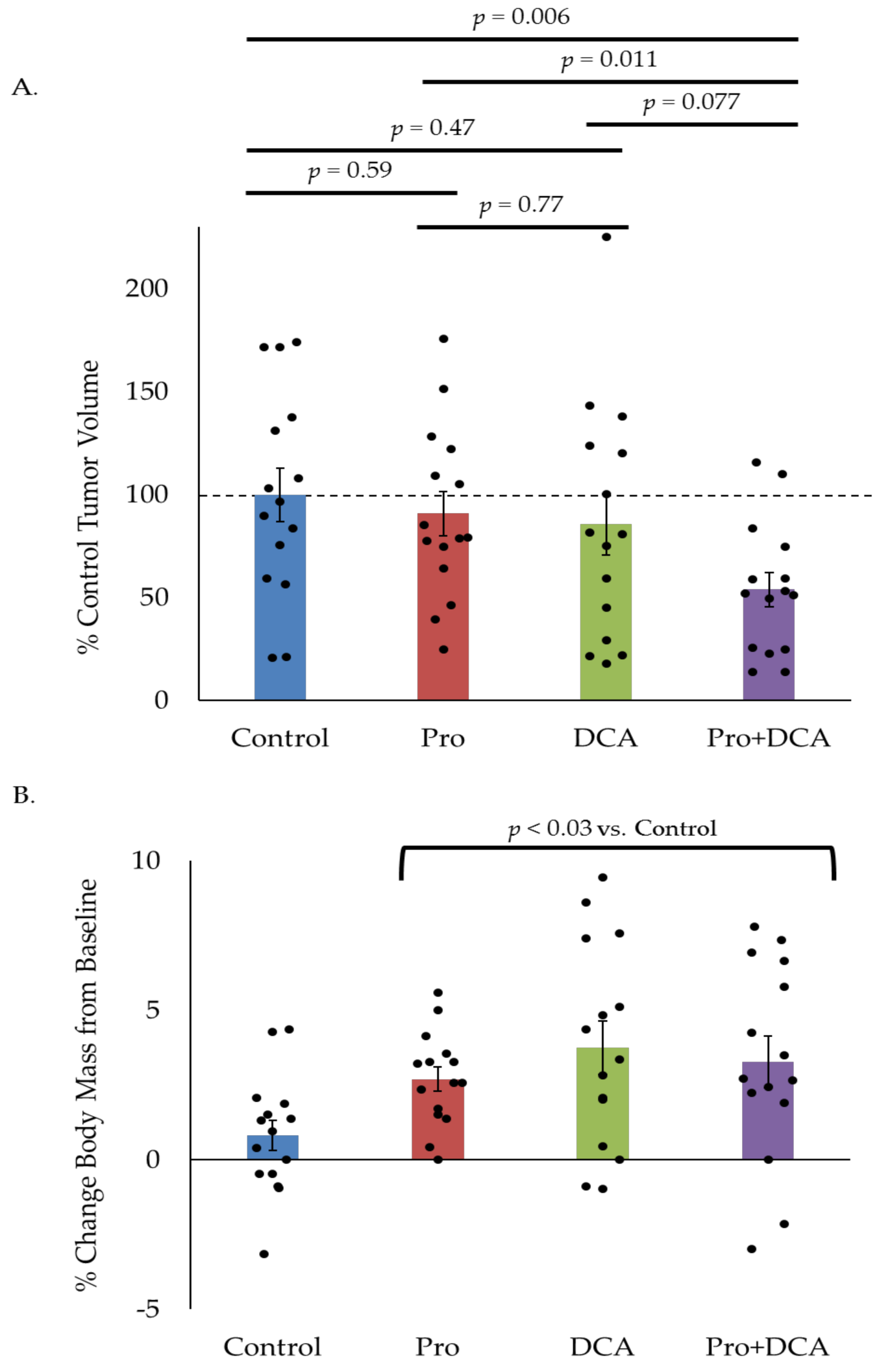
2.5. The Combination of Propranolol and DCA Delays Tumor Growth In Vivo
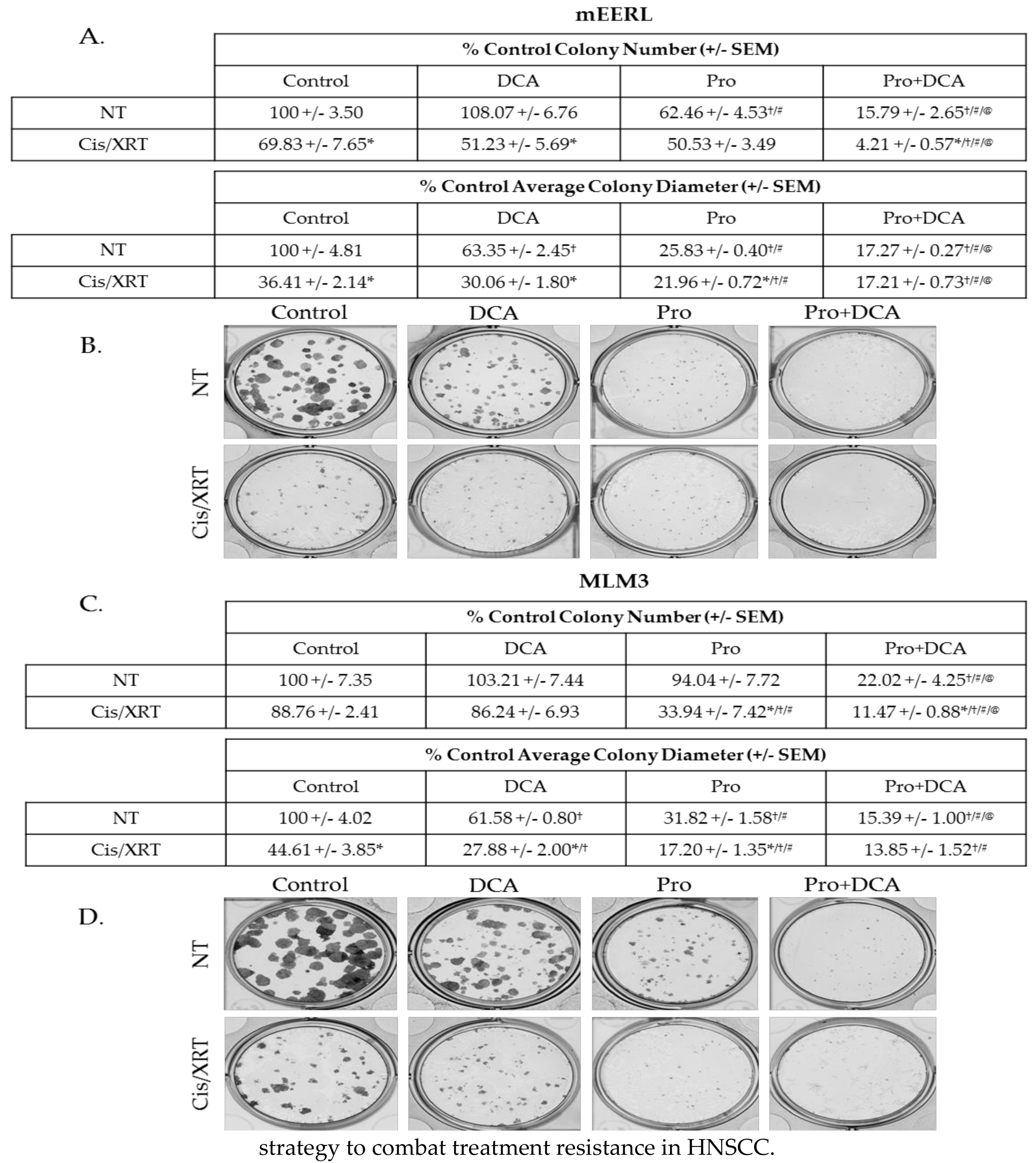
2.6. The Combination of Propranolol and DCA Enhances the Effects of Standard of Care therapeutic Modalities and Sensitizes Resistant Cells to Cisplatin and Radiation.
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Seahorse Extracellular Flux Analysis
4.3. Cellular Proliferation and Viability Assays
4.4. Colony Forming Assays
4.5. Annexin V/Propidium Iodide Apoptosis Assay
4.6. Western Blotting
4.7. Animal Studies
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. The Lancet 2008, 371, 1695–1709. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Marur, S.; Forastiere, A.A. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo. Clin. Proc. 2016, 91, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, C.; Westra, W.H.; Li, S.; Cmelak, A.; Ridge, J.A.; Pinto, H.; Forastiere, A.; Gillison, M.L. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J. Natl. Cancer Inst. 2008, 100, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.G.; Cohen, E.E. Current Treatment Options for Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2015, 33, 3305–3313. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Cohen, E.E. State-of-the-art management of locally advanced head and neck cancer. Br. J. Cancer 2005, 92, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Bernabe, D.G.; Tamae, A.C.; Biasoli, E.R.; Oliveira, S.H. Stress hormones increase cell proliferation and regulates interleukin-6 secretion in human oral squamous cell carcinoma cells. Brain Behav. Immun. 2011, 25, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.C.; Li, J.M.; Lee, K.F.; Huang, Y.C.; Wang, K.C.; Lai, H.C.; Cheng, C.C.; Kuo, Y.H.; Shi, C.S. Selective beta2-AR Blockage Suppresses Colorectal Cancer Growth Through Regulation of EGFR-Akt/ERK1/2 Signaling, G1-Phase Arrest, and Apoptosis. J. Cell. Physiol. 2016, 231, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Choy, C.; Raytis, J.L.; Smith, D.D.; Duenas, M.; Neman, J.; Jandial, R.; Lew, M.W. Inhibition of beta2-adrenergic receptor reduces triple-negative breast cancer brain metastases: The potential benefit of perioperative beta-blockade. Oncol. Rep. 2016, 35, 3135–3142. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.; Moz, M.; Correia, G.; Teixeira, A.; Medeiros, R.; Ribeiro, L. Antiproliferative effects of beta-blockers on human colorectal cancer cells. Oncol. Rep. 2015, 33, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.J.; Liu, K.; Liang, D.F. Expression of beta2-adrenergic receptor in oral squamous cell carcinoma. J. Oral Pathol. Med. 2009, 38, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Wolter, N.E.; Wolter, J.K.; Enepkides, D.J.; Irwin, M.S. Propranolol as a Novel Adjunctive Treatment for Head and Neck Squamous Cell Carcinoma. Otolaryngol. Head Neck Surg. 2012, 41, 334–344. [Google Scholar] [CrossRef]
- Zhang, D.; Ma, Q.; Wang, Z.; Zhang, M.; Guo, K.; Wang, F.; Wu, E. Beta2-adrenoceptor blockage induces G1/S phase arrest and apoptosis in pancreatic cancer cells via Ras/Akt/NF B pathway. Mo. Cancer 2011, 10. [Google Scholar] [CrossRef]
- Zhang, D.; Yong Ma, Q.; Hu, H.-T.; Zhang, M. β2-adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting CREB, NF-κB and AP-1. Cancer Biol. Ther. 2014, 10, 19–29. [Google Scholar] [CrossRef]
- Watkins, J.L.; Thaker, P.H.; Nick, A.M.; Ramondetta, L.M.; Kumar, S.; Urbauer, D.L.; Matsuo, K.; Squires, K.C.; Coleman, R.L.; Lutgendorf, S.K.; et al. Clinical impact of selective and nonselective beta-blockers on survival in patients with ovarian cancer. Cancer 2015, 121, 3444–3451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powe, D.G.; Voss, M.J.; Zanker, K.S.; Habashy, H.O.; Green, A.R.; Ellis, I.O.; Entschladen, F. Beta-Blocker Drug Therapy Reduces Secondary Cancer Formation in Breast Cancer and Improves Cancer Specific Survival. Oncotarget 2010, 1, 628–638. [Google Scholar] [PubMed]
- De Giorgi, V.; Grazzini, M.; Benemei, S.; Marchionni, N.; Botteri, E.; Pennacchioli, E.; Geppetti, P.; Gandini, S. Propranolol for Off-Label Treatment of Patients With Melanoma: Results From a Cohort Study. JAMA Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lucido, C.T.; Callejas-Valera, J.L.; Colbert, P.L.; Vermeer, D.W.; Miskimins, W.K.; Spanos, W.C.; Vermeer, P.D. Beta2-Adrenergic receptor modulates mitochondrial metabolism and disease progression in recurrent/metastatic HPV(+) HNSCC. Oncogenesis 2018, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B. Teaching the basics of cancer metabolism: Developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox. Biol. 2017, 12, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Kishton, R.J.; Rathmell, J.C. Novel therapeutic targets of tumor metabolism. Cancer J. 2015, 21, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Yoon, J.H. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse Warburg effect and its therapeutic implication. World J. Biol. Chem. 2015, 6, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Hoover, A.C.; Strand, G.L.; Nowicki, P.N.; Anderson, M.E.; Vermeer, P.D.; Klingelhutz, A.J.; Bossler, A.D.; Pottala, J.V.; Hendriks, W.J.; Lee, J.H. Impaired PTPN13 phosphatase activity in spontaneous or HPV-induced squamous cell carcinomas potentiates oncogene signaling through the MAP kinase pathway. Oncogene 2009, 28, 3960–3970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spanos, W.C.; Hoover, A.; Harris, G.F.; Wu, S.; Strand, G.L.; Anderson, M.E.; Klingelhutz, A.J.; Hendriks, W.; Bossler, A.D.; Lee, J.H. The PDZ binding motif of human papillomavirus type 16 E6 induces PTPN13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth. J. Virol. 2008, 82, 2493–2500. [Google Scholar] [CrossRef] [PubMed]
- Spanos, W.C.; Nowicki, P.N.; Lee, D.W.; Hoover, A.; Hostager, B.S.; Gupta, A.; Anderson, M.E.; Lee, J.H. Immune Response During Therapy With Cisplatin or Radiation for Human Papillomavirus-Related Head and Neck Cancer. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.; Lee, D.W.; Elzey, B.D.; Anderson, M.E.; Hostager, B.S.; Lee, J.H. Preclinical models of HPV+ and HPV- HNSCC in mice: An immune clearance of HPV+ HNSCC. Head Neck 2009, 31, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, D.W.; Coppock, J.D.; Zeng, E.; Lee, K.M.; Spanos, W.C.; Onken, M.D.; Uppaluri, R.; Lee, J.H.; Vermeer, P.D. Metastatic model of HPV+ oropharyngeal squamous cell carcinoma demonstrates heterogeneity in tumor metastasis. Oncotarget 2016, 7, 24194–24207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: New home for an orphan drug? Biochim. Biophys. Acta 2014, 1846, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Coppock, J.D.; Vermeer, P.D.; Vermeer, D.W.; Lee, K.M.; Miskimins, W.K.; Spanos, W.C.; Lee, J.H. mTOR inhibition as an adjuvant therapy in a metastatic model of HPV+ HNSCC. Oncotarget 2016, 7, 24228–24241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppock, J.D.; Wieking, B.G.; Molinolo, A.A.; Gutkind, J.S.; Miskimins, W.K.; Lee, J.H. Improved Clearance during Treatment of HPV-Positive Head and Neck Cancer through mTOR Inhibition. Neoplasia 2013, 15, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Sandulache, V.C.; Myers, J.N. Altered metabolism in head and neck squamous cell carcinoma: An opportunity for identification of novel biomarkers and drug targets. Head Neck 2012, 34, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Ottosson, S.; Zackrisson, B.; Kjellen, E.; Nilsson, P.; Laurell, G. Weight loss in patients with head and neck cancer during and after conventional and accelerated radiotherapy. Acta Oncol. 2013, 52, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Hager, K.K. Management of Weight Loss in People With Cancer. J. Adv. Pract. Oncol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.D.; Pereira, J.R.; Chen, J.; Liu, J.; Guba, S.C.; John, W.J.; Orlando, M.; Scagliotti, G.; Bonomi, P.D. Relationship between efficacy outcomes and weight gain during treatment of advanced, non-squamous, non-small-cell lung cancer patients. Ann. Oncol. 2016, 27, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, P.; Batus, M.; Fidler, M.J.; Borgia, J.A. Practical and theoretical implications of weight gain in advanced non-small cell lung cancer patients. Ann. Transl. Med. 2017, 5, 152. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell. 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell. Res. 2017, 28, 265–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell. Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burtness, B.; Bauman, J.E.; Galloway, T. Novel targets in HPV-negative head and neck cancer: Overcoming resistance to EGFR inhibition. The Lancet Oncol. 2013, 14, e302–e309. [Google Scholar] [CrossRef]
- Lo Nigro, C.; Denaro, N.; Merlotti, A.; Merlano, M. Head and neck cancer: Improving outcomes with a multidisciplinary approach. Cancer Manag. Res. 2017, 9, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.E.; Patel, M.R.; Couch, M.E.; Hayes, D.N. Molecular biology of head and neck cancer: Risks and pathways. Hematol. Oncol. Clin. North. Am. 2008, 22, 1099–1124. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Somashekar, B.S.; Kamarajan, P.; Danciu, T.; Kapila, Y.L.; Chinnaiyan, A.M.; Rajendiran, T.M.; Ramamoorthy, A. Magic angle spinning NMR-based metabolic profiling of head and neck squamous cell carcinoma tissues. J. Proteome Res. 2011, 10, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Kamarajan, P.; Somashekar, B.S.; MacKinnon, N.; Chinnaiyan, A.M.; Kapila, Y.L.; Rajendiran, T.M.; Ramamoorthy, A. Delineating metabolic signatures of head and neck squamous cell carcinoma: Phospholipase A2, a potential therapeutic target. Int. J. Biochem. Cell. Biol. 2012, 44, 1852–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brohee, L.; Peulen, O.; Nusgens, B.; Castronovo, V.; Thiry, M.; Colige, A.C.; Deroanne, C.F. Propranolol sensitizes prostate cancer cells to glucose metabolism inhibition and prevents cancer progression. Sci. Rep. 2018, 8, 7050. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Bouche, G.; Sukhatme, V.; Meheus, L.; Rooman, I.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-Propranolol as an anti-cancer agent. Ecancermedicalscience 2016, 10, 680. [Google Scholar] [CrossRef] [PubMed]
- Brenner, J.C.; Graham, M.P.; Kumar, B.; Saunders, L.M.; Kupfer, R.; Lyons, R.H.; Bradford, C.R.; Carey, T.E. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck 2010, 32, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Hoover, A.C.; Spanos, W.C.; Harris, G.F.; Anderson, M.E.; Klingelhutz, A.J.; Lee, J.H. The role of human papillomavirus 16 E6 in anchorage-independent and invasive growth of mouse tonsil epithelium. Arch. Otolaryngol. Head Neck Surg. 2007, 133, 495–502. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucido, C.T.; Miskimins, W.K.; Vermeer, P.D. Propranolol Promotes Glucose Dependence and Synergizes with Dichloroacetate for Anti-Cancer Activity in HNSCC. Cancers 2018, 10, 476. https://doi.org/10.3390/cancers10120476
Lucido CT, Miskimins WK, Vermeer PD. Propranolol Promotes Glucose Dependence and Synergizes with Dichloroacetate for Anti-Cancer Activity in HNSCC. Cancers. 2018; 10(12):476. https://doi.org/10.3390/cancers10120476
Chicago/Turabian StyleLucido, Christopher T., W. Keith Miskimins, and Paola D. Vermeer. 2018. "Propranolol Promotes Glucose Dependence and Synergizes with Dichloroacetate for Anti-Cancer Activity in HNSCC" Cancers 10, no. 12: 476. https://doi.org/10.3390/cancers10120476
APA StyleLucido, C. T., Miskimins, W. K., & Vermeer, P. D. (2018). Propranolol Promotes Glucose Dependence and Synergizes with Dichloroacetate for Anti-Cancer Activity in HNSCC. Cancers, 10(12), 476. https://doi.org/10.3390/cancers10120476